Introduction
Bladder cancer is the fourth most common cancer in
men and the ninth most common cancer in women worldwide (1). The majority of bladder cancers are
urothelial cell carcinoma, which originate from the epithelial
lining of the bladder wall (2).
More than half of patients with bladder cancer are diagnosed with
advanced stage cancer and have very poor prognosis (3). Despite improvement in treatment of
bladder cancer, the incidence of the disease is still increasing
(4). Although environmental and
genetic factors have been demonstrated to have important roles in
the development of bladder cancer, the molecular mechanisms
involved in the initiation and progression of the disease remain
unclear. Investigating the underlying mechanisms of bladder cancer
is instrumental for providing better treatment and development of
novel therapeutic agents.
The tumor necrosis factor receptor superfamily
member 14 (TNFRSF14) gene, also known as herpes virus entry
mediator, is located on the short arm of chromosome 1p36 and
encodes a type I trans membrane molecule that serves as a molecular
switch by interacting with different ligands to regulate a series
of immune responses (5).
Activation of TNFRSF14 is involved in the development of various
tumor types, and may be used for assessing prognosis as it is
closely associated with cancer growth and metastasis (6,7). In
a previous study, reduced TNFRSF14 expression was suggested to
contribute to the pathobiology of classical Hodgkin lymphoma
(8). Another previous study
indicated that TNFRSF14 deficiency promotes development of
follicular lymphoma in vivo and establishes a favorable
immune environment (9). Cheung
et al (10) reported that
somatic TNFRSF14 mutations are associated with worse prognosis of
follicular lymphoma. However, the expression levels of TNFRSF14 in
other types of cancer types have been inconsistent. To the best of
our knowledge, there is no published evidence on the expression of
TNFRSF14 in bladder cancer tissues and its association with
clinicopathological parameters remains poorly understood.
In the present study, the aim was to investigate the
expression levels of TNFRSF14 in bladder cancer and evaluate its
clinical significance. In addition, the molecular mechanism of
TNFRSF14 in regulating progression of bladder cancer was examined.
The results indicated that increased expression of TNFRSF14
inhibited bladder cancer proliferation by promoting apoptosis.
Materials and methods
Database
The Cancer Genome Atlas (TCGA) database was utilized
to obtain clinical information and Level 3 RNA sequencing (RNA-Seq)
data. The search terms used in TCGA contained the following
keywords: ‘cases’, ‘primary site’, ‘bladder’ and ‘TCGA-BLCA’. Based
on the above search terms, the clinical information and Level 3
RNA-Seq data were obtained from 404 bladder cancer samples and 28
normal bladder tissue samples (11). TNFRSF14 expression levels in normal
and bladder cancer tissue were analyzed using the limma package
(version 3.32.8) from Bioconductor (12).
Cell cultures
T24 and SW780 bladder cancer cell lines, and
SV-HUC-1 normal human bladder epithelium cell line were purchased
from the Shanghai Cell Bank at the Chinese Academy of Sciences
(Shanghai, China). SV-HUC-1 cells were cultured in Corning™
cellgro™ F12K (Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 10% fetal bovine serum (FBS; Gibco; Thermo Fisher
Scientific, Inc.). T24 and SW780 bladder cancer cell lines, and
SV-HUC-1 cells were cultured in a humidified atmosphere of 5%
CO2 at 37°C. The EJ-M3 cell line was established from
the EJ cell line, which has been demonstrated to be a T24
derivative/contaminants. EJ-M3 cells are essentially highly
invasive T24 derivative cells (13) and were obtained from EJ cells
(14). EJ-M3 cells were generated,
according to the methods described by Girnita et al
(15). Briefly, 24-well plates
were filled with media containing chemokine, and Transwell inserts
coated with 100 µg Matrigel were placed into the wells and
3×105 EJ cells were added onto the membrane. After 7 h
in culture, the insert was removed and cells invading the lower
chamber were further cultured. The medium was changed after 4–6
days. After reaching lamellar fusion, the cells were digested and
subcultured. This process was repeated three times and each time
the amount of Matrigel coating on the insert membrane was increased
by 50 µg. Cells that were retained following this selection process
were identified highly invasive EJ-M3 cells. These cancer cells
were cultured in RPMI-1640 (HyClone; GE Healthcare, Chicago, IL,
USA) supplemented with 10% FBS (Gibco; Thermo Fisher Scientific,
Inc.), 100 U/ml penicillin, and 0.1 mg/ml streptomycin
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany). Cells were
cultured at 37°C in a humidified atmosphere containing 5%
CO2 until 70–80% confluence was reached. Non-adherent
cells were washed away after 3 days of culture and adherent cells
were fed with fresh complete medium. The cells were subcultured at
a 1:2 split ratio after reaching confluence.
Transient transfection and small
interfering RNA (siRNA) transfection
Cells were cultured in 6-well plates in medium
without antibiotics for 24 h prior to transfection, resulting in
70–80% confluence. For transient transfection, the TNFRSF14 gene
was subcloned into a pSG5-HA vector, generating pSG5-HA-TNFRSF14.
The empty vector was used as a negative control for transfection.
Cells were transfected by Lipofectamine® 2000
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. Specifically, cells were cultured in fresh
medium without antibiotics, and when the T24 cells reached 70–80%
confluence, 4 µg DNA (pSG5-HA-TNFRSF14) was combined with 250 µl
Opti-MEM® medium (Invitrogen; Thermo Fisher Scientific,
Inc.) and 10 µl Lipofectamine® 2000 was diluted in 240
µl Opti-MEM®, which were incubated at room temperature
for 5 min. DNA/Opti-MEM® and Lipofectamine®
2000/Opti-MEM® were combined and incubated at room
temperature for 20 min. A total of 500 µl
plasmid/Lipofectamine® complex was added to the cells,
which were cultured at 37°C. Following 24 h, the transfection
medium was replaced with fresh complete medium and cells were
harvested for analysis of proliferation and other experiments.
siRNA against TNFRSF14 (si-TNFRSF14; cat. no.
stQ0003897-1) and negative control (si-NC; cat. no. siN05815122147)
were produced and purchased from Guangzhou RiboBio Co., Ltd.
(Guangzhou, China). The negative control siRNA does not match any
known mammalian GenBank sequences. Cells were seeded at a density
of 3×105/well in 6-well plates overnight and transfected
with siRNA or si-NC at a final concentration of 100 nM using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.). Following 24 h of transfection, reverse
transcription-quantitative polymerase chain reaction (RT-qPCR) was
used to determine transfection efficiency.
RNA extraction and RT-qPCR
RNA was extracted from cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.). cDNA was generated using PrimeScript RT reagent kit (cat.
no. RR037A; Takara Biotechnology Co., Ltd., Dalian, China) at 37°C
for 15 min. Synthesized first-strand cDNA was used as template, and
GAPDH was applied for normalization. Primer sequences were used as
follows: TNFRSF14 forward, 5′-CCAAGTGCAGTCCAGGTTAT-3′ and reverse,
5′-ATTGAGGTGGGCAATGTAGG-3′; GAPDH forward,
5′-GGTGTGAACCATGAGAAGTATGA-3′ and reverse,
5′-GAGTCCTTCCACGATACCAAAG-3′. The RT-qPCR reaction was performed at
95°C for 5 min, followed by 40 cycles of 95°C for 30 sec, 60°C for
45 sec and 72°C for 30 min using an ABI 7500 real-time PCR system
(Applied Biosystems; Thermo Fisher Scientific, Inc.) using SYBR
Select Master Mix (Applied Biosystems; Thermo Fisher Scientific,
Inc.) following the manufacturer's protocol The relative expression
levels of TNFRSF14 were analyzed and normalized to GAPDH using the
2−ΔΔCq method (16).
Cell proliferation analysis
The Cell Counting kit-8 (CCK-8) assay is a sensitive
and accurate method for determining cell viability. In the present
study, cell proliferation was evaluated using the CCK-8 assay at
24, 48 and 72 h after transfection, according to a previous study
(17). Briefly, cells in the
TNFRSF14 or si-TNFRSF14 transfection group and control transfection
group were cultured in 100 µl RPMI-1640 supplemented with 10% FBS
in 96-well plates and incubated at 37°C for 3 days. Subsequently,
CCK-8 dye (BestBio, Shanghai, China), diluted 1:10 in cell culture
medium, was added to each well and incubated for 2 h at 37°C. The
absorbance was read using a microplate reader set at 450 nm. The
450 nm optical density value is proportional to the total number of
live cells and was used to assess cell viability in transfected vs.
control transfected cells. Triplicate wells were used and
experiments were repeated at least three times.
Western blot analysis
Total protein was extracted from transfected and
control transfected cells using 1 mM phenylmethylsulfonyl fluoride
in 1 ml ice-cold radioimmunoprecipitation assay buffer (Beyotime
Institute of Biotechnology, Haimen, China). Subsequently, protein
centration was measured using the bicinchoninic acid assay.
Denatured proteins (20 µg) were separated using 12% SDS-PAGE and
transferred onto Immobilon-P polyvinylidene fluoride membranes (EMD
Millipore, Billerica, MA, USA). Membranes were blocked for 2 h with
5% skimmed milk powder in Tris-buffered saline containing 0.1%
Tween-20 (TBST) and incubated overnight at 4°C with the following
primary antibodies: Anti-protein kinase B (AKT; 1:1,000; cat. no.
SAB4500797), anti-phosphorylated (p)-AKT (1:1,000; cat. no.
SAB4301414), anti-P70 S6 kinase (P70; 1:1,000; cat. no.
SAB4502683), anti-active caspase3-p17 (1:1,000; cat. no.
SAB4503294) and anti-GAPDH (1:5,000; cat. no. SAB2108266;
Sigma-Aldrich; Merck KGaA). Subsequently, membranes were washed
three times with TBST at room temperature, followed by incubation
with an horseradish peroxidase-conjugated anti-rabbit IgG secondary
antibody (1:5,000; cat. no. sc-2004; Santa Cruz Biotechnology,
Inc., Dallas, TX, USA) at room temperature for 2 h. Membranes were
washed a final time with TBST. GAPDH was used as the loading
control. All bands were detected using Enhanced Chemiluminescent
Western Blotting Detection kit (GE Healthcare Life Sciences, Little
Chalfont, UK). Imaging and quantification of protein bands was
conducted using the Bio-Rad Quantity One 1-D Analysis software
version 4.6.9 (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Apoptosis assay
Following 24 h of transfection, cells were harvested
by trypsinization, washed with pre-cooled PBS and fixed overnight
with 70% ethanol at −20°C. T24 or EJ-M3 cells were washed twice,
resuspended in PBS containing 0.1 mg/ml RNase A and 0.1% Triton
X-100 in the dark for 30 min at 37°C. A total of 1×106
cells were double stained with Annexin V-fluorescein isothiocyanate
(FITC) and propidium iodide (PI) using the Apoptosis Detection kit
(BD Biosciences, Franklin Lakes, NJ, USA), according to the
manufacturer's protocol. The stained cells were acquired on a
FACScan flow cytometer (BD Biosciences) equipped with a 488 nm
argon laser and the data were analyzed using Cell Quest software
version 6.0 (BD Biosciences). Cells were categorized into viable
(Annexin V-FITC−/PI−), dead (Annexin
V-FITC−/PI+), early apoptotic (Annexin
V-FITC+/PI−) or apoptotic cells (Annexin
V-FITC+/PI+). The percentage of apoptotic
cells in the experiment group was compared with the control
transfection group. All the samples were measured in
triplicate.
Statistical analysis
In the present study, SPSS software version 18.0
(SPSS, Inc., Chicago, IL, USA) was used to conduct all statistical
analyses. Each assay was performed at least three times. The data
are presented as the mean ± standard deviation. The means between
two groups were compared using Student's t-test. One-way analysis
of variance followed by Student-Newman-Keuls post hoc test was used
to compare the means of multiple groups. The Kaplan-Meier method
was used to evaluate the prognostic value of TNFRSF14 in bladder
cancer, and the Mantel-Cox log-rank test was used to determine the
statistical significance of difference between survival curves.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Downregulation of TNFRSF14 expression
levels in human bladder cancer tissues is associated with poor
prognosis
The association between TNFRSF14 expression levels
and prognosis of bladder cancer was assessed using RNA-Seq data
from the TCGA database. The results indicated that TNFRSF14
expression levels were downregulated in the bladder tissues of
patients with bladder cancer compared with healthy controls
(Fig. 1A; P<0.05). Based on the
median value of TNFRSF14 expression, patients were divided into
TNFRSF14 low and TNFRSF14 high expression groups. As shown in
Fig. 1B, the overall survival was
significantly longer in the TNFRSF14 high expression group compared
with in the TNFRSF14 low expression group.
TNFRSF14 expression levels are
downregulated in bladder cancer cell lines
TNFRSF14 expression levels in three human bladder
cancer cell lines (T24, SW780 and EJ-M3) and a normal human bladder
epithelium cell line (SV-HUC-1) were measured by RT-qPCR. The
results demonstrated TNFRSF14 expression levels were lower in all
bladder cancer cell lines compared with in the SV-HUC-1 cell line
(Fig. 2). Notably, the expression
levels of TNFRSF14 in T24 cells were lower compared with in SW780
and EJ-M3 cells. Therefore, T24 cells were used to perform
overexpression experiments, whereas EJ-M3 cells were used to
conduct silencing assays.
CCK-8 analysis of cell viability
To investigate the role of TNFRSF14 in T24 or EJ-M3
cell proliferation, overexpression and silencing experiments were
conducted using pSG5-HA-TNFRSF14 and si-TNFRSF14, respectively.
RT-qPCR analysis was conducted 72 h after transfection to measure
transfection efficacy. The results in Fig. 3A indicated that TNFRSF14 expression
levels were significantly increased in T24 cells after transfection
(P<0.05). As demonstrated in Fig.
4A, TNFRSF14 expression levels were decreased in EJ-M3 cells
after gene silencing.
Subsequently, the effect of TNFRSF14 overexpression
on cell proliferation was evaluated using the CCK-8 assay at 24, 48
and 72 h after transfection. As demonstrated in Fig. 3B, TNFRSF14 transfection led to a
reduction in cell viability of T24 cells, which was statistically
significant compared with control transfection group after 72 h of
transfection (P<0.05; Fig. 3B).
The results indicated TNFRSF14 overexpression decreased cell
proliferation of bladder cancer cells.
In addition, CCK-8 assay was used to measure the
effect of TNFRSF14 silencing on cell proliferation of EJ-M3 cells
after 24, 48 and 72 h. Compared with control transfected cells,
TNFRSF14 silencing resulted in increased cell viability of EJ-M3
cells, particularly at 72 h after transfection (P<0.05; Fig. 4B). The results indicated that
TNFRSF14 silencing increased cell proliferation of bladder cancer
cells.
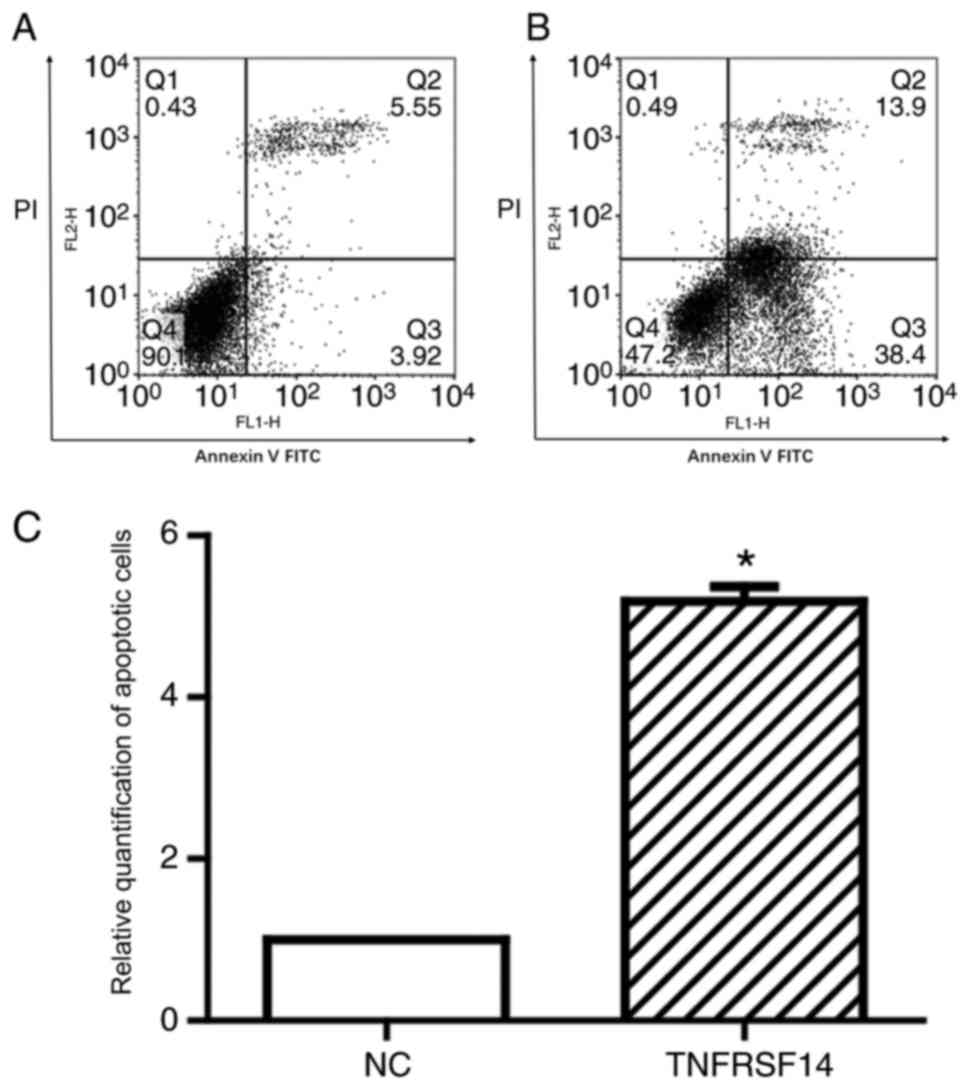
Apoptosis assay
To evaluate the effect of alterations in TNFRSF14
expression levels on apoptosis of T24 or EJ-M3 cells, Annexin
V-FITC and PI double staining was utilized to detect apoptotic
cells, including early (Annexin V-FITC+/PI−)
and late apoptotic (Annexin V-FITC+/PI+)
cells. As presented in Fig. 5,
TNFRSF14 overexpression promoted increased apoptosis in transfected
cells compared with in control cells (5.52 vs. 1; P<0.05),
indicating that TNFRSF14 overexpression may have a direct
therapeutic effect against the human T24 bladder cancer cell line.
As demonstrated in Fig. 6,
TNFRSF14 silencing decreased apoptosis in transfected cells
compared with in control cells (0.51 vs. 1; P<0.05). The results
suggested that suppression of cell viability following TNFRSF14
overexpression was associated with induction of cell apoptosis.
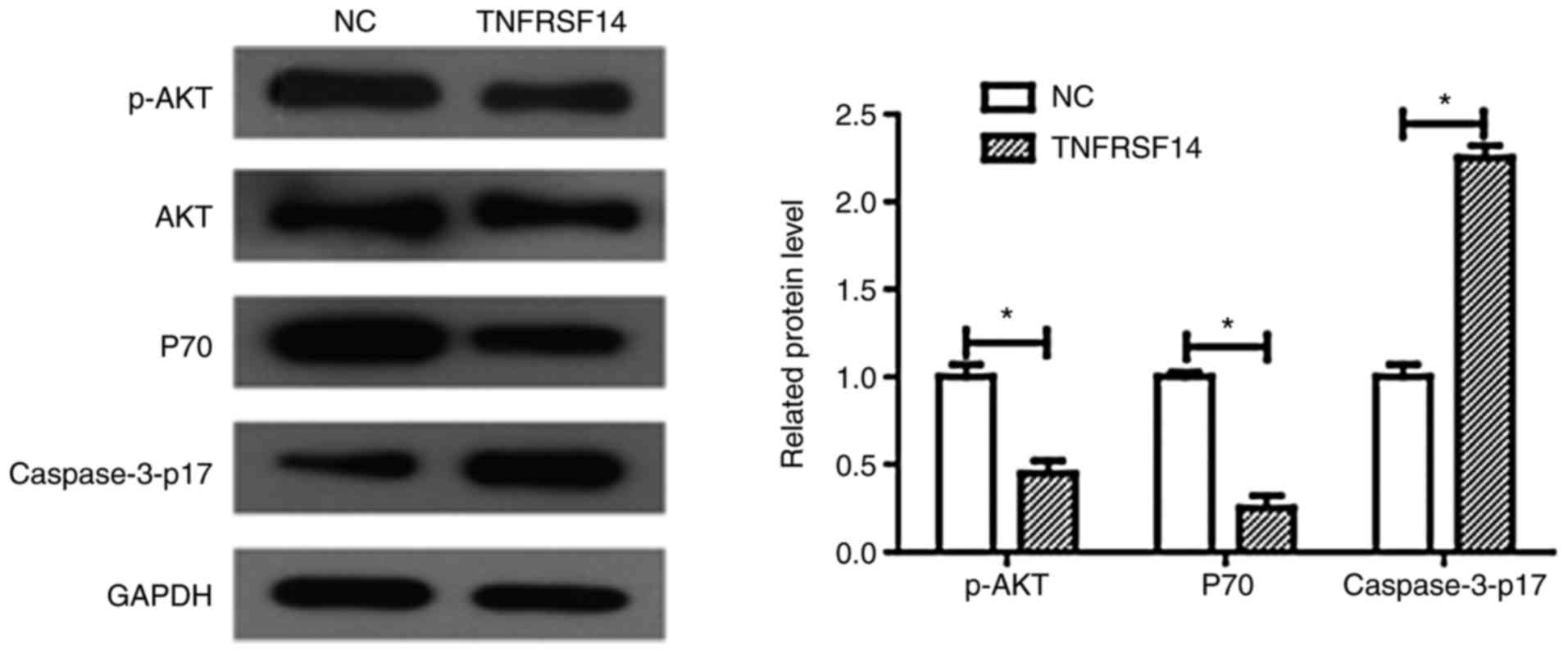
Phosphatidylinositol 3-kinase (PI3K)
signaling pathway
To determine whether abnormalities in TNFRSF14
expression levels affect PI3K signaling, numerous proteins were
detected using western blot analysis, including p-AKT, caspase3-p17
and P70. As demonstrated in Fig.
7, TNFRSF14 overexpression significantly increased caspase3-p17
expression levels in T24 cells (P<0.05), which was in line with
the cell apoptosis results obtained in the present study.
Furthermore, TNFRSF14 overexpression significantly decreased p-AKT
and P70 expression levels in the T24 bladder cancer cell line
(Fig. 7B, P<0.05). Conversely,
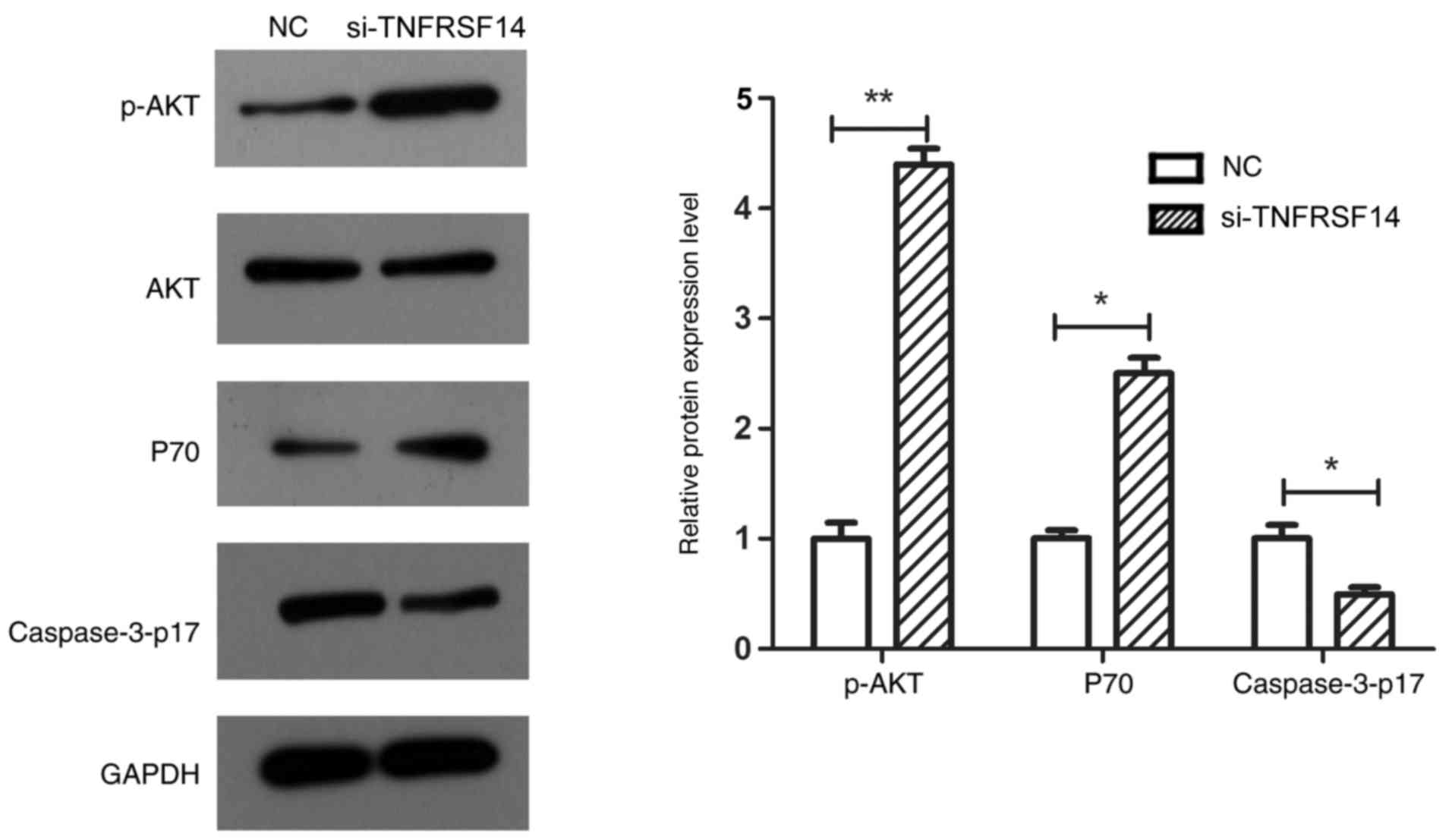
TNFRSF14 silencing significantly decreased caspase3-p17 expression
levels in EJ-M3 cells (P<0.05), which was again in line with the
cell apoptosis results. Furthermore, TNFRSF14 silencing
significantly increased p-AKT (P<0.01) and P70 (P<0.05)
expression levels in the EJ-M3 bladder cancer cell line (Fig. 8).
Discussion
Human bladder cancer is one of the most lethal human
cancers worldwide. Systematic chemotherapy and surgery are the
primary treatment options for bladder cancer (18,19);
however, bladder cancer cells frequently develop drug resistance
and ~50% of patients with advanced bladder cancer do not respond to
chemotherapy. Chemopreventative drugs may inhibit and delay the
initiation of cancer types via various mechanisms, including
anti-proliferation or pro-apoptosis. Notably, TNFRSF14 activation
has been demonstrated to inhibit proliferation of adenocarcinoma
cells (20) and regulate apoptosis
(21), suggesting a potential
tumor suppressive role. To the best of our knowledge, the role of
TNFRSF14 in bladder cancer has not yet been examined, and
investigating its underlying mechanisms of action may aid in the
development of novel diagnostic and therapeutic approaches to
bladder cancer.
In the present study, it was demonstrated that
expression levels of TNFRSF14 were decreased in bladder cancer
tissue compared with negative control tissue, using the TCGA
database. Patients with bladder cancer exhibiting low expression
levels of TNFRSF14 had poorer prognosis compared with those
exhibiting high expression levels of TNFRSF14. Similarly, TNFRSF14
expression levels were additionally reduced in bladder cancer cell
lines. Overexpression of TNFRSF14 in T24 cells stimulated apoptosis
and inhibited proliferation in vitro, which may indicate the
potential protective effects of TNFRSF14 in bladder cancer.
Conversely, TNFRSF14 knockdown in EJ-M3 cells enhanced cell
proliferation, inhibited cell apoptosis, increased the expression
levels of p-AKT and P70, and decreased the expression levels of
caspase3-p17. Overall, the results demonstrated that TNFRSF14 may
act as a tumor suppressor gene in bladder cancer, which is in line
with a previous study (20).
Apoptosis is a form of programmed cell death, which
serves a key role in tissue homeostasis and development in
multicellular organisms (22). An
imbalance between cell proliferation and apoptosis may lead to
fatal diseases, including cancer (23). In various cell-based models of
cancer, cell death triggered by a stimulus may be switched off by
suppressing control points in the cell death pathway; for example,
inhibition of caspase activation (24). Activation of caspase-3 results in
cleavage of numerous proteins, including poly(ADP-ribose)
polymerase (PARP). PARP is a nuclear DNA-binding zinc finger
protein, which is important for DNA repair as well as other
cellular processes, including cell differentiation, proliferation
and apoptosis (25). Cleaved PARP
is a primary indicator of apoptosis. In the present study, the
results demonstrated that TNFRSF14 overexpression in T24 cells
increased the expression levels of caspase-3 and corresponded to an
increased number of apoptotic cells. Therefore, TNFRSF14 may block
bladder cancer progression by increasing the expression levels of
caspase-3, thereby promoting apoptosis and suppressing
proliferation of bladder cancer cells.
The PI3K pathway has a pivotal role in cell growth,
proliferation and survival (26),
and is upregulated in a number of different types of cancer
(27). Notably, the PI3K pathway
is activated in bladder cancer (28). AKT occupies an important regulatory
node in the PI3K pathway, below which the pathway branches
significantly to influence a wide range of cellular processes that
promote cell-cycle progression, cell growth and resistance to
apoptosis. PI3K-AKT signaling has been demonstrated to have an
important role in transforming growth factor β-mediated
epithelial-to-mesenchymal transition (29), which is a key process in bladder
cancer development (30). A
previous study suggested that downregulation of p-AKT induces
caspase-3-dependent apoptosis in cancer cells (31,32),
which is consistent with the results obtained in the present study.
TNFRSF14 overexpression significantly increased the expression
levels of caspase-3, but significantly decreased the expression
levels of p-AKT and P70 in T24 bladder cancer cells. Silencing of
TNFRSF14 yielded opposite results. The results suggested that
TNFRSF14-mediated apoptosis is regulated by a caspase-dependent
cascade that blocks PI3K/AKT signaling and activates other specific
signaling pathways.
In conclusion, to the best of our knowledge, this is
the first study of its kind to investigate the prognostic value of
TNFRSF14 in bladder cancer. The results indicated TNFRSF14
expression levels were decreased in bladder cancer tissue compared
with in normal control tissues using the TCGA database. Patients
with bladder cancer exhibiting low expression levels of TNFRSF14
had worse prognosis compared with those exhibiting high expression
levels of TNFRSF14. Therefore, TNFRSF14 may act as a tumor
suppressor in bladder cancer and serve as a novel prognostic
biomarker. Nevertheless, there are limitations to the present
study. The association between TNFRSF14 expression levels and
prognosis of bladder cancer was determined using RNA-Seq data from
the TCGA database. Further studies using patient tissues are
required to validate the present findings.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YZ performed the experiments, analyzed the data and
drafted the manuscript; M-YL conceived the study and revised the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Luo M, Li Z, Wang W, Zeng Y, Liu Z and Qiu
J: Long non-coding RNA H19 increases bladder cancer metastasis by
associating with EZH2 and inhibiting E-cadherin expression. Cancer
Lett. 333:213–221. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bo J, Yang G, Huo K, Jiang H, Zhang L, Liu
D and Huang Y: microRNA-203 suppresses bladder cancer development
by repressing bcl-w expression. FEBS J. 278:786–292. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Stein JP, Grossfeld GD, Ginsberg DA, Esrig
D, Freeman JA, Figueroa AJ, Skinner DG and Cote RJ: Prognostic
markers in bladder cancer: A contemporary review of the literature.
J Urol. 160:645–659. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Laufer M, Ramalingam S, Schoenberg MP,
Haisfieldwolf ME, Zuhowski EG, Trueheart IN, Eisenberger MA, Nativ
O and Egorin MJ: Intravesical gemcitabine therapy for superficial
transitional cell carcinoma of the bladder: A phase I and
pharmacokinetic study. J Clin Oncol. 21:697–703. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shui JW, Steinberg MW and Kronenberg M:
Regulation of inflammation, autoimmunity and infection immunity by
HVEM-BTLA signaling. J Leukoc Biol. 89:517–523. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bolyard C, Ji YY, Wang PY, Saini U, Rath
KS, Cripe T, Zhang J, Selvendiran K and Kaur B: Doxorubicin
synergizes with 34.5 ENVE to enhance antitumor efficacy against
metastatic ovarian cancer. Clin Cancer Res. 20:6479–6494. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shui JW, Larange A, Kim G, Vela JL, Zahner
S, Cheroutre H and Kronenberg M: HVEM signalling at mucosal
barriers provides host defence against pathogenic bacteria. Nature.
488:222–225. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Salipante SJ, Adey A, Thomas A, Lee C, Liu
YJ, Kumar A, Lewis AP, Wu D, Fromm JR and Shendure J: Recurrent
somatic loss of TNFRSF14 in classical Hodgkin lymphoma. Genes
Chromosomes Cancer. 55:278–287. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kotsiou E, Okosun J, Besley C, Iqbal S,
Matthews J, Fitzgibbon J, Gribben JG and Davies JK: TNFRSF14
aberrations in follicular lymphoma increase clinically significant
allogeneic T-cell responses. Blood. 128:72–81. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cheung KJ, Johnson NA, Affleck JG,
Severson T, Steidl C, Ben-Neriah S, Schein J, Morin RD, Moore R,
Shah SP, et al: Acquired TNFRSF14 mutations in follicular lymphoma
are associated with worse prognosis. Cancer Res. 70:9166–9174.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cancer Genome Atlas Research Network:
Comprehensive molecular characterization of urothelial bladder
carcinoma. Nature. 507:315–322. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: Limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Capesdavis A, Theodosopoulos G, Atkin I,
Drexler HG, Kohara A, MacLeod RA, Masters JR, Nakamura Y, Reid YA,
Reddel RR and Freshney RI: Check your cultures! A list of
cross-contaminated or misidentified cell lines. Int J Cancer.
127:1–8. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yang D, Wang H, Wang J, Zhang C and Xu H:
Establishment of a fluorescent implantation metastasis model of
bladder cancer and real-time microscopic detection in nude mice.
Asian Pac J Cancer Prev. 12:393–396. 2011.PubMed/NCBI
|
|
15
|
Girnita A, All-Ericsson C, Economou MA,
Astrom K, Axelson M, Seregard S, Larsson O and Girnita L: The
insulin-like growth factor-I receptor inhibitor picropodophyllin
causes tumor regression and attenuates mechanisms involved in
invasion of uveal melanoma cells. Clin Cancer Res. 12:1383–1391.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang X, Qi H, Wang S, Feng L, Ji Y, Tao
L, Li S and Wei Y: Cellular responses of aniline oligomers: A
preliminary study. Toxicol Res. 1:201–205. 2012. View Article : Google Scholar
|
|
18
|
van Rhijn BW, Burger M, Lotan Y, Solsona
E, Stief CG, Sylvester RJ, Witjes JA and Zlotta AR: Recurrence and
progression of disease in non-muscle-invasive bladder cancer: From
epidemiology to treatment strategy. Eur Urol. 56:430–442. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Geng W, Ng KTP, Sun CKW, Yau WL, Liu XB,
Cheng Q, Poon RT, Lo CM, Man K and Fan ST: The role of proline rich
tyrosine kinase 2(Pyk2) on cisplatin resistance in hepatocellular
carcinoma. PLoS One. 6:e273622011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Harrop JA, Mcdonnell PC, Brigham-Burke M,
Lyn SD, Minton J, Tan KB, Dede K, Spampanato J, Silverman C,
Hensley P, et al: Herpesvirus entry mediator ligand (HVEM-L), a
novel ligand for HVEM/TR2, stimulates proliferation of T cells and
inhibits HT29 cell growth. J Biol Chem. 273:27548–27556. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gaur U and Aggarwal BB: Regulation of
proliferation, survival and apoptosis by members of the TNF
superfamily. Biochem Pharmacol. 66:1403–1408. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hengartner MO: Biochemistry of apoptosis.
Nature. 407:770–776. 2016. View
Article : Google Scholar
|
|
23
|
Hail N Jr, Carter BZ, Konopleva M and
Andreeff M: Apoptosis effector mechanisms: A requiem performed in
different keys. Apoptosis. 11:889–904. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hartmann A, Troadec J, Hunot S, Kikly K,
Faucheux B, Mouatt-Prigent A, Ruberg M, Agid Y and Hirsch E:
Caspase-8 is an effector in apoptotic death of dopaminergic neurons
in parkinson's disease, but pathway inhibition results in neuronal
necrosis. J Neurosci. 21:2247–2255. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Isabelle M, Moreel X, Gagné JP, Rouleau M,
Ethier C, Gagné P, Hendzel MJ and Poirier GG: Investigation of
PARP-1, PARP-2 and PARG interactomes by affinity-purification mass
spectrometry. Proteome Sci. 8:222010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cantley LC: The phosphoinositide 3-kinase
pathway. Science. 296:1655–1657. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shaw RJ and Cantley LC: Ras, PI(3)K and
mTOR signalling controls tumour cell growth. Nature. 441:424–430.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Knowles MA, Platt FM, Ross RL and Hurst
CD: Phosphatidylinositol 3-kinase (PI3K) pathway activation in
bladder cancer. Cancer Metastasis Rev. 28:305–316. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bakin AV, Tomlinson AK, Bhowmick NA, Moses
HL and Arteaga CL: Phosphatidylinositol 3-kinase function is
required for transforming growth factor β-mediated epithelial to
mesenchymal transition and cell migration. J Biol Chem.
275:368032000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mcconkey DJ, Choi W, Marquis L, Martin F,
Williams MB, Shah J, Svatek R, Das A, Adam L, Kamat A, et al: Role
of epithelial-to-mesenchymal transition (EMT) in drug sensitivity
and metastasis in bladder cancer. Cancer Metastasis Rev.
28:335–344. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang YB, Qin J, Zheng XY, Bai Y, Yang K
and Xie LP: Diallyl trisulfide induces Bcl-2 and
caspase-3-dependent apoptosis via downregulation of Akt
phosphorylation in human T24 bladder cancer cells. Phytomedicine.
17:363–368. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tang C, Lu YH, Xie JH, Wang F, Zou JN,
Yang JS, Xing YY and Xi T: Downregulation of survivin and
activation of caspase-3 through the PI3K/Akt pathway in ursolic
acid-induced HepG2 cell apoptosis. Anticancer Drugs. 20:249–258.
2009. View Article : Google Scholar : PubMed/NCBI
|