Introduction
Gout was often referred to as an affliction of kings
due to its association with a lavish diet and excess alcohol
consumption. In fact, Henry VIII was reported to have suffered from
gout (1). The last few decades has
seen a global rise in new patient diagnoses (2) and the prevalence of the disease has
increased particularly in the UK (3). Gout is an inflammatory form of
crystalline arthritis characterised by the deposition of excess
circulating uric acid in the form of monosodium monohydrate urate
(MSU) crystals into intra-articular joints and tissue spaces
(4). Patients experience recurrent
acute flares of severe joint inflammation. Such attacks involve
inflammation caused by the interaction between MSU crystals and the
local tissue environment including intra-articular cells, monocytes
and macrophages. The active stage of gout is defined by an
inflammatory innate response involving monocytes and neutrophils.
This response is driven in part by NLRP3 inflammasome activation
via NOD-like receptors which activate interleukin-1β (IL-1β)
release (5–7). Studies in vitro suggest that
MSU crystals may induce tumour necrosis factor α (TNFα), IL-1β,
IL-6, platelet-activating factor (PAF) secretion in
undifferentiated monocytes which in turn may promote endothelial
E-selectin expression and secondary neutrophil adhesion (8–10).
Conversely, differentiated macrophages are able to uptake MSU
crystals and release transforming growth factor β1 (TGFβ1), a
powerful inhibitor of inflammation (11).
Therapeutic options for gout management remain
limited to the use of anti-inflammatory treatments such as
non-steroidal anti-inflammatory drugs (NSAIDs), IL-1β antagonists,
adrenocorticotrophic hormone (ACTH), colchicine, allopurinol, or
hydrocortisone injections following acute flares. A study reviewing
patients with tophaceous gout and the risk of complications
reported that for all patients there was at least a 25% casual risk
of arterial hypertension, hyperlipidaemia, diabetes, renal function
impairment and cardiovascular issues. Even after treatment these
patients had at least three gout flares per year (6,7).
Mononuclear cells are a vital part of innate immunity and have been
identified to secrete multiple enzymes into the microenvironment.
However, information regarding the role of enzymes secreted in gout
afflicted joints is limited. Recently published guidelines by the
European League Against Rheumatism have highlighted the potential
of enzymes such as febuxostat (xanthine oxidase inhibitor) and
pegloticase (acts as an uricase) which target uric acid metabolism,
as therapeutic agents in gout (12,13).
Platelet-activating factor acetyl hydrolase (PAF-AH)
has multiple roles in numerous pathologies and synthesis and
secretion of PAF-AH are increased during monocyte to macrophage
differentiation (14–16). Two PAF-AH isoforms have been
identified; plasma PAF-AH and intracytoplasmic PAF-AHII. Plasma
PAF-AH is a polypeptide with molecular weight 40 kDa, mostly
produced by macrophages and hepatocytes. It is an anti-inflammatory
enzyme since it is predominantly involved in degrading inflammatory
phospholipids and is found mainly bound to low density lipoprotein
(LDL) in the circulation (17–19).
Plasma PAF-AH deficiency has been linked to inflammatory reactions
such as sepsis, cardiovascular disease and anaphylaxis (20,21).
PAF-AH resolves inflammation through the
inactivation of PAF, a potent, inflammatory lipid mediator. Serum
PAF concentrations are rigorously controlled by tight regulation of
synthesis and degradation. The acetyl sn-2 group on the backbone of
PAF is required for biological activity and is the target for
PAF-AH action via esterification converting PAF to its inactive
lysosomal-PAF form (22). We have
previously shown that immature monocytes pre-treated with
recombinant PAF-AH showed a dose-dependent inhibition of TNFα
synthesis (12). The mechanism by
which PAF-AH is regulated is still not fully understood and so we
have investigated the effect of common gout therapeutics on PAF-AH
expression. The aim of this study was to investigate the effect of
MSU crystal stimulation on macrophage PAF-AH secretion in
vitro and to determine the factors and pathways that may be
involved in this process.
Materials and methods
Materials
Hydrocortisone (HC) actin D, lipopolysaccharide
(LPS) Dulbecco's modified Eagle medium (DMEM),
dimethyl-ethyl-sulphonyl-oxide, HANKS balanced salt solution,
histopaque, phosphate buffer saline (PBS), Colchicine and methanol
were purchased from Sigma Aldrich, Poole, UK. Proteasome inhibitor
(MG132) and c-Jun N-terminal kinase (JNK) inhibitor (AEG3482) were
purchased from Tocris Bioscience UK. Cyclic adenosine monophosphate
inhibitor (cAMPi) and MSU crystals were purchased from Enzo Life
Sciences, Germany. Monoclonal mouse IgG1 clone 9016, mouse
anti-human TGFβ1 (MAB240), recombinant PAF-AH, IL-1β and IL-6 ELISA
kits and cell lysis solution were purchased from R&D Systems,
Abingdon, UK. Manufacturer's instructions were followed for all
listed kits and reagents. Mouse anti-human IgG1 monoclonal antibody
(MAB3832) was used at 100 µg/ml, mouse anti-human CD163 IgG1
monoclonal antibody (clone 215927) was used at 2.5 µg per
105 cells and were both purchased from R&D Systems,
Abingdon, UK. Goat ant-human IgG whole molecule FITC was used at a
1:64 dilution in PBS and purchased from Sigma, Aldrich, Poole,
UK.
Monocyte and macrophage
preparation
The present study incorporated a series of in
vitro cell cultures of differentiated monocytes isolated from
human blood cones purchased from the National Health Service Blood
Bank (Colindale, London, England). Ethical approval was granted by
the Middlesex University Institutional Ethics Committee, Department
of Natural Sciences (London, England) and this was also a purchase
requirement from the blood bank. The present study complied with
the ethical standards established in 1964 in the Declaration of
Helsinki.
Leucocyte rich blood cones and blood group AB
positive serum were purchased from the NHS Cord blood and
transplant bank (Colindale, London, UK). The cones were washed with
PBS to and centrifuged on histopaque at 620 × g for 20 mins to
harvest leucocytes. The monocyte enriched fraction was obtained
from the interface, washed in HANKS balanced solution without
calcium or magnesium, after which cells were counted and cultured
into 24 well plates at 4×106 per ml in DMEM containing
1% penicillin and streptomycin at 37°C in an atmosphere containing
5% CO2. The mononuclear cells were allowed to adhere for
1 h at 37°C after which any non-adherent cells were removed by
aspiration and washing with PBS. The adherent cells were then
cultured in Dulbecco's media containing 10% AB serum either for
seven days to obtain macrophages or overnight for use as monocytes
(9–11). These methods were established
previously and macrophage differentiation was determined by light
microscopy and flow cytometry phenotypic analysis using the
macrophage differentiation marker CD163 (10). Cell culture media changes were
carried out at days 1, 3, 5 and 7 days of culture replacing with
fresh DMEM containing 10% AB serum.
Differentiated macrophages were stimulated with LPS,
(10 µg/ml) or MSU crystals (0.5 mg/ml) with or without
pre-treatment for 30 min with HC (1, 2 and 4 µM) Stimulation was
stopped after 24 h of incubation in all experiments. The 0.5 mg/ml
MSU concentration and time point (24 h) was used for all
experiments since this concentration and time point were previously
identified as optimum conditions from previous studies of
mononuclear and macrophage cell stimulation (10–13).
Macrophage stimulation
Macrophages were stimulated MSU crystals (0.5 mg/ml)
or LPS (10 µg/ml) with or without pre-treatment for 30 min at 37°C.
This was followed by the addition of pathway inhibitors: AEG3482, a
JNK inhibitor (12.5, 25 and 50 µM), MG132 a proteasome inhibitor
(50 and 100 µM), actin D (2 µM). Incubation was stopped after 24 h
in all experiments.
Treatment with colchicine or
hydrocortisone or anti-TGFβ1
Colchicine was prepared by dissolving in 1 ml
ethanol as recommended by manufacturer's instructions. Further
dilutions were carried out in DMEM media at concentrations of 125,
250 or 500 µg/ml. Hydrocortisone was dissolved in 1 ml DMEM and
further diluted in media to concentrations of 1, 2 and 4 µM. Mouse
anti-TGFβ1 and an isotype matched control IgG1 were both dissolved
in PBS and used at 2.5, 5, 10 ng/ml (anti-TGFβ1) and 10 µg/ml
(control IgG1) respectively. Macrophages were pre-incubated with
colchicine, hydrocortisone, anti-TGFβ1 or IgG1 control for 30 min
prior at 37°C to addition of MSU crystals for 24 h after which
supernatants were collected and analysed for PAF-AH content.
Monocyte stimulation with recombinant
PAF-AH
Recombinant PAF-AH was dissolved in 0.5 ml of PBS
and further diluted in DMEM media (2.5–10 ng/ml). Freshly isolated
monocytes were pre-treated with recombinant PAF-AH for 20 min prior
to the addition of MSU crystals and then incubated at 37°C for 24 h
after which supernatants were collected for cytokine analysis.
Enzyme linked immunosorbent assays
(ELISAs)
IL-1β and IL-6 concentrations in cell culture
supernatants were determined by sandwich ELISA using matched
antibodies (DuoSet: R&D Systems, Abingdon, UK). PAF-AH levels
were determined using a solid phase quantikine ELISA (DPLG70 from
R&D Systems) with sensitivity (0.8–50 ng/ml) in conditioned
cell culture supernatants. All samples were measured in duplicates
using manufacturer's instructions without any deviations. Results
were expressed as the mean ± SEM cytokine concentration (ng/ml)
from at least 4 experiments.
Statistical analysis
All samples were measured in duplicate from at least
three experiments, with results expressed as the mean ± standard
deviation. Statistical analysis was carried out by applying one-way
analysis of variance with Dunnett's post hoc test or Student's
t-test where appropriate. P<0.05 was considered to indicate a
statistically significant difference. All data were analysed using
Excel (2016 version; Microsoft Corporation, Redmond, WA, USA).
Results
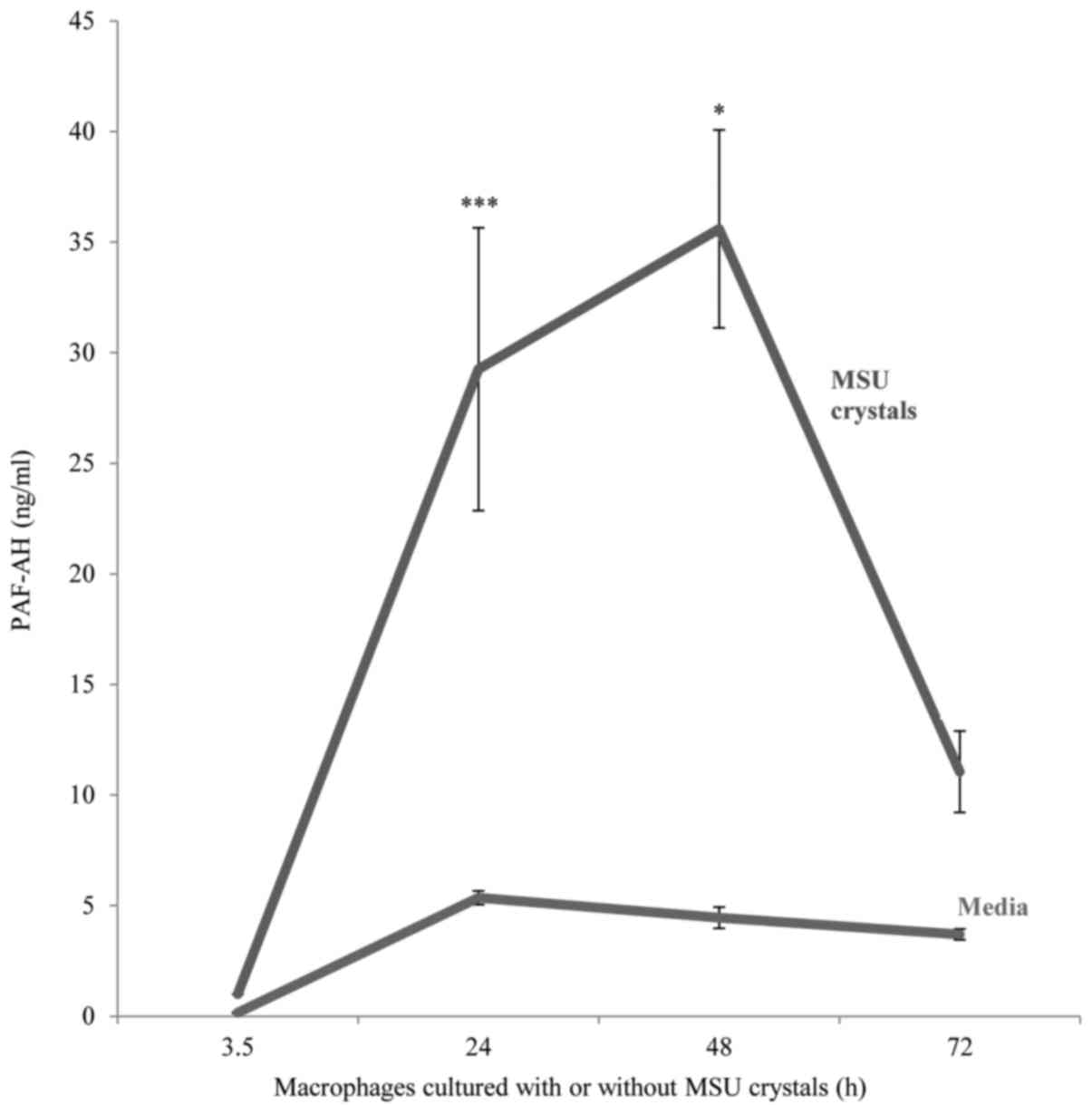
PAF-AH secretion by MSU crystal
stimulated macrophages
MSU crystal stimulation resulted in the upregulation
of PAF-AH secretion by in vitro differentiated macrophages
compared to unstimulated macrophages. Peak enzyme release was
detected at an incubation time of 24–48 h which was approximately
six-fold higher than unstimulated macrophages (mean ± SD; 29.3±6.3
ng/ml vs. 5.36±0.3 ng/ml respectively, P<0.001) after which a
decline in enzyme secretion occurred with levels reverting to near
baseline levels after 72 h (Fig.
1).
Effect of hydrocortisone, colchicine,
LPS, anti-TGFβ1 and pathway inhibitors on macrophage PAF-AH
secretion
Co-incubation of macrophages with hydrocortisone
(1–4 µM) significantly decreased MSU crystal stimulated PAF
secretion (Fig. 2A).
Interestingly, LPS-stimulated macrophages also release PAF-AH after
24 h of LPS stimulation. However when we added equivalent
concentration of HC to LPS stimulated macrophages we did not
observe a significant decrease in PAF-AH secretion (Fig. 2). Also colchicine did not
significantly alter the levels of PAF-AH at the doses used. In the
next stage of our investigation we tested a variety of pathway
inhibitors to investigate the signalling mechanisms involved in
this model. Notably, the JNK inhibitor AEG3482 did not decrease MSU
crystal-mediated PAF-AH secretion. In fact, PAF-AH release was
enhanced following incubation with this inhibitor (Fig. 3). Inclusion of a proteasome
inhibitor to the cultures significantly reduced PAF-AH release at
the doses used (Fig. 4). In
contrast, PAF-AH secretion was not significantly altered by the
addition of cyclic AMP inhibitor (Fig.
5) implying that this mode of intracellular signalling was not
involved in MSU-mediated PAF-AH secretion. The addition of actin D
into the co-cultures resulted in complete amelioration of PAF-AH
detection in this pathway as seen in Fig. 4 suggesting that MSU crystal de novo
synthesis of PAF-AH is required in macrophages. Addition of
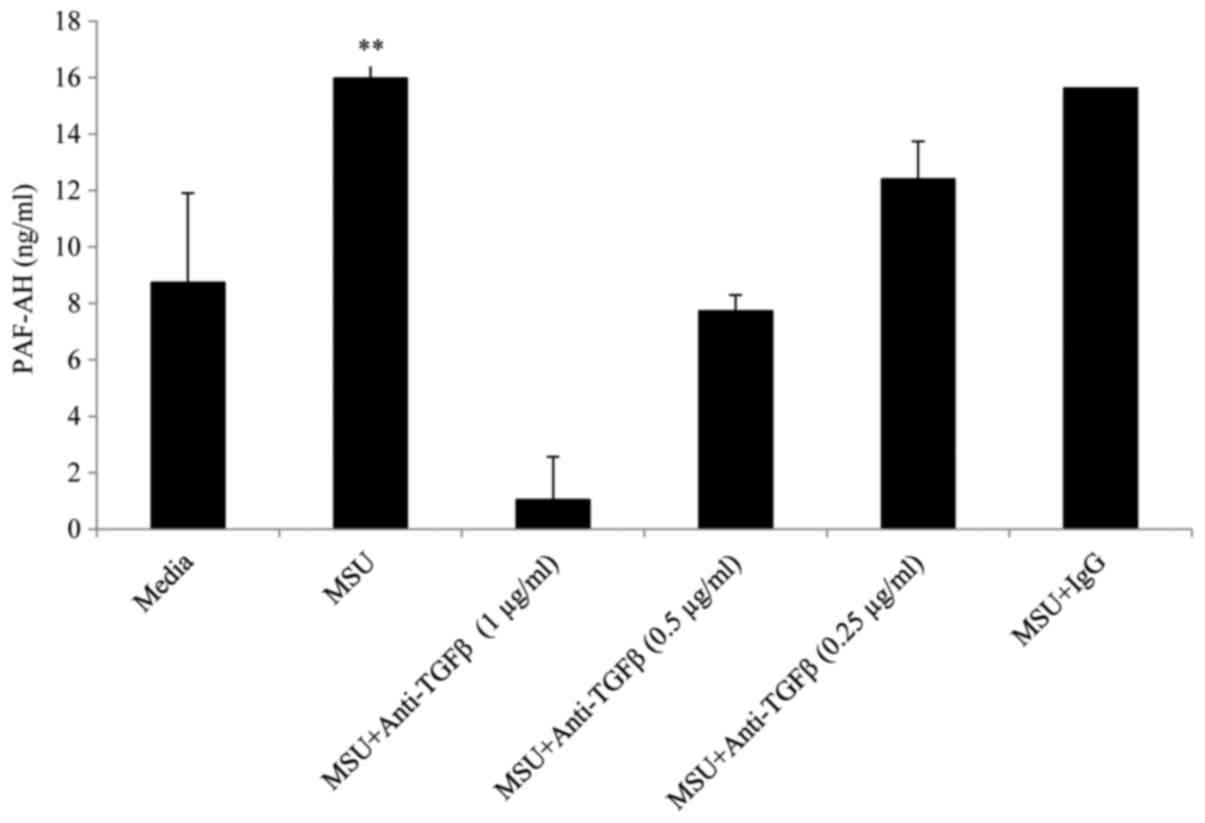
neutralising anti-TGFβ1 resulted in a dose dependent reduction in
PAF-AH at concentrations of 0.25, 0.5 and 1 µg/ml whereas an
isotype matched IgG control antibody at the same concentrations had
no effect on PAF-AH secretion (Fig.
5).
Treatment with recombinant PAF-AH
inhibits pro-inflammatory cytokine secretion by immature monocytes
stimulated with MSU crystals
We investigated whether the activity of recombinant
PAF-AH could also modify IL-1β and IL-6 secretion. Previously, we
had reported that recombinant PAF-AH decreases TNFα cytokine
secretion from immature human monocytes in a similar experimental
model (9).
Pre-treatment of undifferentiated, inflammatory
monocytes with recombinant PAF-AH at doses of 2.5, 5 and 10 ng/ml
and then MSU crystals for 24 h resulted in a dose dependent
decrease in IL-1β and IL-6 secretion. The highest inhibition was
achieved at 10 ng/ml recombinant PAF-AH (P=0.02 for IL-1β and
P=0.003 for IL-6 respectively, Fig.
6).
Discussion
To the best of our knowledge this is the first study
to identify that macrophage uptake of MSU crystals leads to an
upregulation in secretion of the enzyme PAF-AH. Little is known
about the enzymes involved in the inflammation resolution phase of
gout with research focussing mainly on caspase-1 activating NALP3
(23,24). PAF-AH is actually an
anti-inflammatory phospholipase that is found as a plasma isoform
complexed mainly with LDLs (25).
Low levels of PAF-AH seem to correlate with a number of modalities.
A study involving patients with acute allergy reactions reported
that those patients with the lowest levels of plasma PAF-AH were at
high risk of severe anaphylaxis (26). Also, mast cells derived from rat
bone marrow secrete PAF-AH upon direct IgE activation indicating
PAF-AH may have a potentially crucial function in pathological and
physiological defence. It is postulated that the likely function of
PAF-AH may be as a safety biosensor during inflammation. This is
due to its ability to neutralise the effects of inflammatory PAF
and oxidised phospholipids which are upregulated in pathologies
such as thrombosis, allergy, sepsis as well as gout (26,27).
A local increase in harmful lipids often accompanies sustained
inflammation. Thus upregulation of PAF-AH takes place in cells to
deactivate and degrade PAF. In fact joint inflammation in gout is
accompanied by a local increase in PAF, prostaglandin E2 and
leukotrienes (28,29). In this study we have demonstrated
that recombinant PAF-AH treatment may tip the inflammatory balance
in favour of inflammation resolution as it was able to decrease
monocyte derived IL-1β and IL-6 secretion due to MSU crystal
stimulation. These results are consistent with our previous study
in which PAF was upregulated by monocytes in response to MSU
crystals (9) and recombinant
PAF-AH administration downregulated TNFα secretion. The exact form
of PAF was not characterised; we propose that this was probably
inert lysosomal PAF. Physiologically PAF-AH functions on active
PAF, converting it into lysosomal-PAF (the inert form of PAF).
Active PAF is a potent lipid intermediate that has immunomodulatory
effects and a pivotal role in the pathogenesis of inflammatory
disorders such as cardiovascular disease rendering this molecule
inactive is important in healing (30). Given this function, it is not
surprising that administration of exogenous PAF-AH to animals with
systemic inflammatory response syndromes or sepsis resulted in
decreased leukocyte accumulation as well as pro-inflammatory
cytokine levels and increased bacterial clearance in septic mice.
Indeed PAF-AH enhances sepsis clearance by hydrolysing acetyl
groups attached to other lipid substrates besides PAF (31,32).
This function of the enzyme in neutralising inflammatory mediators
could be particularly crucial in inflammatory gout joints in which
a number of lipid mediators such as Prostaglandin E2 and PAF have
been located (28). In
endotoxaemic rats, an upregulation of plasma PAF-AH was accompanied
by a direct increase in ability to inactivate PAF and oxidised
phospholipids (33).
We discovered that blocking TGFβ1 activity using a
neutralising antibody resulted in a dose dependant decrease in
PAF-AH secretion. The anti-inflammatory activity of TGFβ1 is well
established in gout inflammation resolution (13). Our results suggest that this effect
may be attributed in part by the upregulation of PAF-AH.
Essentially both PAF-AH and TGFβ1 share a transcription binding
site involved in macrophage regulation, mediating effects through
canonical Sp1 (16,34). Hydrocortisone is known for its
anti-inflammatory properties through suppression of vascular
permeability, vasodilation and leukocyte migration into inflamed
sites (35). Unexpectedly in our
study hydrocortisone decreased PAF-AH secretion by MSU crystal
stimulated macrophages. However, there is evidence that
hydrocortisone may also have pro-inflammatory properties. For
example, hydrocortisone can interact with anti-inflammatory drugs
such as aspirin hydrolysis where it mediates esterase activity
(36). Furthermore, when normal
subjects were injected with 300 mg of hydrocortisone, there was an
increase in toll like inflammatory receptor 2, 5, 9, high mobility
group box-1 and matrix metalloproteinase-9 expression occurred
(37). Perhaps the long term use
of hydrocortisone therapy in gout or arthritis could result in
inadvertently driving the inflammatory response rather than
limiting it.
Co-incubation of MSU-stimulated macrophages with the
proteasome inhibitor MG132 resulted in a significant decrease in
PAF-AH secretion. This suggests that this pathway involves protein
degradation (38). Conversely,
colchicine had no significant effect. Colchicine is routinely
prescribed as a prophylactic treatment for gout at similar doses
tested in our study. It has recognized, powerful anti-inflammatory
properties in gout affecting cytokine levels; increasing levels of
TGFβ1, prostaglandins and lowering pro-inflammatory factor levels
(39).
Our results could be relevant to other arthropathic
disorders such as rheumatoid arthritis (RA) as well as gout.
Recently, it has been shown that PAF-AH levels are significantly
decreased in the sera of RA patients with active disease
highlighting that PAF-AH probably plays a protective role in the
development of RA (40). Moreover
analysis of the hypercoagulable state in RA patients showed
significant reduction in anti-inflammatory serum PAF-AH along with
a decrease in IL-4 and IL-10 but a rise in inflammatory mediators
such as IL-6, IL-17 and PAF (41).
Therefore future studies should examine the expression profile of
PAF-AH levels in the synovial fluid samples obtained from gout
patients during active disease compared to quiescent joints. The
efficacy of PAF-AH treatment for gout patients should also be
explored. In conclusion, we have demonstrated that PAF-AH enzyme
secretion is upregulated by macrophages following MSU crystal
uptake and involves anti-inflammatory TGFβ1. This research expands
our understanding of an important anti-inflammatory enzyme which
could be functioning as a biosensor responding to local
microenvironmental conditions.
Acknowledgements
Not applicable.
Funding
The present study was funded by the Middlesex
University Department of Natural Sciences Research Initiative
(2014).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
DY designed all of the experiments, collected and
analyzed the data, performed statistical analysis and wrote the
manuscript. FH contributed to data analysis and writing the
manuscript.
Ethics approval and consent to
participate
The present study incorporated a series of in
vitro cell cultures of differentiated monocytes isolated from
human blood cones purchased from the National Health Service Blood
Bank (Colindale, London, England). Ethical approval was granted by
the Middlesex University Institutional Ethics Committee, Department
of Natural Sciences (London, England) and this was also a purchase
requirement from the blood bank. The present study complied with
the ethical standards established in 1964 in the Declaration of
Helsinki.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hippocrates: The Genuine Works of
Hippocrates. I and II. Adams F: New York: William Wood and Company;
1886
|
|
2
|
Copeman WS: A short history of the gout
and the rheumatic diseases. Berkeley and Los Angeles: University of
California Press; 1964
|
|
3
|
Kuo CF, Grainge MJ, Mallen C, Zhang W and
Doherty M: Rising burden of gout in the UK but continuing
suboptimal management: A nationwide population study. Ann Rheum
Dis. 72:661–667. 2015. View Article : Google Scholar
|
|
4
|
di Giovine FS, Malawista SE, Thornton E
and Duff GW: Urate crystals stimulate production of tumour necrosis
factor alpha from human blood monocytes and synovial cells.
Cytokine mRNA and protein kinetics, and cellular distribution. J
Clin Invest. 87:1375–1381. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Schiltz C, Lioté F, Prudhommeaux F,
Meunier A, Champy R, Callebert J and Bardin T: Monosodium urate
monohydrate crystal induced inflammation in vivo: Quantitative
histomorphometric analysis of cellular events. Arthritis Rheum.
46:1643–1650. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Daoussi D, Andonpoulos I and Andonopoulos
AP: ACTH as a treatment for acute crystal induced arthritis: Update
on clinical evidence and mechanisms of action. Semin Arthritis
Rheum. 43:648–653. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Vanja SK, Rathinam VA and Fitzgerald KA:
Mechanisms of inflammasome activation: Recent advances and novel
insights. Trends Cell Biol. 25:308–315. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yagnik DR, Hillyer P, Marshall D, Smythe
CD, Krausz T, Haskard DO and Landis RC: Noninflammatory
phagocytosis of monosodium urate monohydrate crystals by mouse
macrophages. Implications for the control of joint inflammation in
gout. Arthritis Rheum. 43:1779–1789. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yagnik D: Macrophage derived platelet
activating factor implicated in the resolution phase of gouty
inflammation. Int J Inflam. 2014:5264962014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Landis RC, Yagnik DR, Florey O,
Philippidis P, Emons V, Mason JC and Haskard DO: Safe disposal of
inflammatory monosodium urate monohydrate crystals by
differentiated macrophages. Arthritis Rheum. 46:3026–3033. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yagnik DR, Evans BJ, Florey O, Mason JC,
Landis RC and Haskard DO: Macrophage release of transforming growth
factor beta1 during resolution of monosodium urate monohydrate
crystal-induced inflammation. Arthritis Rheum. 50:2273–2280. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Howard KM, Abdel-Al M, Ditmyer M and Patel
N: Lipopolysaccharide and platelet-activating factor stimulate
expression of platelet-activating factor acetylhydrolase via
distinct signaling pathways. Inflam Res. 60:735–744. 2011.
View Article : Google Scholar
|
|
13
|
Richette P, Doherty M, Pascual E, Barskova
V, Becce F, Castañeda-Sanabria J, Coyfish M, Guillo S, Jansen TL,
Janssens H, et al: 2016 updated EULAR evidence-based
recommendations for the management of gout. Ann Rheum Dis.
76:29–42. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Elstad MR, Stafforini DM, McIntyre TM,
Prescott SM and Zimmerman GA: Platelet-activating factor
acetylhydrolase increases during macrophage differentiation. A
novel mechanism that regulates accumulation of platelet-activating
factor. J Biol Chem. 25:8467–8470. 1989.
|
|
15
|
Narahara H and Johnston JM: Effects of
endotoxins and cytokines on the secretion of platelet-activating
factor-acetylhydrolase by human decidual macrophages. Am J Obstet
Gynecol. 169:531–537. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wu X, Zimmerman GA, Prescott SM and
Stafforini DM: The p38 MAPK pathway mediates transcriptional
activation of the plasma platelet-activating factor acetylhydrolase
gene in macrophages stimulated with lipopolysaccharide. J Biol
Chem. 279:36158–36165. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hattori K, Hattori M, Adachi H, Tsujimoto
M, Arai H and Inoue K: Purification and characterization of
platelet-activating factor acetylhydrolase II from bovine liver
cytosol. J Biol Chem. 270:22308–22313. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Stafforini DM, McIntyre TM, Zimmerman GA
and Prescott SM: Platelet-activating factor acetylhydrolases. J
BiolChem. 272:17895–17898. 1997.
|
|
19
|
Stafforini DM, Prescott SM, Zimmerman GA
and McIntyre TM: Mammalian platelet-activating factor
acetylhydrolases. Biochim Biophys Acta. 1301:161–173. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Stafforini DM, Prescott SM and McIntyre
TM: Human plasma platelet-activating factor acetylhydrolase.
Purification and properties. J Biol Chem. 262:4223–4230.
1987.PubMed/NCBI
|
|
21
|
Neto Castro Faria HC, Stafforini DM,
Prescott SM and Zimmerman GA: Regulating inflammation through the
anti-inflammatory enzyme platelet-activating
factor-acetylhydrolase. Mem Inst Oswaldo Cruz. 100 Suppl 1:S83–S91.
2005. View Article : Google Scholar
|
|
22
|
Blank ML, Lee T, Fitzgerald V and Snyder
F: A specific acetylhydrolase for
1-alkyl-2-acetyl-sn-glycero-3-phosphocholine (a hypotensive and
platelet-activating lipid). J Biol Chem. 256:175–178.
1981.PubMed/NCBI
|
|
23
|
Akira S, Misawa T, Satoh T and Saitoh T:
Macrophages control innate inflammation. Diabetes Obes Metab. 15
Suppl 3:S10–S18. 2013. View Article : Google Scholar
|
|
24
|
So AK and Martinon F: Inflammation in
gout: Mechanisms and therapeutic targets. Nat Rev Rheumatol.
13:639–647. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Triggiani M, Granata F, Giannattasio G and
Marone G: Secretory phospholipases A2 in inflammatory and allergic
diseases: Not just enzymes. J Allergy Clin Immunol. 116:1000–1006.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Perelman B, Adil A and Vadas P:
Relationship between platelet activating factor acetylhydrolase
activity and apolipoprotein B levels in patients with peanut
allergy. Allergy Asthma Clin Immunol. 10:202014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nakajima K, Murakami M, Yanoshita R,
Samejima Y, Karasawa K, Setaka M, Nojima S and Kudo I: Activated
mast cells release extracellular type platelet-activating factor
acetylhydrolase that contributes to autocrine inactivation of
platelet-activating factor. J Biol Chem. 272:19708–19713. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Vadas P, Browning J, Edelson J and
Pruzanski W: Extracellular phospholipase A2 expression and
inflammation: The relationship with associated disease states. J
Lipid Mediat. 8:1–30. 1993.PubMed/NCBI
|
|
29
|
Miguélez R, Palacios I, Navarro F,
Gutierrez S, Sanchez-Pernaute O, Egido J, González E and
Herrero-Beaumont G: Anti-inflammatory effect of a PAF receptor
antagonist and a new molecule with antiproteinase activity in an
experimental model of acute urate crystal arthritis. J Lipid Mediat
Cell Signal. 13:35–49. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tjolker LW, Eberhardt C, Unger J, Trong
HL, Zimmerman GA, McIntyre TM, Stafforini DM, Prescott SM and Gray
PW: Plasma platelet activating factor acetylhydrolase is a secreted
phospholipase A2 with a catalytic triad. J Biol Chem.
270:25481–25487. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gomes RN, Bozza FA, Amâncio RT, Japiassú
AM, Vianna RC, Larangeira AP, Gouvêa JM, Bastos MS, Zimmerman GA,
Stafforini DM, et al: Exogenous platelet activating factor
acetylhyhrolase reduces mortality in mice with systemic
inflammatory response syndrome and sepsis. Shock. 26:41–49. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Opal S, Laterre PF, Abraham E, Francois B,
Wittebole X, Lowry S, Dhainaut JF, Warren B, Dugernier T, Lopez A,
et al: Recombinant human platelet-activating factor acetylhydrolase
for treatment of severe sepsis: Results of a phase III,
multicenter, randomized, double-blind, placebo-controlled, clinical
trial. Crit Care Med. 32:332–341. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Howard KM and Olson MS: The expression and
localization of plasma platelet-activating factor acetylhydrolase
in endotoxemic rats. J Biol Chem. 275:19891–19896. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li JM, Datto MB, Shen X, Hu PP, Yu Y and
Wang XF: Sp1, but not Sp3, functions to mediate promoter activation
by TGF-beta through canonical Sp1 binding sites. Nucleic Acids Res.
26:2449–2456. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Coutinho AE and Chapman KE: The
anti-inflammatory and immunosuppressive effects of glucocorticoids,
recent developments and mechanistic insights. Mol Cell Endocrinol.
335:2–13. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhou G, Marathe GK, Hartiala J, Hazen SL,
Allayee H, Tang WH and McIntyre TM: Aspirin hydrolysis in plasma is
a variable function of butyrylcholinesterase and
platelet-activating factor acetylhydrolase 1b2 (PAFAH1b2). J Biol
Chem. 288:11940–11948. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Dandona P, Ghanim H, Sia CL, Green K,
Abuaysheh S, Dhindsa S, Chaudhuri A and Makdissi A: A mixed
anti-inflammatory and pro-inflammatory response associated with a
high dose of corticosteroids. Curr Mol Med. 14:793–801. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Guo N and Peng Z: Mg132, a proteasome
inhibitor, induces apoptosis in tumour cells. Asia Pac J Clin
Oncol. 9:6–11. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Dalbeth N, Lauterio TJ and Wolfe HR:
Mechanism of action of colchicine in the treatment of gout. Clin
Ther. 36:1465–1479. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Łuczaj W, Gindzienska-Sieskiewicz E,
Jaroka-karpowicz I, Andisic L, Sierakowski S, Zarkovic N, Waeg G
and Skrzydlewska E: The onset of lipid peroxidation in rheumatoid
arthritis: Consequences and monitoring. Free Radic Res. 50:304–313.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhang P, Liu J, Tan B, Zhu F and Fang L:
Hypercoagulable state is associated with NF-kappa B activation and
increased inflammatory factors in patients with rheumatoid
arthritis. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. 32:364–368.
2016.(In Chinese). PubMed/NCBI
|