Introduction
Thoracic aortic dissection (TAD) is a highly lethal
cardiovascular disease, which is characterized by the separation of
thoracic aortic medial layer along the length of the vessel
(1). Typical TAD begins with the
sudden initial tear in the aortic intima, and intraluminal
pulsatile blood enters into the medial layer through an intimal
tear, resulting in rapid aortic dilation and rupture. In spite of
the improvement of diagnostic and therapeutic techniques over the
years, the overall mortality of TAD remains high (2). The understanding of the pathogenesis
of this serious illness may produce a better outcome in the future.
Many studies have been performed to explore the pathogenesis of
TAD. Most studies were mainly focused on the genetic diversity,
clinical pathology, and hemodynamics. However, the potential
molecular mechanism of TAD remains unclear.
Long non-coding RNAs (lncRNAs) are defined as more
than 200 nt in length and found to regulate protein-coding gene
expression at both the transcriptional and post-transcriptional
levels. Studies showed that lncRNAs play critical roles in
cardiovascular physio-pathological processes (3). For example, lncRNA Braveheart (Bvht)
was associated with cardiovascular development (4). Upregulated myocardial
infarction-associated transcript 1 (Mirt1) and Mirt2 can promote
cardiac contractile function and decrease left ventricular
remodeling (5). Myosin heavy
chain-associated RNA transcript (Mhrt) can protect the heart from
pathological cardiac hypertrophy (6). Although an increasing number of
lncRNAs have been characterized, the role of lncRNAs in TAD has not
been investigated. A recent study revealed that the overexpressed
lncRNA HIF1 alpha-antisense RNA 1 (HIF1A-AS1) in the
thoraco-abdominal aortic aneurysm (TAAA) promoted the proliferation
and apoptosis of vascular smooth muscle cells (VSMCs), which may
contribute to the pathogenesis of TAAA (7). Wang et al (8) reported that the interaction between
BRG1 and HIF1A-AS1 may be involved in the pathogenesis of thoracic
aortic aneurysms. Since TAA and TAD possesses similar pathological
basis, these results suggested the potential role of lncRNAs in the
pathogenesis of TAD. However, until now, there was no study on the
expression profile of lncRNAs in TAD.
In the present study, we demonstrated expression
profile of lncRNAs between TAD and normal thoracic aorta (NTA)
using third-generation lncRNA microarray techniques. These results
will help provide further insight into the pathogenesis of TAD.
Materials and methods
Acquisition of clinical specimens
Ascending aorta specimens were obtained from TAD
patients undergoing surgical repair (TAD group, n=6; mean age,
51.4±13.4 years) at Fuwai Hospital and organ donors without aortic
diseases (NTA group, n=6; mean age, 49.6±12.6 years). No
significant difference in the age was found between TAD and NTA
(P>0.05). All patients were confirmed to have acute Stanford
type I aortic dissection within 14 days of the symptom onset before
surgery. All subjects have not any history of Marfan syndrome,
bicuspid aortic valve, or any other connective tissue disease. The
clinical characteristics of patients and donors were in Table I. The study protocol was approved
by the international review board of Beijing Yuho Rehabilitation
Hospital, (Beijing, China). Written informed consent was obtained
from each of the patients. Aortic media tissues were sectioned into
smaller sizes and briefly stored at −80°C until RNA extraction.
 | Table I.Clinical characteristics. |
Table I.
Clinical characteristics.
| Variables | NTA (n=6) | TAD (n=6) |
|---|
| Age (years) | 49.6±12.6 | 51.4±13.4 |
| Males/females | 6/0 | 6/0 |
| Hypertension | 2 (33.3%) | 3 (50.0%) |
| Atherosclerosis | 1 (16.7%) | 2 (33.3%) |
| Smoking | 2 (33.3%) | 3 (50.0%) |
| Aortic size (mm) | 31.4±9.7 | 63.4±15.2 |
RNA isolation and lncRNA microarray
analysis
Total RNA was extracted using TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
following the manufacturer's instruction. For the global profiling
of human lncRNAs and protein-coding transcripts, we utilized the
third-generation lncRNA microarray (Arraystar v3.0; KangChen,
Shanghai, China), which contains more than 30,000 capture probes,
covering all lncRNAs from authoritative databases (UCSC Knowngenes,
RefSeq and Ensembl) and their coding proteins. The microarray
hybridization was performed based on manufacturer's instruction.
Quantile normalization and subsequent data analysis was performed
using GeneSpring GX 11.5.1 software (Agilent Technologies, Inc.,
Santa Clara, CA, USA).
Validation by reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
To confirm the microarray data, expressions of
selected lncRNA and coding proteins were tested using RT-qPCR with
the GoTaq qPCR Master Mix (Promega Corporation, Madison, WI, USA)
on an Mx3005P real-time PCR System (Agilent Technologies, Inc.).
The primers used for RT-qPCR were listed in Table II. Each RNA sample was evaluated
in triplicate. Gene expression results were analyzed with the
2−∆∆Cq method and normalized to GAPDH expression.
| Table II.Primers used in the present study. |
Table II.
Primers used in the present study.
| A, lncRNA |
|---|
|
|---|
| Gene name | Forward (5′-3′) | Reverse (3′-5′) |
|---|
| CERKL |
TGTAAAGTTGAATAAAGCGG |
TGCCTTGGGCTGTCGTAG |
| RP11-395B7.4 |
AGGACTTGGAACTATGGG |
AAGGGAGAATTAAAGGCTA |
| RP11-887P2.1 |
TGTGCCTGGCTGCCTTAG |
TAGCCGGAGGGTTAGGGAT |
| lncP2RX7 |
GGGGGTTGAATGTGAGATGA |
GTTAGGGAATGTTCTGAAGG |
| CDKN2B-AS1 |
AGATGCAGAGGACATGCACGG |
GGCAAGGCAAGTGAGGAG |
| RP11-796E2.4 |
CGCCTGTAATCCCAGCAC |
TCCGGGTTCACGCCATTC |
| HIF1A-AS2 |
TTCAACCCTGCTTCCTGG |
TGCTACCTTCTGTCCTGG |
| HOXD-AS2 |
CCTCAGTACATAAACTTCCTAA |
CTTTCACCTTCTCACAGG |
| AC007254.3 |
ACGACCAGGCTTACGGAG |
TCATTTCCCGTGTTGGGC |
| OGFR-AS1 |
ACTGCCCTTGTTAATGTCA |
TGGTCGTATTGTTCTTGC |
| AX746823 |
TCGATTTCGTCAAGCATT |
GAGGGCAGAAAGATCACA |
| RP11-69I8.3 |
GAAGTCAGAAATACTGTGGGTA |
CAAATGTGTGTTCGTGGG |
| RP11-317J10.2 |
CTCCAAACTGTCTCACCC |
AAAACCACCGTTACCAAA |
| RP11-318K15.2 |
GCTCTTACCGCTGGGTGT |
CCTTGGGGAGGGAAGTTAG |
| RP11-536K7.5 |
CACAGTAGAGGCGAAACC |
CAGGGAATAAGTGGACAGAT |
| NPPA-AS1 |
GTGAAGAAAGACTTCAT |
GTAGGGAAGGCAGTAGGGTGGAG |
|
| B,
mRNAs |
|
| Gene
name | Forward
(5′-3′) | Reverse
(3′-5′) |
|
| ITGA4 |
TGGACAGCTAGAATTGGT |
GTAGCTCTTAGATCCGGTG |
| MUC12 |
CATGTACGTCTGCTATCCAGG |
CTCCTTAGTCATACCACGATG |
| SOCS2 |
TATAGGTGCAGGAGATGGTTC |
GCTGTTATGGGTGAAACTCTG |
| P2RX7 |
GGCGTCGTGGTGAATGATG |
CCTAACTTGGCGTCCTATGC |
| CDKN2B |
TCAACGCTCGATTCTATTGTGG |
TCAACGAGATTCTGATTGTGGG |
| BTG1 |
GGGGGCGGATACTAATCCC |
CACGACAAAAGCGTACTCTGT |
| HIF1A |
TCTCTGACGGCTATCCAAAGAA |
GCAGCAGTTCTGGGTTAGTTG |
| HOXD3 |
CTTGGAGGACGGCGATTA |
CCGTCACGAACTTCTTCGT |
| SLC8A1 |
GCTAGAAGCCGTGACGGCT |
CTGACGGTACTCTAGTGGAG |
| OGFR |
TCGTCGTCTAGACATTA |
CTCTAGTGGGTGACTC |
| RUNX1 |
GACGGCTGTCTATCCAAA |
GAAAGACGTCTCTTC |
| CTGF |
AAGGTCAAGCTAGACTG |
GGAGACTAAGGCAGTG |
| CA2 |
ATTGCTTATGAGTGAACGCC |
AGGCATAGGTAGGCAGAG |
| LYN |
GGTGGTGAAGATTAATCGC |
ATGGGACATTTAAGGCAATC |
| IL2RA |
GGAAGTAATACAATAGA |
GCTGTTAACAACGTAAACTC |
| NPPA |
AGCATGTAGGAGAGTTAACA |
TAGAGGGGACCTTACAAG |
| GAPDH |
GCCGTCTAGACCTGACCT |
AGGATTGTCGCGTGGTGG |
Gene ontology (GO) and pathway
analysis
The GO analysis (www.geneontology.org) and Kyoto Encyclopedia of Gene
and Genomes database (KEGG, www.genome.jp/kegg) were used to analyze the main
function and pathway of differentially expressed lncRNAs and
mRNAs.
LncRNA and mRNA interaction network
analysis
MetaCoreTM software (Auto Expand Algorithm) was used
to analyze the regulatory network of differentially expressed genes
associated with lncRNAs.
Statistical analysis
All statistical data were analyzed by SPSS v20.0
software (IBM Corp., Armonk, NY, USA). Data were presented as the
mean ± standard deviation of at least 3 independent experiments.
The results were analyzed by analysis of variance with a Bonferroni
post hoc test, or by Student's t-test. P<0.05 was considered to
indicate a statistically significant difference.
Results
LncRNA microarray profiling
Arraystar lncRNA Microarray (v3.0), which can detect
about 30,586 lncRNAs and 26,109 coding transcripts, was used for
the comprehensive analysis of core genes associated with the
pathogenesis of TAD. Using third-generation lncRNA microarray, we
detected a total of 25,348 lncRNAs and 24,852 mRNAs. The
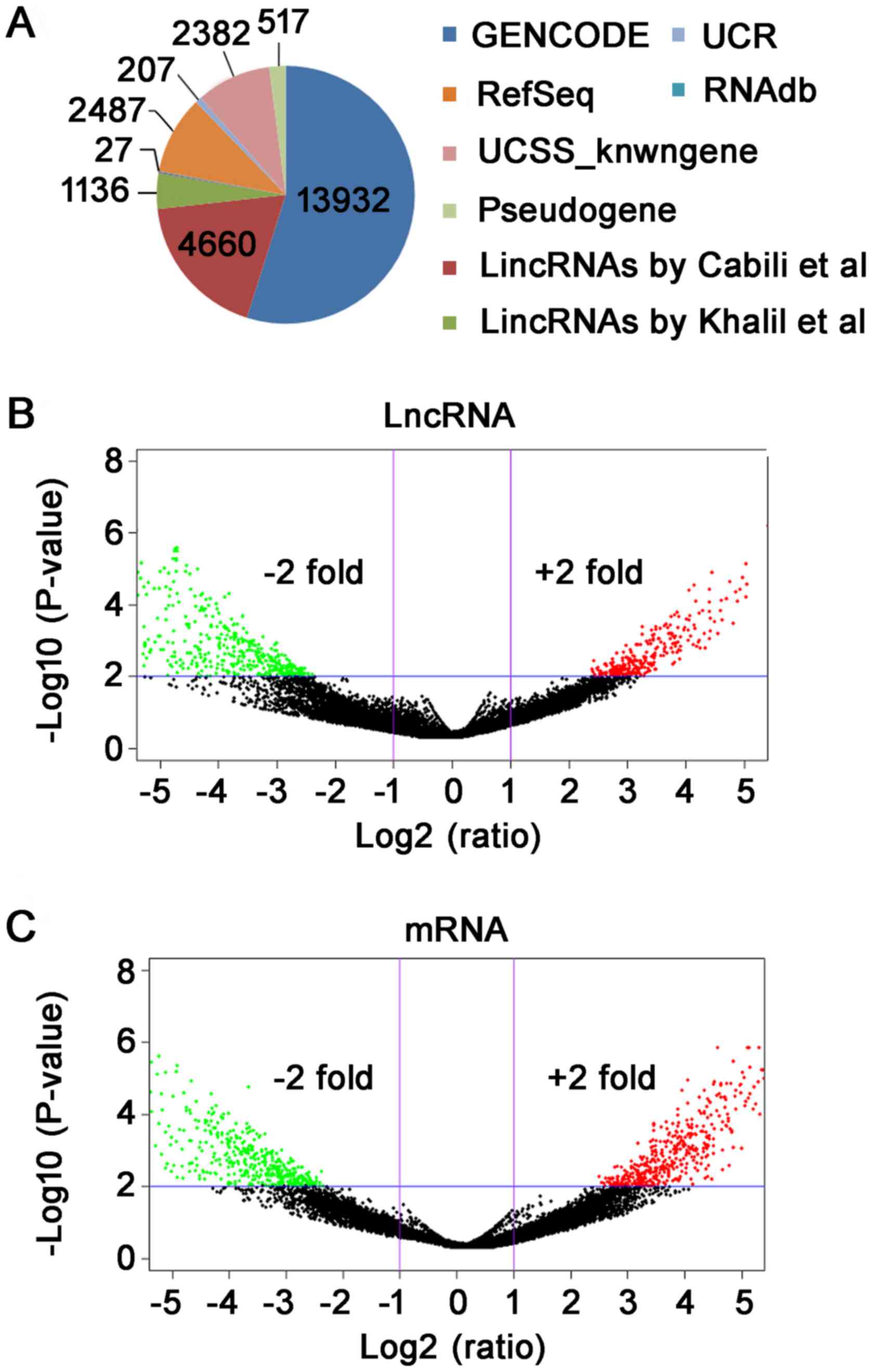
statistical information of lncRNAs was shown in Fig. 1A. Furthermore, the differentially
expressed lncRNAs and mRNAs were screened as per the fold-change
and P-value. The screening criteria for differentially expressed
lncRNAs and mRNAs is fold-change ≥2 and P-value <0.01.
Subsequently, we have used log2 (fold-change) as the X-axis and
log10 (P-value) as the Y-axis to show the distribution of lncRNAs
and mRNAs in Volcano plot (Fig. 1B and
C). In total, 765 differentially expressed lncRNA were
identified between TAD and NTA, including 289 up-regulated and 476
down-regulated (fold-change ≥2, P<0.01). Through microarray, we
found 619 differentially expressed mRNA (fold-change ≥2,
P<0.01). Among mRNAs, 265 were up-regulated and 354 were
down-regulated in TAD compared with NTA.
Expression signatures of dysregulated
lncRNAs between TAD and NTA
The distribution of relationship types, the sequence
length and chromosomes of differentially expressed lncRNA are shown
in Fig. 2. For those up-regulated
lncRNAs, there were 156 intergenic, 75 natural antisense, 69 exon
sense-overlapping, 57 intronic antisense, 30 intron
sense-overlapping and 26 bidirectional lncRNAs. For those
down-regulated lncRNAs, there were 329 intergenic, 105 natural
antisense, 99 intronic antisense, 39 exon sense-overlapping, 19
bidirectional, and 11 intron sense-overlapping lncRNAs (Fig. 2A). The lncRNAs were mainly
concentrated between 400 and 800 bp in length (Fig. 2B). The chromosome distribution
showed the up-regulated and down-regulated lncRNAs were located at
various chromosomes, respectively (Fig. 2C).
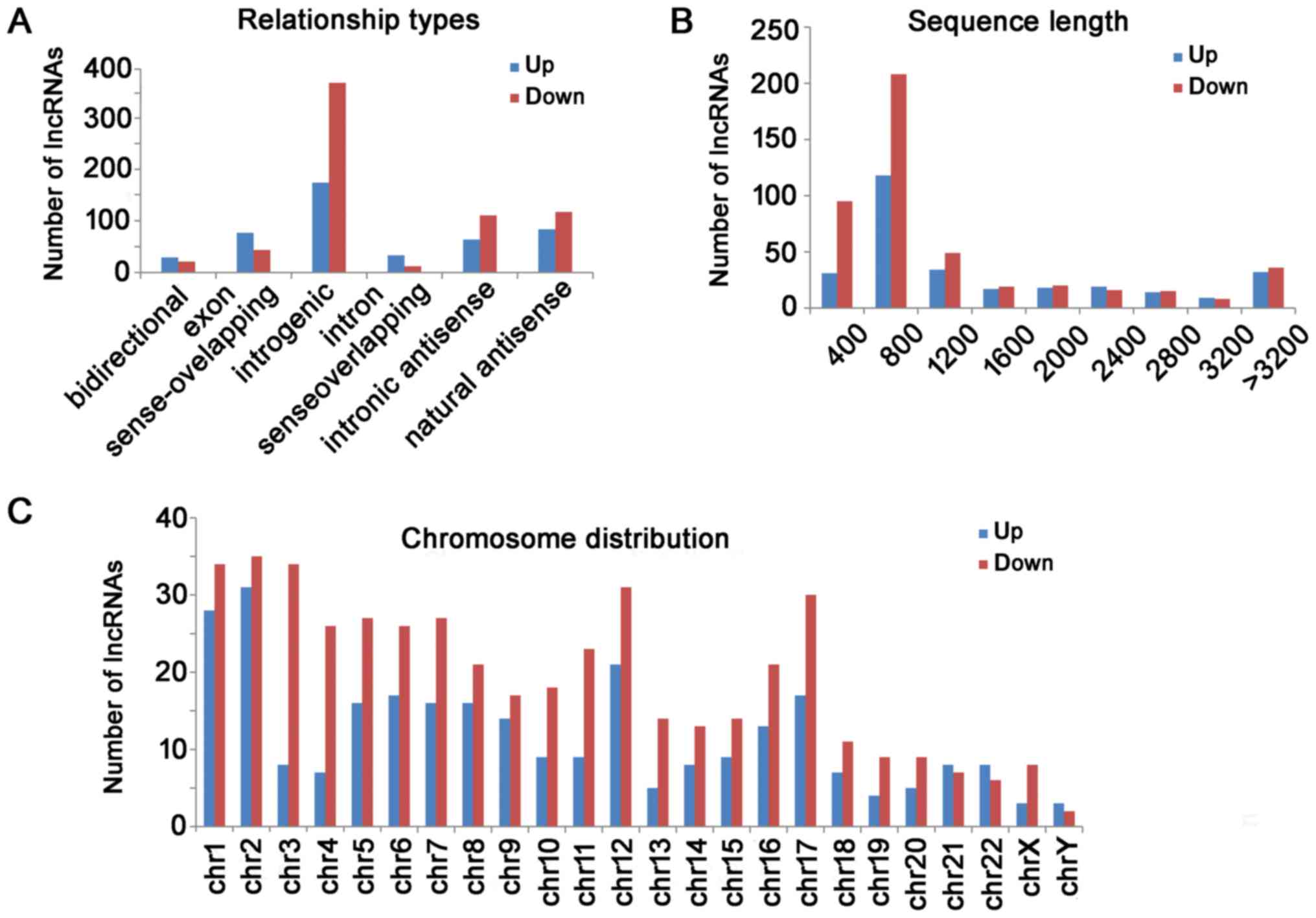 | Figure 2.Differential lncRNAs were classified
according to their distribution. (A) For those upregulated lncRNAs,
there were 156 intergenic, 75 natural antisense, 69 exon
sense-overlapping, 57 intronic antisense, 30 intron
sense-overlapping and 26 bidirectional lncRNAs. For those
downregulated lncRNAs, there were 329 intergenic, 105 natural
antisense, 99 intronic antisense, 39 exon sense-overlapping, 19
bidirectional and 11 intron sense-overlapping lncRNAs. (B) Length
distribution of the dysregulated lncRNAs. The lncRNAs were mainly
between 400 and 1200 bp in length. (C) The chromosome distribution
indicated the numbers of upregulated and downregulated lncRNAs
locations in various chromosomes. LncRNA, long non-coding RNA; Chr,
chromosome. |
GO and pathway analysis
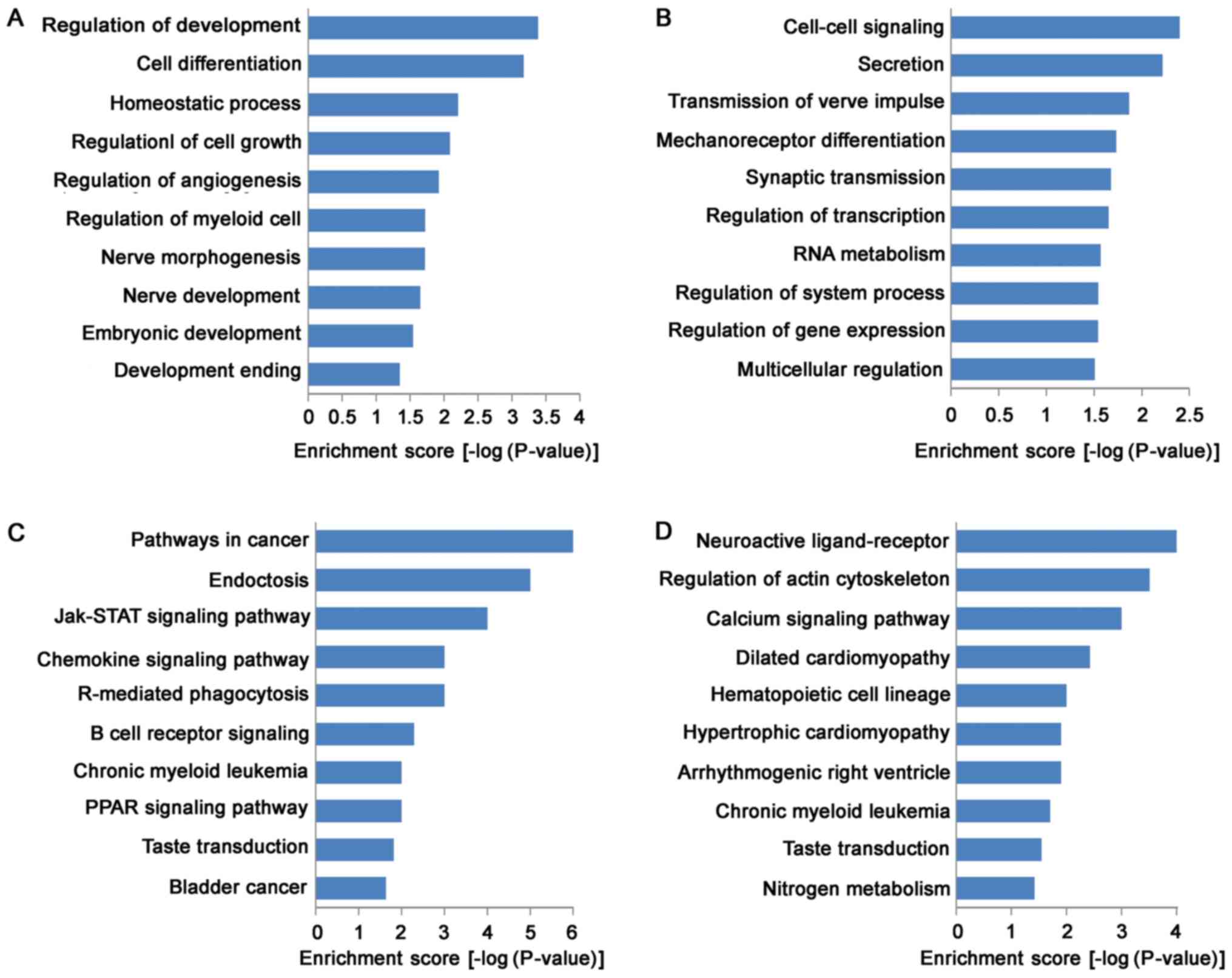
GO analysis indicated that the functions of
up-regulated mRNAs were involved in a variety of biological
processes, including VSMC development and vascular homeostasis,
such as in cell growth (GO:0001558), cell differentiation
(GO:0045597), homeostatic process (GO:0032844) and angiogenesis
(GO:0045766; Fig. 3A). Meanwhile,
the functions of down-regulated mRNAs were mainly involved in
cell-cell signaling (GO:0007267), transcription regulation
(GO:0045893), RNA metabolism (GO:0051254), and gene expression
(GO:0010628; Fig. 3B). KEGG
pathway analysis indicated that 31 pathways were down-regulated and
24 pathways were up-regulated. Most of up-regulated pathways were
enriched in Jak-STAT signaling pathway (KEGG: hsa04630), chemokine
signaling pathway (KEGG: hsa04062), PPAR (KEGG: hsa03320) signaling
pathway and B cell receptor signaling pathway (KEGG: hsa04662),
suggested that up-regulated pathways in TAD were closely associated
with signal transduction (Fig.
3C). Moreover, down-regulated pathways were enriched in calcium
signaling pathway (KEGG: hsa04020), arrhythmogenic right
ventricular cardiomyopathy (ARVC) (KEGG: hsa05412), hypertrophic
cardiomyopathy (HCM) (KEGG: hsa05410) and dilated cardiomyopathy
(KEGG: hsa05414; Fig. 3D).
Selection of core genes in TAD
To reduce the lncRNAs for further investigation and
to enrich those potentially involved in TAD, we first selected
candidate lncRNAs with a significant expression (fold-change >4,
P<0.01) that were associated with an annotated protein-coding
gene through the GO term enrichment and scientific literatures.
After functional annotation and enrichment analysis, we found: i) 8
protein-coding genes (BTG1; CA2; CDKN2B; HIF1A; IL2RA; LYN; RUNX1;
HOXD3) are associated with cell differentiation; ii) 5 genes (CA2,
HIF1A, IL2RA, LYN, P2RX7) are related to homeostasis; iii) 6) genes
(CTGF, SOCS2, NPPA, MUC12, OGFR, BTG1) are correlated with cell
growth; iv) 3 genes (BTG1, HIF1A, RUNX1) have positive regulation
on angiogenesis and v) 2 genes (ITGA4, SLC8A1) are associated with
cardiac disease. Finally, 16 lncRNAs associated with important
genes were selected as candidates for the pathogenesis of TAD
(Table III).
| Table III.Details of the 16 candidate lncRNAs
in thoracic aortic dissection. |
Table III.
Details of the 16 candidate lncRNAs
in thoracic aortic dissection.
| Gene symbol | Regulation | RNA length | Chromosome | Strand | Relationship | mRNA | TAD/NTA | P-value |
|---|
| CERKL | Down | 3,030 | Chr2 | − | Natural
antisense | ITGA4 | 0.04 | 0.005 |
| RP11-395B7.4 | Up | 372 | Chr7 | − | Natural
antisense | MUC12 | 8.58 | 0.014 |
| RP11-887P2.1 | Up | 3,325 | Chr12 | − | Intronic
antisense | SOCS2 | 7.75 | 0.017 |
| lncP2RX7 | Up | 3,604 | Chr12 | + | Exon
sense-overlapping | P2RX7 | 12.48 | 0.009 |
| CDKN2B-AS1 | Up | 1,469 | Chr9 | + | Intronic
antisense | CDKN2B | 10.92 | <0.001 |
| RP11-796E2.4 | Down | 1,898 | Chr12 | + | Natural
antisense | BTG1 | 5.49 | 0.003 |
| HIF1A-AS2 | Up | 2,051 | Chr14 | − | Natural
antisense | HIF1A | 12.67 | <0.001 |
| HOXD-AS2 | Up | 692 | Chr2 | − | Natural
antisense | HOXD3 | 6.63 | 0.012 |
| AC007254.3 | Down | 344 | Chr2 | + | Intronic
antisense | SLC8A1 | 0.01 | 0.008 |
| OGFR-AS1 | Down | 668 | Chr20 | − | Intronic
antisense | OGFR | 11.00 | <0.001 |
| AX746823 | Up | 2,943 | Chr21 | − | Intron
sense-overlapping | RUNX1 | 9.80 | 0.023 |
| RP11-69I8.3 | Up | 495 | Chr6 | + | Natural
antisense | CTGF | 10.29 | 0.019 |
| RP11-317J10.2 | Up | 430 | Chr8 | − | Bidirectional | CA2 | 13.79 | <0.001 |
| RP11-318K15.2 | Up | 651 | Chr8 | + | Intron
sense-overlapping | LYN | 13.34 | 0.004 |
| RP11-536K7.5 | Up | 480 | Chr10 | + | Natural
antisense | IL2RA | 21.43 | 0.006 |
| NPPA-AS1 | Up | 660 | Chr1 | + | Natural
antisense | NPPA | 6.51 | <0.001 |
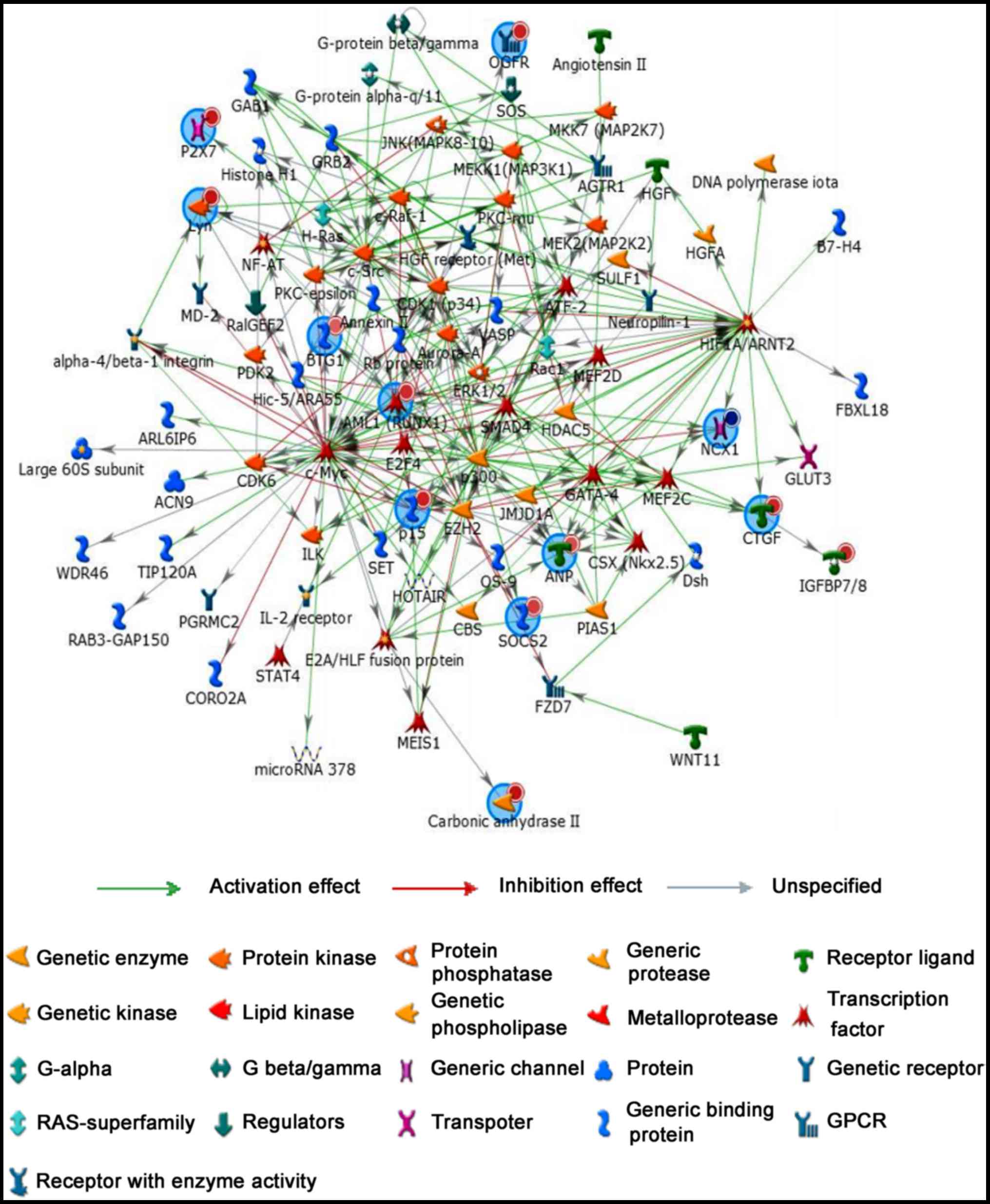
Regulatory network analysis
To further study relationship between coding genes,
regulatory network analysis was performed by MetaCoreTM software to
show network objects of 16 most promising lncRNA candidates
(Fig. 4). Network objects that
were associated with 16 candidate lncRNAs were listed Table IV. Similarly, 16 mRNA candidates
(ITGA4, MUC12, SOCS2, P2RX7, CDKN2B, BTG1, HIF1A, HOXD3, SLC8A1,
OGFR, RUNX1, CTGF, CA2, LYN, IL2RA and NPPA) also played an
important role in the regulatory network. Additionally, more genes
and signal pathways were involved in this network, suggested
regulatory mechanisms of lncRNAs and mRNAs are complex in the
pathogenesis of TAD.
| Table IV.Network objects associated with 16
lncRNA candidates. |
Table IV.
Network objects associated with 16
lncRNA candidates.
| No. | Tag | Gene | Network object |
|---|
| 1 | NM_001122607 | RUNX1 (Runt-related
transcription factor 1) | AML1 RUNX1 |
| 2 | NM_006172 | NPPA (Natriuretic
peptide A) | ANP |
| 3 | NM_001731 | BTG1 (B-cell
translocation gene 1) | BTG1 |
| 4 | NM_000067 | CA2 (Carbonic
anhydrase II) | Carbonic anhydrase
II |
| 5 | NM_001901 | CTGF (Connective
tissue growth factor) | CTGF IGFBP7/8 |
| 6 | NM_181054 | HIF1A (Hypoxia
inducible factor 1, α subunit) | HIF1A |
| 7 | NM_000417 | IL2RA (Interleukin
2 receptor α) | IL-2R α chains
IL2RA |
| 8 | NM_000885 | ITGA4 (Integrin, α
4) | ITGA4 |
| 9 | NM_002350 | LYN (LYN
proto-oncogene, Src family tyrosine kinase) | Lyn |
| 10 | NM_001164462 | MUC12 (Mucin 12,
cell surface associated') | Mucin 12 |
| 11 | NM_001112802 | SLC8A1 (Solute
carrier family 8 (sodium/calcium exchanger), member 1) | NCX1 |
| 12 | NM_007346 | OGFR (Opioid growth
factor receptor) | OGFR |
| 13 | NM_078487 | CDKN2B
(Cyclin-dependent kinase inhibitor 2B) | p15 |
| 14 | NM_002562 | P2RX7 (Purinergic
receptor P2X, ligand gated ion channel, 7) | P2X7 |
| 15 | NM_003877 | SOCS2 (Suppressor
of cytokine signaling 2) | SOCS2 |
| 16 | NM_006898 | HOXD3 (Homeobox
D3) | HOXD3 |
Validation of lncRNA and mRNA
candidates by RT-qPCR
Validation of lncRNAs candidates by RT-qPCR was
shown in Fig. 5A. The detection of
the expression level of selected 16 lncRNAs demonstrated a good
consistency with the microarray results. Among them, lncP2RX7,
HIF1A-AS2, AX746823, RP11-69I8.3 and RP11-536K7.5 increased
dramatically in the TAD group compared with the NTA group
(P<0.01, respectively). Validation of 16 mRNAs candidates by
RT-qPCR was shown in Fig. 5B.
Among mRNAs, P2RX7, CDKN2B, HIF-1A, RUNX1, CTGF and IL2RA increased
significantly in the TAD group compared with the NTA group
(P<0.01, respectively). These genes may play critical roles in
the pathogenesis of TAD.
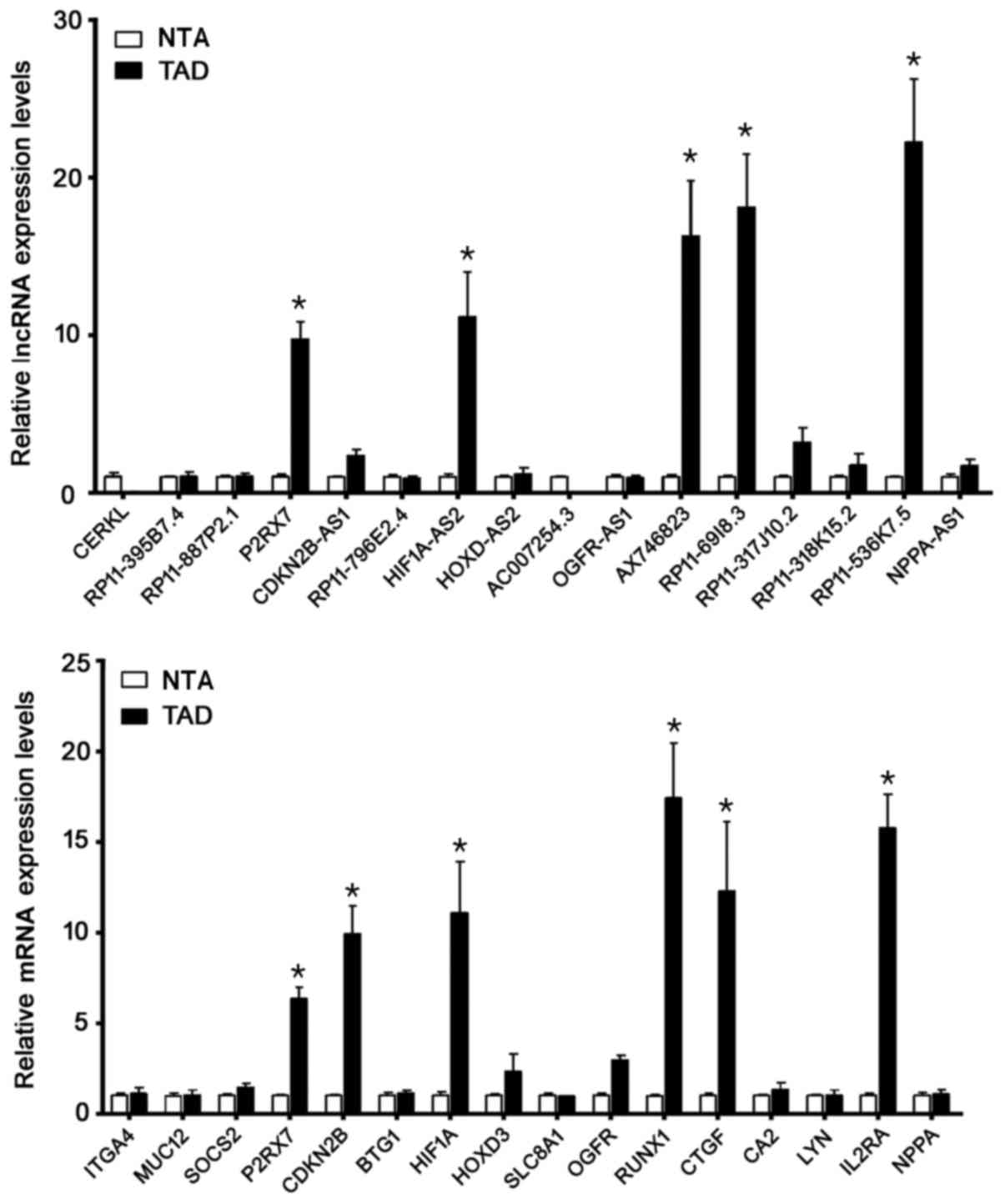 | Figure 5.Candidate genes were validated by
reverse transcription-quantitative polymerase chain reaction. Among
the lncRNAs, lncP2RX7, HIF1A-AS2, AX746823, RP11-69I8.3 and
RP11-536K7.5 increased markedly in the TAD group when compared with
the control group. Among the mRNAs, P2RX7, CDKN2B, HIF-1A, RUNX1,
CTGF and IL2RA increased markedly in the TAD group when compared
with the NTA group. *P<0.05 vs. NTA. LncRNA, long non-coding
RNA; P2RX7, purinergic receptor P2X7; HIF, hypoxia inducing factor;
TAD, thoracic aortic dissection; CDKN2B, cyclin dependent kinase
inhibitor 2B; RUNX1, runt-related transcription factor 1; CTGF,
connective tissue growth factor; IL2RA, interleukin 2 receptor a
chain; NTA, normal thoracic aorta. |
Discussion
TAD is a life-threatening vascular disease that
involves the separation of the layers within the aortic wall. Its
formation, progression, and rupture cannot be reliably prevented by
pharmacological therapies because the molecular mechanisms of the
pathogenesis are still currently unclear (9). LncRNAs are a newly discovered class
of non-coding RNAs, and they have important functions in regulating
a variety of physio-pathological processes at transcriptional and
post-transcriptional levels (10).
Comprehensive comparison study of lncRNA expression profiles may
help to identify candidate genes for the pathogenesis of TAD. In
this study, the microarray that contains more than 30,000 known
human lncRNAs and coding genes was utilized to detect fold-changes
in the expressions of lncRNAs and mRNAs in TAD compared with those
of NTA. After detailed analysis of data, we identified 765
differentially expressed lncRNAs, in which 289 were up-regulated
and 476 were down-regulated. In 619 differentially expressed mRNA,
265 mRNAs were up-regulated and 354 mRNAs were down-regulated.
Through bioinformatics approaches of GO, pathways and network
analysis, 16 lncRNAs and their coding genes were selected as
candidates for the pathogenesis of TAD. These candidates were
verified by RT-qPCR. Results showed that 5 lncRNAs (lncP2RX7,
HIF1A-AS2, AX746823, RP11-69I8.3 and RP11-536K7.5) and 6 mRNAs
(P2RX7, CDKN2B, HIF-1A, RUNX1, CTGF and IL2RA) were significant
expressed in dissected thoracic aortas, suggested they may be core
genes and play critical roles in the pathogenesis of TAD.
So far, exploratory studies in the cardiovascular
setting have identified several lncRNAs associated cardiovascular
diseases (11). For example,
lncRNA Bvht was identified as a key regulator of cardiovascular
commitment from nascent mesoderm, suggested the potential
implication in cardiovascular development (12). Both Mirt1 and Mirt2 lncRNAs were
up-regulated in a mouse model of myocardial infarction. Increased
expression of both Mirt1 and Mirt2 can promote cardiac contractile
function and decrease left ventricular remodeling (5). The H19 lncRNA is a novel negative
regulator of cardiomyocyte hypertrophy (13). A cardiac specific lncRNA Mhrt has
been demonstrated to protect the heart from pathological cardiac
hypertrophy (6). However, specific
studies looking at lncRNAs in TAD are still lacking. Recent studies
showed some exciting reports on lncRNAs in aortic aneurysm. The
inhibition of lncRNA HIF1A-AS1 in VSMCs suppressed cell apoptosis
and enhanced cell proliferation, which may participate in the
pathogenesis of thoraco-abdominal aorta aneurysm (TAAA) (7,14).
BRG1 expression is increased in TAA and regulates proliferation and
apoptosis of VSMCs through the HIF1A-AS1 (6). These results may shed light on
important functions of lncRNAs in TAD. Using third-generation
lncRNA microarray, we revealed differential expression profiles of
lncRNAs between TAD and NTA. The fact that some lncRNAs were
differentially expressed in the developing or diseased heart
provides a strong indication for their involvement in cardiac
physio-pathology (15–17). Our results may provide important
insights into the pathogenesis of TAD disease.
GO term and KEGG pathway analyses were utilized to
gain insight into the function of differentially expressed genes
(18). GO analysis indicated that
the functions of dysregulated mRNAs were involved in a variety of
biological processes, including VSMC development and vascular
homeostasis. These both processes were closely associated with the
formation and development of TAD. The aortic media is mainly
composed of VSMCs, which are the source of extracellular matrix
(ECM) proteins such as collagen and elastin. Medial degeneration is
the main histopatholocgic characteristics of dissected thoracic
aorta (18). GO results may imply
the therapeutic potential of TAD by modulating lncRNAs. Moreover,
KEGG pathway analysis indicated that 31 pathways were
down-regulated and 24 pathways were up-regulated. Most of
up-regulated pathways were enriched in Jak-STAT, chemokine, PPAR
and B cell receptor signaling pathways, suggested that up-regulated
pathways in TAD were closely associated with signal transduction.
In addition, down-regulated pathways were mainly enriched in ARVC,
HCM and dilated cardiomyopathy, indicated that these diseases may
have a common pathogenic mechanism. To reduce the lncRNAs for
further investigation and to find those potentially involved in
TAD, we selected 16 lncRNAs as candidate genes for TAD through the
significant expression (fold-change >4, P<0.01) and GO term
enrichment. Regulatory network analysis demonstrated complex
regulatory mechanisms of 16 mRNAs in the pathogenesis of TAD.
Microarray results of lncRNAs and mRNAs was validated by RT-qPCR.
Among lncRNAs, lncP2RX7, HIF1A-AS2, AX746823, RP11-69I8.3 and
RP11-536K7.5 increased dramatically in the TAD group compared with
the NTA group (P<0.01, respectively). With the exception of
HIF1A-AS2 reported in TAAA, other lncRNAs were first found in the
dissected thoracic aorta. According to protein-coding genes,
lncP2RX7, AX746823, RP11-69I8.3 and RP11-536K7.5 were related to
the activation of nuclear receptor (P2RX7), nuclear transcription
(RUNX1), connective tissue development (CTGF) and inflammation
(IL2RA). These genes may play important roles in the pathogenesis
of TAD. Whether therapeutic modulation of these lncRNAs could
decrease TAD development and whether this effect translates into
improved survival after TAD remains to be determined.
PCR results showed that P2RX7, CDKN2B, HIF-1A,
RUNX1, CTGF and IL2RA were significant expressed in dissected
thoracic aortas, suggested they may be core genes and play critical
roles in the pathogenesis of TAD. To date, HIF-1A has been involved
in the proliferation, migration and morphological changes of VSMCs.
Mechanically, hypoxia promoted the expression of HIF-1A by PI3K-AKT
pathway in human aortic SMCs; HIF-1A further suppressed the
expressions of AEG-1, a-SMA and SM22a, and promoted osteopontin
(OPN) expression. Functionally, HIF-1A inhibited the proliferation
and migration of human aortic SMCs (19). Moreover, CTGF is a matricellular
protein expressed in the vascular wall, which regulates diverse
cellular functions. Ungvari et al (20) showed that the expression of CTGF
was increased in the abdominal aorta of ApoE−/− mice and
in the adventitial region of the abdominal aorta in human AAA. CTGF
is principally regulated at the level of transcription and is
induced by mechanical stresses and a number of cytokines and growth
factors, including TGF-β (21).
Additionally, used vascular injury models, Leeper et al
(22) found that CDKN2B knockout
mice displayed reduced neointimal lesions and developed larger
aortic aneurysms. In situ and in vitro studies
suggested that these effects were attributable to increased smooth
muscle cell apoptosis (23).
Besides, P2X7 receptor activation is the initial
event leading to vascular dysfunction following lipopolysaccharide
(LPS) treatment. Activation of P2X7 receptor amplifies LPS-induced
hyporeactivity in mouse endothelium-intact aorta, which is
associated with IL-1β-mediated release of nitric oxide by iNOS
(24). And then, Pahl et al
(25) reported that mRNA and
protein expression of transcription factor RUNX1 in human abdominal
aortic aneurysm. Furthermore, an integral-membrane protein, soluble
IL2RA has been isolated and determined to result from extracellular
proteolysis. This suggested the pathogenesis and development of
human TAD may be related to inflammation of the local environment
in the aorta (26). However, the
molecular mechanisms of these genes are not completely understood
in TAD.
lncRNAs typically show tissue-specific and vascular
disease-specific patterns of expression (27,28).
Give this specificity, lncRNAs may be favorable biomarkers than
current coding proteins for TAD. In addtion, lncRNAs are functional
molecules, thus their expressions may be a better indicator of
disease states. Because of the limited sample size, the screening
biomarker had some limitations, hoping to expand the sample size in
the future. Future studies would also be needed to reveal whether
P2RX7, CDKN2B, HIF-1A, RUNX1, CTGF and IL2RA expressions were
associated to TAD and which cells expressed these proteins in TAD
lesion.
This is first report about the differentially
expressed lncRNA profiles between TAD and NTA. These results may
provide important insights into the pathogenesis of TAD disease.
Expanding our understanding about differential expression profiles
of lncRNAs will assist into novel diagnostics and therapeutics,
which will ultimately improve outcomes for patients with TAD.
Acknowledgements
The authors would like to thank the Department of
Cardiovascular Surgery, The Army General Hospital (Beijing,
China).
Funding
The present study was supported by the Project
Supported by the National Natural Science Foundation of China
(grant no. 81400854) and Beijing Postdoctoral Research Foundation
(grant nos. 2015ZZ-50 and 2016ZZ-44).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YL and NY conceived and designed the experiments. XZ
and XB conducted RNA sequencing and analysis. GQ and MZ performed
the remaining experiments. HL and DL conducted data analysis. YL
and NY produced the manuscript. All authors have read and approved
the final manuscript.
Ethics approval and consent to
participate
The study protocol was approved by the international
review board of Beijing Yuho Rehabilitation Hospital, (Beijing,
China). Written informed consent was obtained from each of the
patients.
Consent for publication
Written informed consent was obtained from each of
the patients.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Clouse WD, Hallett JW Jr, Schaff HV,
Spittell PC, Rowland CM, Ilstrup DM and Melton LJ III: Acute aortic
dissection: Population-based incidence compared with degenerative
aortic aneurysm rupture. Mayo Clin Proc. 79:176–180. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Leitman IM, Suzuki K, Wengrofsky AJ,
Menashe E, Poplawski M, Woo KM, Geller CM, Lucido D, Bernik T,
Zeifer BA and Patton B: Early recognition of acute thoracic aortic
dissection and aneurysm. World J Emerg Surg. 8:472013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Batista PJ and Chang HY: Long noncoding
RNAs: Cellular address codes in development and disease. Cell.
152:1298–1307. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kornienko AE, Guenzl PM, Barlow DP and
Pauler FM: Gene regulation by the act of long non-coding RNA
transcription. BMC Biol. 11:592013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ishii N, Ozaki K, Sato H, Mizuno H, Saito
S, Takahashi A, Miyamoto Y, Ikegawa S, Kamatani N, Hori M, et al:
Identification of a novel non-coding RNA, MIAT, that confers risk
of myocardial infarction. J Hum Genet. 51:1087–1099. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Miner GH, Faries PL, Costa KD, Hanss BG
and Marin ML: An update on the etiology of abdominal aortic
aneurysms: Implications for future diagnostic testing. Expert Rev
Cardiovasc Ther. 13:1079–1090. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
He Q, Tan J, Yu B, Shi W and Liang K: Long
noncoding RNA HIF1A-AS1A reduces apoptosis of vascular smooth
muscle cells: Implications for the pathogenesis of thoracoabdominal
aorta aneurysm. Pharmazie. 70:310–315. 2015.PubMed/NCBI
|
|
8
|
Wang S, Zhang X, Yuan Y, Tan M, Zhang L,
Xue X, Yan Y, Han L and Xu Z: BRG1 expression is increased in
thoracic aortic aneurysms and regulates proliferation and apoptosis
of vascular smooth muscle cells through the long non-coding RNA
HIF1A-AS1 in vitro. Eur J Cardiothorac Surg. 47:439–446. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Duggirala A, Delogu F, Angelini TG, Smith
T, Caputo M, Rajakaruna C and Emanueli C: Non coding RNAs in aortic
aneurysmal disease. Front Genet. 6:1252015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
ENCODE Project Consortium, . Birney E,
Stamatoyannopoulos JA, Dutta A, Guigó R, Gingeras TR, Margulies EH,
Weng Z, Snyder M, Dermitzakis ET, et al: Identification and
analysis of functional elements in 1% of the human genome by the
ENCODE pilot project. Nature. 447:799–816. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dudzinski DM and Isselbacher EM: Diagnosis
and management of thoracic aortic disease. Curr Cardiol Rep.
17:1062015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Papait R, Kunderfranco P, Stirparo GG,
Latronico MV and Condorelli G: Long noncoding RNA: A new player of
heart failure? J Cardiovasc Transl Res. 6:876–883. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu L, An X, Li Z, Song Y, Li L, Zuo S,
Liu N, Yang G, Wang H, Cheng X, et al: The H19 long noncoding RNA
is a novel negative regulator of cardiomyocyte hypertrophy.
Cardiovasc Res. 111:56–65. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhao Y, Feng G, Wang Y, Yue Y and Zhao W:
Regulation of apoptosis by long non-coding RNA HIF1A-AS1 in VSMCs:
Implications for TAA pathogenesis. Int J Clin Exp Pathol.
7:7643–7652. 2014.PubMed/NCBI
|
|
15
|
Magistri M, Faghihi MA, St Laurent G III
and Wahlestedt C: Regulation of chromatin structure by long
noncoding RNAs: Focus on natural antisense transcripts. Trends
Genet. 28:389–396. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Leung A, Trac C, Jin W, Lanting L, Akbany
A, Sætrom P, Schones DE and Natarajan R: Novel long noncoding RNAs
are regulated by angiotensin II in vascular smooth muscle cells.
Circ Res. 113:266–278. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Freedman JE and Miano JM: National Heart,
Lung, and Blood Institute Workshop Participants*: Challenges and
opportunities in linking long noncoding RNAs to cardiovascular,
lung, and blood diseases. Arterioscler Thromb Vasc Biol. 37:21–25.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Huang DW, Sherman BT and Lempicki RA:
Bioinformatics enrichment tools: Paths toward the comprehensive
functional analysis of large gene lists. Nucleic Acids Res.
37:1–13. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu K, Fang C, Shen Y, Liu Z, Zhang M, Ma
B and Pang X: Hypoxia-inducible factor 1a induces phenotype switch
of human aortic vascular smooth muscle cell through PI3K/AKT/AEG-1
signaling. Oncotarget. 8:33343–33352. 2017.PubMed/NCBI
|
|
20
|
Ungvari Z, Valcarcel-Ares MN, Tarantini S,
Yabluchanskiy A, Fülöp GA, Kiss T and Csiszar A: Connective tissue
growth factor (CTGF) in age-related vascular pathologies.
Geroscience. 39:491–498. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sachdeva J, Mahajan A, Cheng J, Baeten JT,
Lilly B, Kuivaniemi H and Hans CP: Smooth muscle cell-specific
Notch1 haploinsufficiency restricts the progression of abdominal
aortic aneurysm by modulating CTGF expression. PLoS One.
12:e01785382017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Leeper NJ, Raiesdana A, Kojima Y, Kundu
RK, Cheng H, Maegdefessel L, Toh R, Ahn GO, Ali ZA, Anderson DR, et
al: Loss of CDKN2B promotes p53-dependent smooth muscle cell
apoptosis and aneurysm formation. Arterioscler Thromb Vasc Biol.
33:e1–e10. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chiao CW, Tostes RC and Webb RC: P2X7
receptor activation amplifies lipopolysaccharide-induced vascular
hyporeactivity via interleukin-1 beta release. J Pharmacol Exp
Ther. 326:864–870. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chiao CW, da Silva-Santos JE, Giachini FR,
Tostes RC, Su MJ and Webb RC: P2X7 receptor activation contributes
to an initial upstream mechanism of lipopolysaccharide-induced
vascular dysfunction. Clin Sci (Lond). 125:131–141. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pahl MC, Erdman R, Kuivaniemi H, Lillvis
JH, Elmore JR and Tromp G: Transcriptional (ChIP-Chip) analysis of
ELF1, ETS2, RUNX1 and STAT5 in human abdominal aortic aneurysm. Int
J Mol Sci. 16:11229–11258. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li L, Yang SH, Yao Y, Xie YQ, Yang YQ,
Wang YH, Yin XY, Ma HD, Gershwin M and Lian ZX: Block of both TGF-β
and IL-2 signaling impedes Neurophilin-1+ regulatory T cell and
follicular regulatory T cell development. Cell Death Dis.
7:e24392016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fatica A and Bozzoni I: Long non-coding
RNAs: New players in cell differentiation and development. Nat Rev
Genet. 15:7–21. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jiang X and Ning Q: The emerging roles of
long noncoding RNAs in common cardiovascular diseases. Hypertens
Res. 38:375–379. 2015. View Article : Google Scholar : PubMed/NCBI
|