Introduction
Chemotherapy continues to be the most widely
employed therapeutic option for cancer treatment. Chemotherapeutic
drugs were previously assumed to suppress body's immune system and
detrimental to the efficacy of immunotherapy because of their
nonspecific cytostatic and cytotoxic effects. However, an
increasing number of studies have reported that some cancer
chemotherapeutic drugs stimulate anticancer immune responses under
certain conditions. Doxorubicin, cyclophosphamide, gemcitabine, and
paclitaxel have been proved to induce immunogenic cell death (ICD)
in cancer through surface exposure of calreticulin and release of
adenosine triphosphate (ATP) and high-mobility group protein box-1
(HMGB-1) (1–7).
Although ICD is the most important way to trigger a
chemotherapy-induced immune response against cancer, it is not the
only way (8,9). Recent studies have demonstrated that
some chemotherapeutic drugs and agents can enhance antitumor
immunity by eliminating and inactivating immunosuppressive cells,
such as myeloid-derived suppressor cells (MDSCs), T regulatory
cells (Tregs), and tumor-associated macrophages (TAMs) (10–17).
Paradoxically, some chemotherapy agents, such as cyclophosphamide,
melphalan and doxorubicin, have been shown to induce MDSCs or TAMs
that inhibit immune responses (18–20).
Oxaliplatin (OXP) is effective against many solid
tumors and is commonly used in the treatment of colorectal cancer,
gastric cancer, pancreatic cancer and ovarian cancer. The major
antitumor mechanism is the induction of tumor cell apoptosis by
inhibition of DNA synthesis through their covalent binding to DNA
in cells (21). Apart from its the
cytotoxic properties, recent studies have shown that OXP has the
ability to increase the immunogenicity of cancer cells and induce
ICD (22). The effects of OXP on
the tumor immunosuppressive microenvironment are not clearly
understood, and the cellular identity of OXP-induced suppressor
cells has not been well studied.
Peritoneal metastasis occurs on 40% of patients with
colorectal cancer. It is one of the most common metastasis pathways
and compromises the long-term survival of patients with colorectal
cancer. OXP is a primarily used chemotherapeutic agent in the
intraperitoneal (i.p.) chemotherapy for peritoneal metastasis in
colorectal cancer patients (23,24).
In our previous study, we have reported the antitumor effect of OXP
plus IL-7 and showed that combined treatment inhibits tumor cell
growth partly by immunoregulation. In that study, we found that OXP
alone did not show antitumor effects or induce any anti-immune
reactions when intravenously administered for the treatment of lung
metastasis of colon cancer. However, in the abdominal implantation
model, OXP was intraperitoneally administered and significant
antitumor effect was observed. Interestingly, Buhtoiarov et
al (25) found that i.p. and
intravenous (i.v.) chemotherapy regimens induce different antitumor
effects and immune reactions. After reviewing the relevant studies,
we found that there had been no research on changes in the tumor
immune microenvironment when i.p. OXP administration was performed
to treat peritoneal metastasis of colon cancer. We believed that
the tumor immune microenvironment in the peritoneal metastasis
model might be exposed to a very high concentration of OXP
following i.p. administration and might affect immune cells. Thus,
in the current study, we evaluated the antitumor activity and
changes in the tumor immune microenvironment following OXP
administration in the abdominal implantation model.
Materials and methods
Cell line
The murine CT26 colon carcinoma line was purchased
from American Type Culture Collection (ATCC). CT26 cells were
inoculated in 75 cm2 culture flasks and maintained in
RPMI-1640 media supplemented with 10% FBS and 100 U/ml
penicillin.
Mouse model of colon
tumorigenesis
Pathogen-free female BALB/c mice were obtained from
Vital River Laboratory Animal Technology Co., Ltd., Beijing and
used at the age of 6–8 weeks. The mice were kept under specific
pathogen-free conditions stated in the institution guidelines. All
experiments were approved by the Animal Ethics Committee of Sichuan
University (Sichuan, China). 2×105 CT26 colon cancer
cells were injected into peritoneal cavities to build an abdominal
metastasis model. Five days after tumor inoculation, the mice were
randomized into control group and an OXP-treated group, each with
ten mice. Six of each group were used to detect the number and
weight of tumor nodules. Three of each group were used to do flow
cytometric analysis. Further, 5 mg/kg OXP or phosphate buffer
solution (PBS) was intraperitoneally injected on days 6, 9, 12, 15
after tumor inoculation. Thirteen days after the first treatment,
the animals were euthanized and tumor nodules in the peritoneal
cavity were counted and weighed.
Reagents
OXP was purchased from Sanofi S.A. (Paris, France).
Fluorescent-conjugated flow cytometry anti-mouse antibodies (Abs)
cluster of differentiation (CD)8 (cat. no. 553030; 1:100), CD69
(cat. no. 551113; 1:100), CD11b (cat. no. 553312; 1:100), Gr-1
(cat. no. 551460; 1:100) for flow cytometry were purchased from BD
Pharmingen. Antibody F4/80 (cat. no. 123110; 1:100; BD Biosciences,
Franklin Lakes, NJ, USA) for flow cytometry was purchased from
BioLegend, Inc. (San Diego, CA, USA). The mouse regulatory T cell
staining kit (cat. no. 88-8118-40; 1:100) for flow cytometry was
obtained from eBioscience.
Flow cytometric analysis
The tumor cells and spleen cells were harvested from
the mice. Tumor cells were minced and digested in a cocktail
containing collagenase i.v. (1 mg/ml) and DNase (0.1 mg/ml) in
RPMI-1640 for 1 h at 37°C. Osmotic lysis was used to remove the
erythrocytes from the spleen cells. Next, single-cell suspensions
of tumor cells and spleen cells were stained using
fluorochrome-labeled antibodies, CD4, CD8, CD69, CD11b, F4/80, and
matched isotype control antibodies. The mouse regulatory T cell
staining kit was used to stain intercellular FoxP3. After staining,
the cells were analyzed using the FACSCalibur flow-cytometer (BD
Biosciences) and the data were analyzed using the FlowJo software
version 7.6 (FlowJo LLC, Ashland, OR, USA).
Statistical analysis
GraphPad Prism version 5.0 software (GraphPad
Software, Inc., La Jolla, CA, USA) was used to perform statistical
analysis and create figures. Results are presented as the mean ±
standard error. The unpaired Student's t-test was used for paired
comparisons. P<0.05 was considered to indicate a statistically
significant difference.
Results
Inhibitory effect of i.p
OXP administration against abdominal metastasis of
colon cancer. To investigate the inhibitory effect of i.p. OXP
administration on tumor growth in case of abdominal metastasis in
mice, we established the abdominal metastasis model as mentioned
above. The mice were randomized into PBS and OXP (5
mg/kg/injection) groups and, each with 10 mice. The tumor nodules
of each mouse were cut, counted, and weighed after euthanasia. As
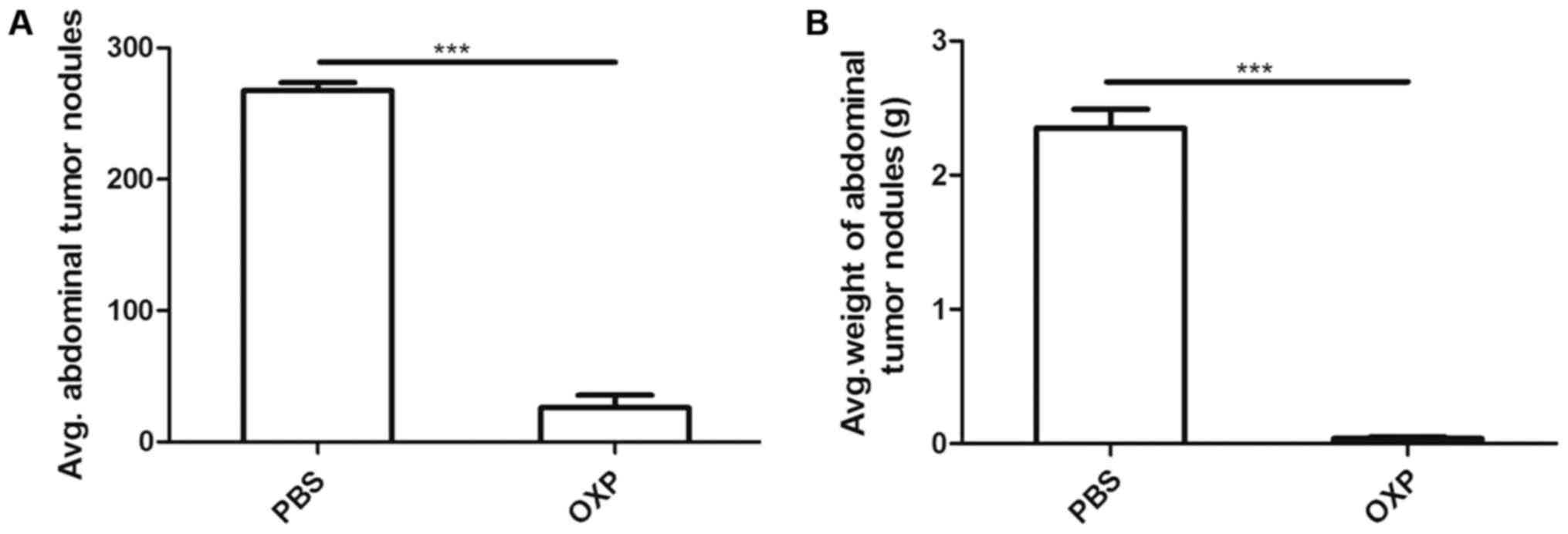
shown in Fig. 1 (n=6), i.p. OXP
administration significantly reduced the weight (0.04±0.012) g and
number (26.00±9.539) of tumor nodules compared with PBS
administration (2.350±0.142) g and (267.7±5.897) (P<0.0001).
i.p. OXP administration significantly increased
activated T cells in the tumors of the abdominal implantation
model. Tumor cells were harvested from the OXP and PBS groups and
single-cell suspensions were made. Further, activated CD8+ T cells
in each group were analyzed by flow cytometry. CD69 is a marker of
T-cell activation. We determined whether i.p. OXP administration
increases the number of activated CD8+ T cells (CD8+CD69+
population) in the tumors. As shown in Fig. 2 (n=3), i.p. OXP administration
significantly increased the number of CD8+CD69+ positive T cells in
tumor tissue compared with PBS administration (P<0.05). The
percent of CD8+CD69+T cells in the two groups were 6.135±0.125 and
4.275±0.135%, respectively.
i.p. OXP administration had no effect on Tregs
(CD4+Foxp3+T cells) of spleen in the abdominal implantation model.
The fork-head/winged helix transcription factor, FoxP3, is a
relatively specific marker for Treg cells (26). The Treg cells in the tumor are very
few and flow cytometry is difficult to detect. We detected the
tumor for Treg cells and we failed. We evaluated whether i.p. OXP
administration had effect on Treg cell populations (CD4+FoxP3+
population) of spleens in the abdominal metastasis mice. Spleens
from the two groups of mice were harvested, and the percentage of
CD4+FoxP3+ population was analyzed. The percentages of Tregs in
spleens was 22.37±3.132% in the group with i.p. administration and
23.37±2.748% in the control group with PBS administration, which
was not a significant difference (P>0.05) (Fig. 3) (n=3).
i.p. OXP administration reduced TAMs
(CD11b+F4/80high) of the tumor in the abdominal
implantation mode. We examined the effects of i.p. administration
on the TAMs (CD11b+ F4/80high) (27). Single-cell suspensions of tumor
tissue from the OXP group and PBS group were harvested and analyzed
for CD11b+F4/80high macrophages cells by flow cytometry.
As shown in Fig. 4 (n=3), OXP
administration significantly decreased the number of
CD11b+F4/80high cells in tumor tissue compared with PBS
administration (P<0.01). The percentages of
CD11b+F4/80high cells in the OXP and control groups were
0.797±0.383 and 7.743±0.69%, respectively.
i.p. OXP administration reduced MDSCs (CD11b+Gr-1+
cells) of the spleen in the abdominal implantation model. Spleens
from the two groups of mice were collected, and the percentage of
MDSCs was detected using the CD11b and Gr-1 antibodies by flow
cytometry. The percentage of MDSCs in spleens was 6.12±1.344% in
the group with i.p. OXP administration and 20.73±3.897% in the
control group with PBS administration. Thus, i.p. OXP
administration significantly decreased the number of MDSCs in
spleens compared with the control in the abdominal metastasis model
(P<0.05) (Fig. 5) (n=3).
Discussion
In vivo i.p. administration of OXP inhibited
the growth of tumors in the abdominal metastasis mouse model.
Specifically, OXP treatment significantly increased the number of
tumor-infiltrating activated CD8+ T cells, decreased the number of
CD11b+F4/80high macrophage in tumors, and decreased the
number of MDSCs in spleens. These results suggest that i.p. OXP
administration might weaken the tumor immune inhibitory
microenvironment.
In the tumor microenvironment, tumor cells can
promote immunoinhibitory pathways. This is the main reason by
virtue of which tumor cells can escape from the host immune system
and is impediment of antitumor immunotherapy (28–31).
Tregs, MDSCs, immature dendritic cells (DC), and alternatively
activated macrophages (M2) are the main immunosuppressive cells
(28,32,33).
It is one of the most important aims of cancer research to alter
the immunosuppressive tumor microenvironment by eliminating and/or
inactivating these immunosuppressive cells.
The cytotoxicity of chemotherapeutic agents is
nonspecific and can decrease lymphocytes for a short time.
Short-term lymphocytopenia may be beneficial for cancer patients
(34–36). Lympho-depletion results in a
decrease in the number of immunosuppressive cells, such as Tregs
and MDSCs. Recent studies have demonstrated that, apart from their
direct cytotoxic effects, several cytotoxic chemotherapeutic drugs
may block immunoinhibitory signal networks and enhance antitumor
immunity (31).
MDSCs, a heterogeneous population of
undifferentiated myeloid cells, accumulate in the tumor
microenvironment (37). MDSCs are
the main immunosuppressive cells in tumors, including those of
colorectal cancer (38,39). It has been demonstrated that some
anticancer agents can enhance anticancer immunity by eliminating or
inhibiting MDSCs. Gemcitabine combined with 5-fluorouracil (5FU)
has been reported to induce MDSCs apoptosis and deplete MDSCs
(15). It has also been shown that
Taxanes (docetaxel, paclitaxel) can impair the function of MDSCs
and promote MDSCs differentiation into dendritic cells (16,40).
Doxorubicin was also observed to decrease MDSCs in the spleen and
tumor tissue and weaken the inhibitory function of residual MDSCs
(11,41). These chemotherapeutic agents may
inhibit MDSCs by different mechanisms. 5FU may selectively kill
MDSCs because the expression of thymidylate synthase in MDSCs is
lower (15). Docetaxel can block
Stat3 phosphorylation and then induce development of M1 macrophages
(16). Doxorubicin may eliminate
MDSCs because the proliferation status of MDSCs is higher than that
of T cells or NK cells (11). In
our study, we demonstrated that i.p. OXP administration decreased
MDSCs in the spleen in the abdominal metastasis model. This effect
has been previously observed in patients with advanced colorectal
cancer (19). FOLFOX (folinic
acid, 5FU, and OXP) and FOLFIRI (folinic acid, 5FU, and irinotecan)
are the commonly used treatment regimens for advanced colorectal
cancer. Kanterman et al (19) revealed that FOLFOX and FOLFIRI have
opposite effects on MDSCs and immune status in patients with
advanced colorectal cancer. For example, FOLFOX reduced the
accumulation of MDSCs in blood, whereas FOLFIRI enhanced the
suppressive environment (19).
TAMs are activated by cancer cells. Studies have
shown that the proportion of TAMs is as high as 60% in the tumor
stroma (42). Most of TAMs in
tumor microenvironments are of the M2 phenotype. Macrophages can be
phenotypically polarized by the microenvironment. There are two
main polarized groups: classically activated (M1) and alternatively
activated (M2) macrophages (10).
M1 macrophages are generally considered pro-inflammatory and
secrete interleukin-12, oxygen species, nitric oxide and tumor
necrosis factor. In contrast, M2 macrophages are considered to have
anti-inflammatory effect and induce production of interleukin-10
and transforming growth factor (TGF-β). M1 and M2 macrophages can
transform into each other (43)
IL-10 and TGF-β, which are produced by various of tumor cells in
the tumor microenvironment, induce M2 polarization. TAMs can
promote tumor cell growth and metastasis, angiogenesis, adaptive
immunity and stroma formation by producing various growth factors
(44–46). Previous studies have shown that
TAMs are correlated with poor prognosis in a variety of cancers
(47,48). Thus, TAMs are now considered as a
promising target for anticancer therapy (49–51).
However, little research has been conducted on the influence of
traditional chemotherapy drugs on TAMs. Buhtoiarov et al
(25) found by employing a
combined chemotherapy regimen (vincristine, cyclophosphamide, and
doxorubicin) alone or with immunotherapy, caused the phenotype of
TAMs in tumors to change from M2 to M1. In contrast, Dijkgraaf
et al (20) found that
cisplatin or carboplatin in vitro induce M2 macrophages in
tumor cell lines. The peritoneal cavity normally harbors naïve
macrophages that play essential roles in regulating tissue repair
and inflammatory responses. Cancer cells can polarize peritoneal
macrophages toward an M2 phenotype. M2 macrophages were found to be
increased in malignant ascites in xenograft models (52–54).
Depletion of macrophages was shown to block ovarian tumor
progression and inhibit tumor-associated ascites in vivo
(55). In our study, i.p.
administration of OXP in vivo inhibited peritoneal tumor
growth and ascites accumulation in the abdominal metastasis model
and markedly decreased TAMs in tumors. It suggests that i.p. OXP
might decreased TAMs in the tumor microenvironment and induce
antitumor effects.
In addition, Tregs, another type of
immunosuppressive cell, can control anticancer immune response
(56). Cyclophosphamide,
paclitaxel, and temozolomide at low-doses have been reported to
selectively reduce the number of Tregs and improve the effect of
immunotherapy (14,57,58).
However, in our study, i.p. OXP did not show any effect on the
proportion of Tregs compared with control.
Utilizing chemotherapeutic agents to interfere with
immunosuppressive cells is an appealing strategy. Our study showed
that i.p. OXP administration may transform the immunosuppressive
microenvironment into an immuno-stimulatory microenvironment. These
findings would be useful to design more effective
chemoimmunotherapeutic strategies. In fact, OXP combined with
immunotherapy has demonstrated synergistic antitumor effects in
previous research. For example, Gonzalez-Aparicio et al
(59) found that OXP combined with
liver-specific expression of interleukin-12 reduces the
immunosuppressive microenvironment and inhibited tumor metastasis
in colorectal cancer mice models. In our previous study, we also
found that OXP plus interleukin-7 inhibited tumor growth
accompanying significant infiltration of activated T cells in the
tumor microenvironment (60).
In our previous study, we found that i.v.
administration of OXP alone exerted no antitumor effect and had no
effect on the number of TAMs (CD11b+F4/80high cells) in the lung
metastasis model of colon cancer. In this study, we found that i.p.
administration of OXP alone inhibits the growth of tumor and
decreased the number of TAMs and MDSCs in abdominal metastasis
model of colon cancer. Immune reactions induced by OXP differ with
the different routes of administration. We speculated that i.p. OXP
administration resulted in very high concentrations of OXP in the
tumor environment in the peritoneal metastasis model; hence,
different immune reactions were induced as compared with i.v. OXP
administration. Our results suggest the presence of an unrecognized
underlying mechanism when OXP is administrated via intraepithelial
route; however, further studies are required to obtain the details
of the mechanisms of immuno-stimulatory properties of OXP.
In the current, we showed that i.p. OXP
administration may induce an immune response against tumors by
weakening the immunosuppressive microenvironment in abdominal
metastasis of colon cancer in mice. These findings may aid in the
designing of more effective chemoimmunotherapeutic strategies.
Acknowledgements
Not applicable.
Funding
The present study was supported partly by the
National Natural Science Foundation of China (grant no.
81402358).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
X-CC and H-FG designed the present study, and
analyzed and interpreted the data. H-FG performed flow cytometry
and was a major contributor in writing the manuscript. LZ and JH
performed the animal experiments and flow cytometry. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
All experiments were approved by the Animal Ethics
Committee of Sichuan University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Casares N, Pequignot MO, Tesniere A,
Ghiringhelli F, Roux S, Chaput N, Schmitt E, Hamai A, Hervas-Stubbs
S, Obeid M, et al: Caspase-dependent immunogenicity of
doxorubicin-induced tumor cell death. J Exp Med. 202:1691–1701.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Galluzzi L, Senovilla L, Zitvogel L and
Kroemer G: The secret ally: Immunostimulation by anticancer drugs.
Nat Rev Drug Discov. 11:215–233. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hernandez-Alcoceba R and Berraondo P:
Immunochemotherapy against colon cancer by gene transfer of
interleukin-12 in combination with oxaliplatin. Oncoimmunology.
1:97–99. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zitvogel L and Kroemer G: Anticancer
immunochemotherapy using adjuvants with direct cytotoxic effects. J
Clin Invest. 119:2127–2130. 2009.PubMed/NCBI
|
|
5
|
Lake RA and Robinson BW: Immunotherapy and
chemotherapy-a practical partnership. Nat Rev Cancer. 5:397–405.
2005. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nowak AK, Robinson BW and Lake RA:
Gemcitabine exerts a selective effect on the humoral immune
response: Implications for combination chemo-immunotherapy. Cancer
Res. 62:2353–2358. 2002.PubMed/NCBI
|
|
7
|
Green DR, Ferguson T, Zitvogel L and
Kroemer G: Immunogenic and tolerogenic cell death. Nat Rev Immunol.
9:353–363. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Galluzzi L, Vitale I, Abrams JM, Alnemri
ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry
WS, Fulda S, et al: Molecular definitions of cell death
subroutines: Recommendations of the nomenclature committee on cell
death 2012. Cell Death Differ. 19:107–120. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zitvogel L, Galluzzi L, Smyth MJ and
Kroemer G: Mechanism of action of conventional and targeted
anticancer therapies: Reinstating immunosurveillance. Immunity.
39:74–88. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mosser DM and Edwards JP: Exploring the
full spectrum of macrophage activation. Nat Rev Immunol. 8:958–969.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Alizadeh D, Trad M, Hanke NT, Larmonier
CB, Janikashvili N, Bonnotte B, Katsanis E and Larmonier N:
Doxorubicin eliminates myeloid-derived suppressor cells and
enhances the efficacy of adoptive T-cell transfer in breast cancer.
Cancer Res. 74:104–118. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lutsiak ME, Semnani RT, De Pascalis R,
Kashmiri SV, Schlom J and Sabzevari H: Inhibition of CD4(+)25+ T
regulatory cell function implicated in enhanced immune response by
low-dose cyclophosphamide. Blood. 105:2862–2868. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Madajewicz S, Waterhouse DM, Ritch PS,
Khan MQ, Higby DJ, Leichman CG, Malik SK, Hentschel P, Gill JF,
Zhao L and Nicol SJ: Multicenter, randomized phase II trial of
bevacizumab plus folinic acid, fluorouracil, gemcitabine (FFG)
versus bevacizumab plus folinic acid, fluorouracil, oxaliplatin
(FOLFOX4) as first-line therapy for patients with advanced
colorectal cancer. Invest New Drugs. 30:772–778. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang L, Dermawan K, Jin M, Liu R, Zheng
H, Xu L, Zhang Y, Cai Y, Chu Y and Xiong S: Differential impairment
of regulatory T cells rather than effector T cells by
paclitaxel-based chemotherapy. Clin Immunol. 129:219–229. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Vincent J, Mignot G, Chalmin F, Ladoire S,
Bruchard M, Chevriaux A, Martin F, Apetoh L, Rébé C and
Ghiringhelli F: 5-Fluorouracil selectively kills tumor-associated
myeloid-derived suppressor cells resulting in enhanced T
cell-dependent antitumor immunity. Cancer Res. 70:3052–3061. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kodumudi KN, Woan K, Gilvary DL, Sahakian
E, Wei S and Djeu JY: A novel chemoimmunomodulating property of
docetaxel: suppression of myeloid-derived suppressor cells in tumor
bearers. Clin Cancer Res. 16:4583–4594. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Suzuki E, Kapoor V, Jassar AS, Kaiser LR
and Albelda SM: Gemcitabine selectively eliminates splenic
Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and
enhances antitumor immune activity. Clin Cancer Res. 11:6713–6721.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ding ZC, Lu X, Yu M, Lemos H, Huang L,
Chandler P, Liu K, Walters M, Krasinski A, Mack M, et al:
Immunosuppressive myeloid cells induced by chemotherapy attenuate
antitumor CD4+ T-cell responses through the PD-1-PD-L1 axis. Cancer
Res. 74:3441–3453. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kanterman J, Sade-Feldman M, Biton M,
Ish-Shalom E, Lasry A, Goldshtein A, Hubert A and Baniyash M:
Adverse immunoregulatory effects of 5FU and CPT11 chemotherapy on
myeloid-derived suppressor cells and colorectal cancer outcomes.
Cancer Res. 74:6022–6035. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dijkgraaf EM, Heusinkveld M, Tummers B,
Vogelpoel LT, Goedemans R, Jha V, Nortier JW, Welters MJ, Kroep JR
and van der Burg SH: Chemotherapy alters monocyte differentiation
to favor generation of cancer-supporting M2 macrophages in the
tumor microenvironment. Cancer Res. 73:2480–2492. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Graham J, Mushin M and Kirkpatrick P:
Oxaliplatin. Nat Rev Drug Discov. 3:11–12. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tesniere A, Schlemmer F, Boige V, Kepp O,
Martins I, Ghiringhelli F, Aymeric L, Michaud M, Apetoh L, Barault
L, et al: Immunogenic death of colon cancer cells treated with
oxaliplatin. Oncogene. 29:482–491. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gervais MK, Dubé P, McConnell Y, Drolet P,
Mitchell A and Sideris L: Cytoreductive surgery plus hyperthermic
intraperitoneal chemotherapy with oxaliplatin for peritoneal
carcinomatosis arising from colorectal cancer. J Surg Oncol.
108:438–443. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Elias D, Gilly F, Boutitie F, Quenet F,
Bereder JM, Mansvelt B, Lorimier G, Dubè P and Glehen O: Peritoneal
colorectal carcinomatosis treated with surgery and perioperative
intraperitoneal chemotherapy: Retrospective analysis of 523
patients from a multicentric French study. J Clin Oncol. 28:63–68.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Buhtoiarov IN, Sondel PM, Wigginton JM,
Buhtoiarova TN, Yanke EM, Mahvi DA and Rakhmilevich AL: Anti-tumour
synergy of cytotoxic chemotherapy and anti-CD40 plus CpG-ODN
immunotherapy through repolarization of tumour-associated
macrophages. Immunology. 132:226–239. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hori S, Nomura T and Sakaguchi S: Control
of regulatory T cell development by the transcription factor Foxp3.
Science. 299:1057–1061. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pucci F, Venneri MA, Biziato D, Nonis A,
Moi D, Sica A, Di Serio C, Naldini L and De Palma M: A
distinguishing gene signature shared by tumor-infiltrating
Tie2-expressing monocytes, blood ‘resident’ monocytes, and
embryonic macrophages suggests common functions and developmental
relationships. Blood. 114:901–914. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gabrilovich DI and Nagaraj S:
Myeloid-derived suppressor cells as regulators of the immune
system. Nat Rev Immunol. 9:162–174. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mundy-Bosse BL, Lesinski GB, Jaime-Ramirez
AC, Benninger K, Khan M, Kuppusamy P, Guenterberg K, Kondadasula
SV, Chaudhury AR, La Perle KM, et al: Myeloid-derived suppressor
cell inhibition of the IFN response in tumor-bearing mice. Cancer
Res. 71:5101–5110. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wesolowski R, Markowitz J and Carson WE
III: Myeloid derived suppressor cells-a new therapeutic target in
the treatment of cancer. J Immunother Cancer. 1:102013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Alizadeh D and Larmonier N:
Chemotherapeutic targeting of cancer-induced immunosuppressive
cells. Cancer Res. 74:2663–2668. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zitvogel L, Tesniere A and Kroemer G:
Cancer despite immunosurveillance: Immunoselection and
immunosubversion. Nat Rev Immunol. 6:715–727. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Colombo MP and Piconese S:
Regulatory-T-cell inhibition versus depletion: The right choice in
cancer immunotherapy. Nat Rev Cancer. 7:880–887. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Restifo NP, Dudley ME and Rosenberg SA:
Adoptive immunotherapy for cancer: Harnessing the T cell response.
Nat Rev Immunol. 12:269–281. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Dudley ME, Wunderlich JR, Robbins PF, Yang
JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry R, Restifo NP,
Hubicki AM, et al: Cancer regression and autoimmunity in patients
after clonal repopulation with antitumor lymphocytes. Science.
298:850–854. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wrzesinski C, Paulos CM, Gattinoni L,
Palmer DC, Kaiser A, Yu Z, Rosenberg SA and Restifo NP:
Hematopoietic stem cells promote the expansion and function of
adoptively transferred antitumor CD8 T cells. J Clin Invest.
117:492–501. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gabrilovich DI, Ostrand-Rosenberg S and
Bronte V: Coordinated regulation of myeloid cells by tumours. Nat
Rev Immunol. 12:253–268. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sun HL, Zhou X, Xue YF, Wang K, Shen YF,
Mao JJ, Guo HF and Miao ZN: Increased frequency and clinical
significance of myeloid-derived suppressor cells in human
colorectal carcinoma. World J Gastroenterol. 18:3303–3309.
2012.PubMed/NCBI
|
|
39
|
Kanterman J, Sade-Feldman M and Baniyash
M: New insights into chronic inflammation-induced
immunosuppression. Semin Cancer Biol. 22:307–318. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Michels T, Shurin GV, Naiditch H, Sevko A,
Umansky V and Shurin MR: Paclitaxel promotes differentiation of
myeloid-derived suppressor cells into dendritic cells in vitro in a
TLR4-independent manner. J Immunotoxicol. 9:292–300. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Mattarollo SR, Loi S, Duret H, Ma Y,
Zitvogel L and Smyth MJ: Pivotal role of innate and adaptive
immunity in anthracycline chemotherapy of established tumors.
Cancer Res. 71:4809–4820. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Heusinkveld M and van der Burg SH:
Identification and manipulation of tumor associated macrophages in
human cancers. J Transl Med. 9:2162011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sica A and Mantovani A: Macrophage
plasticity and polarization: In vivo veritas. J Clin Invest.
122:787–795. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kuang DM, Zhao Q, Peng C, Xu J, Zhang JP,
Wu C and Zheng L: Activated monocytes in peritumoral stroma of
hepatocellular carcinoma foster immune privilege and disease
progression through PD-L1. J Exp Med. 206:1327–1337. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Galdiero MR, Garlanda C, Jaillon S, Marone
G and Mantovani A: Tumor associated macrophages and neutrophils in
tumor progression. J Cell Physiol. 228:1404–1412. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lievense LA, Bezemer K, Aerts JG and
Hegmans JP: Tumor-associated macrophages in thoracic malignancies.
Lung Cancer. 80:256–262. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhang H, Wang X, Shen Z, Xu J, Qin J and
Sun Y: Infiltration of diametrically polarized macrophages predicts
overall survival of patients with gastric cancer after surgical
resection. Gastric Cancer. 18:740–750. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhang M, He Y, Sun X, Li Q, Wang W, Zhao A
and Di W: A high M1/M2 ratio of tumor-associated macrophages is
associated with extended survival in ovarian cancer patients. J
Ovarian Res. 7:192014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Sica A, Schioppa T, Mantovani A and
Allavena P: Tumour-associated macrophages are a distinct M2
polarised population promoting tumour progression: Potential
targets of anti-cancer therapy. Eur J Cancer. 42:717–727. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Tang X, Mo C, Wang Y, Wei D and Xiao H:
Anti-tumour strategies aiming to target tumour-associated
macrophages. Immunology. 138:93–104. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Nywening TM, Wang-Gillam A, Sanford DE,
Belt BA, Panni RZ, Cusworth BM, Toriola AT, Nieman RK, Worley LA,
Yano M, et al: Targeting tumour-associated macrophages with CCR2
inhibition in combination with FOLFIRINOX in patients with
borderline resectable and locally advanced pancreatic cancer: A
single-centre, open-label, dose-finding, non-randomised, phase 1b
trial. Lancet Oncol. 17:651–662. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wang X, Deavers M, Patenia R, Bassett RL
Jr, Mueller P, Ma Q, Wang E and Freedman RS: Monocyte/macrophage
and T-cell infiltrates in peritoneum of patients with ovarian
cancer or benign pelvic disease. J Transl Med. 4:302006. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Robinson-Smith TM, Isaacsohn I, Mercer CA,
Zhou M, Van Rooijen N, Husseinzadeh N, McFarland-Mancini MM and
Drew AF: Macrophages mediate inflammation-enhanced metastasis of
ovarian tumors in mice. Cancer Res. 67:5708–5716. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ko SY, Ladanyi A, Lengyel E and Naora H:
Expression of the homeobox gene HOXA9 in ovarian cancer induces
peritoneal macrophages to acquire an M2 tumor-promoting phenotype.
Am J Pathol. 184:271–281. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Bak SP, Walters JJ, Takeya M,
Conejo-Garcia JR and Berwin BL: Scavenger receptor-A-targeted
leukocyte depletion inhibits peritoneal ovarian tumor progression.
Cancer Res. 67:4783–4789. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Shevach EM: Mechanisms of foxp3+ T
regulatory cell-mediated suppression. Immunity. 30:636–645. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Ghiringhelli F, Larmonier N, Schmitt E,
Parcellier A, Cathelin D, Garrido C, Chauffert B, Solary E,
Bonnotte B and Martin F: CD4+CD25+ regulatory T cells suppress
tumor immunity but are sensitive to cyclophosphamide which allows
immunotherapy of established tumors to be curative. Eur J Immunol.
34:336–344. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Banissi C, Ghiringhelli F, Chen L and
Carpentier AF: Treg depletion with a low-dose metronomic
temozolomide regimen in a rat glioma model. Cancer Immunol
Immunother. 58:1627–1634. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Gonzalez-Aparicio M, Alzuguren P, Mauleon
I, Medina-Echeverz J, Hervas-Stubbs S, Mancheno U, Berraondo P,
Crettaz J, Gonzalez-Aseguinolaza G, Prieto J and Hernandez-Alcoceba
R: Oxaliplatin in combination with liver-specific expression of
interleukin 12 reduces the immunosuppressive microenvironment of
tumours and eradicates metastatic colorectal cancer in mice. Gut.
60:341–349. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Gou HF, Huang J, Shi HS, Chen XC and Wang
YS: Chemo-immunotherapy with oxaliplatin and interleukin-7 inhibits
colon cancer metastasis in mice. PLoS One. 9:e857892014. View Article : Google Scholar : PubMed/NCBI
|