Introduction
Autophagy is a membrane-dependent mechanism for the
turnover of subcellular components and represents a major cellular
homeostatic mechanism. It serves as a source of metabolic fuel, it
removes aggregated proteins or dysfunctional mitochondria, and
determines cell fate (1). In
addition to its basic role in the turnover of proteins and
organelles, autophagy has multiple physiological and
pathophysiological functions, including roles in cell
differentiation and immune response (2). Changes in autophagy affect the
removal of aged and defective mitochondria, protein aggregates and
internalized bacteria. The function of autophagy in human disease
appears to be pleiotropic, with implications in cancer, metabolic,
neurodegenerative, cardiovascular diseases, autoimmune, as well as
lung diseases (2,3). More specifically, in the lungs,
enhanced autophagy has been described in epithelial cells from
patients with chronic obstructive pulmonary disease (COPD), leading
to apoptosis (4,5). By contrast, the alveolar macrophages
of patients with COPD present an impairment in autophagy (6).
Idiopathic pulmonary fibrosis (IPF) is a chronic
progressive fibrotic disease of unknown etiology (7). The current dogma in the disease
pathogenesis centers on lung epithelial cell dysfunction in an
aging lung, which leads to fibroblast activation and excessive
extracellular matrix deposition, finally leading to the destruction
of the lung architecture. IPF shares some intriguing similarities
with rheumatoid lung disease, in pathogenesis (8,9), as
well as disease progression (10).
Rheumatoid arthritis (RA) is an autoimmune disease, characterized
by joint inflammation and is thought to be driven by antibodies
against citrullinated proteins (11). Interstitial lung disease (ILD)
commonly complicates RA, in up to 60% in certain studies, and
frequently precedes joint inflammation (12). Smoking, a common risk factor in
both idiopathic- and autoimmune-driven lung disease, is associated
with changes in the autophagy pathway. The accumulation of
autophagosomes is present in lung tissue from patients with COPD
(5) and in lung epithelial cells
exposed to cigarette smoke extract (13).
Aging, a hallmark of IPF pathogenesis, is also
associated with insufficient autophagy (14), while increasing evidence suggests
that the autophagic process is impaired in IPF (15,16).
By contrast, in RA, increased autophagy has been linked with the
enhanced survival of RA fibroblast-like synoviocytes (RA-FLS)
(17), a major source of
pro-inflammatory cytokines. Intriguingly, the induction of
autophagy in antigen-presenting cells can lead to citrullinated
peptide presentation, a hallmark of RA pathogenesis (18). In alveolar macrophages,
citrullinated peptides are increased in anti-CCP-positive RA
patients, as well as in IPF (19).
Thereafter, a contradicting role of autophagy in both diseases has
been proposed and it is likely that the role of autophagy is
cell-specific, as previous studies on COPD have suggested (5,6,13).
Alveolar macrophages are essential for maintaining
the sterility of the lung (20)
and have been proposed to play a role in lung fibrosis (21), both in initiating the inflammatory
response after injury, as well as in resolution and repair. As
regards the autophagy process in macrophages in fibrosis, little is
known. Of note, a study by Drakopanagiotakis et al
identified that alveolar macrophages of patients with IPF had a
decreased apoptotic rate, which may enhance the progression of IPF
(22), while AKT1 has been shown
to mediate apoptosis resistance by enhancing mitophagy (23).
Therefore, the purpose of the current study was to
examine the levels of the major regulatory molecules of the
autophagic pathway in bronchoalveolar lavage fluid (BALF) cells
derived from patients with IPF and RA-ILD. Although RA is an
autoimmune disease mainly associated with inflammatory pathways and
is characterized by a different disease progression and therapeutic
strategies, it is known that lung involvement in RA leading to ILD
and destruction of the lung parenchyma, finally results in the same
survival as IPF. Thus, apart from obvious differences, these two
diseases present some rather intriguing similarities at the
clinical and molecular level (24,25).
It is of great interest to investigate the molecular pathways and
potent pathogenetic mechanisms, not only by focusing on systemic
markers or joint inflammation, but also by evaluating lung
microenvironment.
Materials and methods
Patients
A total of 75 subjects were recruited from the
Department of Thoracic Medicine, University Hospital of Heraklion,
Greece, consisting of patients with IPF (n=55) and patients with
RA-ILD (n=20) between March, 2014 to March, 2017.
The diagnosis of IPF was based on European
Respiratory Society (ERS)/American Thoracic Society (ATS) clinical
guidelines and high resolution computed tomography (HRCT) criteria
or on open or video-assisted thoracoscopic biopsy (n=5), with all
biopsies reviewed by the same two histopathologists (26). In accordance with the
aforementioned criteria, any patient presenting any known cause of
pulmonary fibrosis, such as a systemic connective tissue disorder,
was excluded from this study using both immunologic screening and
rheumatologic clinical evaluation (26). All patients with IPF were newly
diagnosed and had not received any previous treatment. The criteria
for the diagnosis of connective tissue disease CTD included the
American College of Rheumatology (ACR) 1987 revised criteria for
the classification of RA (27).
Patients with RA-ILD had HRCT findings indicative of definite ILD.
Informed consent was obtained from all patients who participated in
this study. This study was approved by the Ethics Committees of the
University Hospital of Heraklion (Crete, Greece; IRB no. 17030)
BALF processing
BALF was obtained from all patients at room
temperature. Briefly, a flexible bronchoscope was wedged into a
sub-segmental bronchus of a predetermined region of interest based
on radiographical findings. A BAL technique was performed by
instilling a total of 180 ml of normal saline in 60-ml aliquots,
each retrieved by low suction. Samples were filtered through
sterile 70-nm cell strainers (BD Biosciences, San Jose, CA, USA)
and centrifuged at 500 × g or 5 min at 4°C. Cell pellets were
washed and re-suspended with cold PBS. Total cell count and cell
viability were subsequently assessed using Trypan blue (ICN
Pharmaceuticals, Costa Mesa, CA, USA). A total of 1–1.5 million
cells were centrifuged and cell pellets were homogenised in
TriReagent™ (MBL) for total RNA, or RIPA buffer (Invitrogen/Thermo
Fisher Scientific, Waltham, MA, USA) containing protease and
phosphatase inhibitors; Pierce/Thermo Fisher Scientific) for
SDS-PAGE/western blot analysis, followed by storage at −80°C.
Differential cell population count was analysed by
May-Grunewald-Giemsa staining as previously described (19).
RNA extraction and mRNA expression
analysis
Total RNA was isolated from BALF cells using the
mirVana™ miRNA isolation kit (Ambion/Thermo Fisher Scientific) with
minor modifications. The quality and quantity of the isolated RNA
was assessed by agarose gel electrophoresis and spectrophotometry
(NanoDrop/Thermo Fisher Scientific), respectively. For gene
expression analyses, 500 ng of total RNA were first treated with
DNA-free (Ambion/Thermo Fisher Scientific) in order to remove
genomic DNA contamination, followed by 1st strand cDNA synthesis
using Maxima RT™ and real-time qPCR analysis using Maxima
SYBR-Green pPCRmix (both from Fermentas/Thermo Fisher Scientific)
on a Mx3005P qPCR system (Agilent Technologies, Santa Clara, CA,
USA). The thermocycling conditions were as follows: 95°C for 5 min,
40 cycles of 95°C for 20 sec, 55°C for 20 sec and 72°C for 20 sec,
followed by melting curve with a ramp speed of 1°C per sec between
60°C and 95°C. The probe and primer sequences are summarized in
Table I. GAPDH levels were used as
endogenous control for the normalization of mRNA expression levels
in BALF samples. Relative expression values for the patient cohort
were calculated by the following equation: Relative gene expression
= Effgoi(Calibrator Ctgoi -
SampleCtgoi)/Effref(Calibrator Ctref
-SampleCtref), where (Eff refers to efficiency, goi to gene
of interest, and ref to reference gene).
 | Table I.The sequences of the primers
used. |
Table I.
The sequences of the primers
used.
| Gene name | Primer
sequences |
|---|
| p62 | F:
agctgccttgtacccacatc |
|
| R:
cagagaagcccatggacag |
| BECLIN1 | F:
tcaccatccaggaactcaca |
|
| R:
tggctcctctcctgagttagtc |
| ULK1 | F:
gccctcgtacccaagctc |
|
| R:
gaggccagggtcttctgc |
| BNIP3 | F:
tgctgctctctcatttgctg |
|
| R:
gactccagttcttcatcaaaaggt |
| PINK1 | F:
ggagtatggagcagtcacttacag |
|
| R:
ggcagcacatcagggtagtc |
| PARKIN | F:
cacctacccagtgaccatga |
|
| R:
cgacctccactgggaaac |
| GAPDH | F:
agccacatcgctcagacac |
|
| R:
gcccaatacgaccaaatcc |
Western blot analysis
Total protein lysates (20 ng) of BALF samples were
lysed in RIPA buffer including protease inhibitor (both from Thermo
Fisher Scientific). Total protein mass was determined by Coomassie
(Bradford protein assay kit) (Thermo Fisher Scientific). The
proteins were separated by 12% SDS-PAGE and transferred onto 0.45
nm nitrocellulose membranes (Bio-Rad, Hercules, CA, USA), followed
by the detection of p62 with an anti-p62 mouse monoclonal antibody,
at a 1/500 dilution (M162-3; MBL, Woburn, MA, USA), and β-actin
using an anti-β-actin mouse monoclonal antibody at a 1/5,000
dilution (A2228; Sigma, St. Louis, MO, USA). Appropriate goat
anti-mouse HRP conjugated secondary antibody at a 1/2,000 dilution
(AP124P; Chemicon/Merck KGaA, Darmstadt, Germany) was used and
immunodetection was performed with enhanced chemiluminescence
reagent Luminata™ (Millipore, Billerica, MA, USA). Bands were
visualised with the ChemiDoc XRS+ system and densitometric analyses
were performed using Image Lab™ software (both from Bio-Rad).
Statistical analysis
Gene expression analysis was performed using Prism 5
software following the incorporation of relative expression values
in average (duplicates) normalized to GAPDH. Group comparisons were
made using the Mann-Whitney U test or the Chi-square testing as
appropriate. A P-value <0.05 was considered to indicate a
statistically significant difference.
Results
Patient characteristics
The data of the patients, including those in the IPF
and RA-ILD group used in this study are summarized in Table II.
| Table II.Patient characteristics. |
Table II.
Patient characteristics.
|
Characteristics | IPF | RA-ILD | P-value |
|---|
| Number | 55 | 20 |
|
| Sex
(male/female) | 47/8 | 7/13 | <0.0001 |
| Packyears | 15 (0–150) | 0 (0–175) | NS |
| Non-smokers | 16 | 10 | NS |
| Ex smokers | 30 | 4 | 0.02 |
| Current
smokers | 5 | 3 | NS |
| Age (years) | 68 (27–86) | 67.50 (46–81) | NS |
Increased expression of BECLIN1 in
BALF cells derived from patients with RA-ILD compared with those
from patients with IPF
The initiation of autophagosome formation requires
specific regulatory complexes, including two major contributors,
BECLIN1 and Unc-51 like autophagy activating kinase 1
(ULK1). The expression levels of these two essential
molecules were examined in BALF cells derived from patients with
IPF and RA-ILD. Statistically significant higher mRNA levels of
BECLIN1 were observed in the BALF cells from patients with
RA-ILD in comparison to those from patients with IPF (Fig. 1), while similar levels of
ULK1 were detected (Table
III).
| Table III.Expression levels of the genes
analyzed. |
Table III.
Expression levels of the genes
analyzed.
| Gene | IPF | RA-ILD | P-value |
|---|
| p62 | 0.90
(0.32–5.47) | 0.95
(0.49–2.22) | NS |
| BNIP3 | 0.98
(0.16–10.37) | 1.07 (0–14.29) | NS |
| BECLIN1 | 0.83
(0.32–1.88) | 1.09
(0.56–1.66) | <0.01 |
| ULK1 | 1.24
(0.40–3.59) | 1.09 (0–8.76) | NS |
| S100A9 | 0.70
(0.28–5.71) | 0.81
(0.35–3.22) | NS |
| PINK1 | 0.99
(0.26–2.33) | 0.83
(0.12–2.68) | NS |
| PARKIN | 0.99 (0–3.64) | 0.88 (0–1.68) | NS |
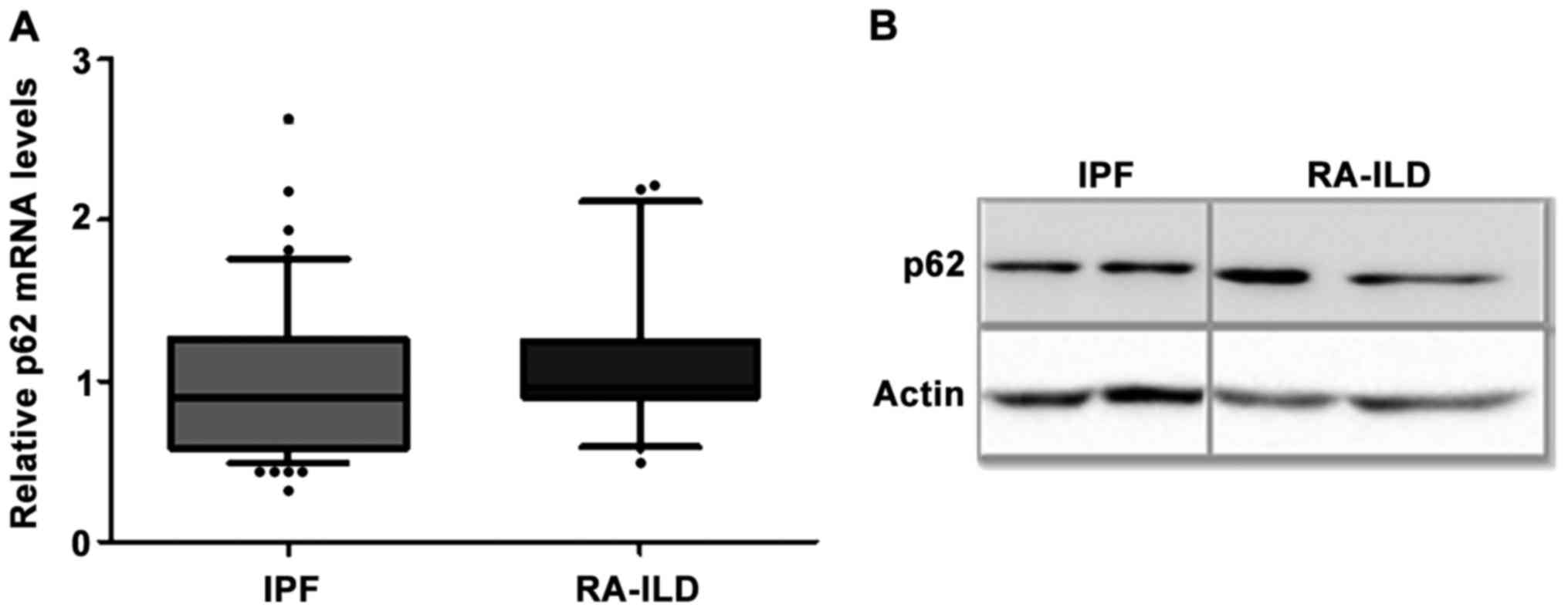
No differences in the mRNA and protein
levels of the adaptor molecule p62 in BALF cells of IPF and RA-ILD
patients
A major role in the selective degradation of
components or organelles is played by the adaptor molecules, mainly
including p62 (also known as sequestosome1) and BCL2
interacting protein 3 (BNIP3). Although the adaptor
molecules are not required for autophagosome formation, they were
evaluated in order to assess the levels of autophagy. The mRNA
levels of p62 (Fig. 2A) and
BNIP3 (Table III) were
measured in BALF cells and no significant difference was observed
between the IPF and RA-ILD groups. Moreover, the measurement of the
p62 protein levels is a widely used indicator of degradation
through autophagy machinery (2,28).
Thus, the protein levels of p62 in fresh BALF cells of a subgroup
of patients were also analyzed and they were found similar in the
IPF and RA-ILD samples (Fig.
2B).
Evaluation of markers of mitochondrial
homeostasis
The role of impaired mitochondrial recycling has
been highlighted in aging lung and in pulmonary disorders including
IPF (14,29,30).
Mitochondrial dysfunction due to insufficient autophagy has been
shown in alveolar epithelial cells (AEC)IIs and fibroblasts of IPF
patients (30–32). The two mostly investigated
molecules that determine mitochondrial degradation are PTEN-induced
putative kinase 1 (PINK1) and PARKIN. No differences
in the expression levels of PINK1 and PARKIN were
observed between the IPF (n=55) and RA-ILD (n=20) groups (Fig. 3A and B). The PINK1 protein levels
were also evaluated in the IPF (n=11) and RA-ILD (n=7) BALF cells
and no significant difference was observed (Mann-Whitney U test, no
significance). A representative western blot is presented in
Fig. 3C.
Discussion
Idiopathic- and autoimmune-related ILD share some
intriguing similarities in clinical presentation (10,33),
as well as in pathogenetic processes (19,34)
and genetic predisposition (35).
In the current study, gene and protein expression of key
autophagy-related molecules were evaluated in BALF cells derived
from patients with IPF and RA-ILD. BECLIN1 expression was
significantly upregulated in the BALF cells derived from patients
with RA-ILD compared to those derived from patients with IPF.
However, additional gene expression analysis of p62, ULK1
and BNIP3, also involved in the autophagy pathway, did not
display any statistically significant differences between the
groups. As regards the selective degradation of mitochondria by
mitophagy, we found the same mRNA expression levels of PINK1
and PARKIN, while no significant differences were observed
in the protein levels of PINK1.
Oxidative stress, endoplasmic reticulum stress and
hypoxia are not only mechanisms implicated in the pathogenesis of
IPF, but also inducers of autophagy (29).A previous study by Patel et
al examined markers of autophagic activity (LC3 and p62) in
human IPF lungs and the number of autophagosomes and it was
detected that autophagy was not induced in human IPF lungs
(15). Another study claimed that
there was insufficient autophagy in alveolar epithelial cells in
lungs of patients with IPF, which may lead to epithelial cell
senescence, whereas in fibroblasts, insufficient autophagy may
induce differentiation into myofibroblasts (36). Bueno et al proposed that
there may be an induction of the autophagy process in alveolar
epithelial cells (AECs) in IPF, but finally the autophagy flux is
impaired (31). Contrariwise, in
RA, increased autophagy has been associated with RA fibroblast-like
synoviocytes (RA-FLS) (37),
leading to the production of citrullinated proteins, a hallmark in
disease pathogenesis (38). Of
note, one commonly used medication for RA treatment is
hydroxychloroquine (Plaquenil), a disease-modifying anti-rheumatic
agent. Notably, hydroxychloroquine is an inhibitor of lysosomal
acidification and mostly accumulates in acidic cytoplasmic
vesicles. Another study demonstrated that the inhibition of the
autophagy-related protein, histone deacetylase 6 (HDAC6), reduces
the inflammatory cytokine secretion from macrophages and FLS, and
ameliorates arthritis disease severity in mouse models (39). Alterations to the regulation of
autophagy have also been identified to contribute to the
progression of other autoimmune diseases (40).
Although autophagy has been well described in IPF
AECs and fibroblasts and in RA sunovium, its role in alveolar
macrophages remains unknown. Alveolar macrophages represent very
important cells in lung composition, since they play a central role
in inflammation, host defense and tissue homeostasis. They
participate in all stages of the fibrotic process, as they serve as
key regulators of fibroblast recruitment, proliferation and
activation. It is currently believed that macrophages within the
lung orchestrate the downstream progression and maintenance of
fibrosis (13). Furthermore,
smoking, related to the pathogenesis of both diseases, has been
linked to a defect in functional autophagy in alveolar macrophages
(6). BAL is a minimally invasive
procedure, providing immune cells from the alveolar compartment and
is an excellent research tool for the investigation of alveolar
macrophages. This study demonstrated that, in BALF cells, the mRNA
levels of major contributors to the selective degradation of
cellular components; p62, BNIP3 and ULK1, were
similarly expressed in idiopathic and autoimmune lung fibrosis.
Of note, BECLIN1, a major autophagic
regulator and tumor suppressor protein (41), was found significantly upregulated
in BALF cells derived from patients with RA-ILD compared to those
from patients with IPF. BECLIN1 is required for the initiation of
autophagosome formation. Hypoxia, a critical component in
interstitial lung diseases (42,43)
regulates autophagy, and this activation requires BECLIN1 (44). Recent data have indicated that
BECLIN1 is downregulated in fibroblasts from patients with
IPF (45). Furthermore,
Nintedanib, an approved medication for the treatment of IPF, has
been demonstrated to induce autophagy in IPF fibroblasts in a
BECLIN1-dependent manner (16). In
this study, although the mRNA levels of BECLIN1 were
significantly lower in BALF cells from patients with IPF compared
to those from patients with RA-ILD, this is not a sufficient marker
alone to imply the decrease in the regulation of autophagy in
IPF.
Mitophagy is a form of macroautophagy that
selectively degrades damaged mitochondria. Mitochondrial kinase
PINK1 senses damage and signals this to the cytosolic E3 ligase,
PARKIN. PINK1 expression decreases with age, leading to the
accumulation of damaged mitochondria. In lungs affected by IPF,
further dysregulation of many of the regulatory mechanisms that
control mitochondrial function has recently been identified in
epithelial cells, fibroblasts and macrophages (30). Dysfunctional mitochondria in IPF
have been associated with the decreased expression of the major
regulatory molecule of mitophagy, PINK1, in AECs from
patients with IPF (31); however,
of note, no difference was observed in fibroblasts (31). PINK1-deficient mice exhibit
susceptibility to apoptosis and spontaneous TGFβ-driven lung
fibrosis and a low expression of PINK1 promotes fibroblast to
myofibroblast transition (46).
Similarly, PARKIN deficiency enhanced myofibroblast
differentiation and pro-fibrotic signaling (32). Notably, in this study, we observed
that, PINK1 and PARKIN were similarly expressed in
BALF cells from patients with IPF and RA-ILD, which suggests a role
of mitophagy in rheumatoid lung disease.
Although the results of this study provide evidence
of similar levels of major autophagy molecules in BALF cells from
patients with IPF and RA-ILD, certain limitations should be noted.
Autophagy is a dynamic process and the evaluation of the expression
of key molecules in the process, is not an efficient marker of the
autophagic levels. A better characterization of the autophagic
pathway in interstitial lung disease requires the evaluation not
only of the mRNA levels of certain regulatory molecules and the
protein levels at a steady state condition, but also an assessment
of the autophagic flux (28). For
instance, using a substance that can block the turnover of
autophagosomes is a widely used method to estimate the potent
accumulation of autophagosomes.
Conclusively, the findings of this study provide
insight into the autophagic pathway in BALF cells derived from
patients with both idiopathic- and autoimmune-related ILD. Further
research is required to better define the autophagy machinery in
BALF cells from patients with ILD, with regards to understanding
the disease pathogenesis and establishing novel therapeutic
targets.
Acknowledgements
Not applicable.
Funding
This study was funded by an annual research grand of
the Hellenic Thoracic Society HTS-2013.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
EV and SS performed the experiments and analyzed and
interpreted the data. EB, AT and GM also contributed to the
acquisition of human samples and interpretation of the data. EV and
AT wrote the manuscript. DAS, NT and KA made substantial
contributions to the conception and design of this study. All
authors were involved in the drafting of the manuscript, and all
have read and approved the final version of the manuscript.
Ethics approval and consent to
participate
Informed consent was obtained from all patients who
participated in this study. This study was approved by the Ethics
Committees of the University Hospital of Heraklion (Crete, Greece;
IRB no. 17030).
Patient consent for publication
Not applicable.
Competing interests
DAS is the Editor-in-Chief for the journal, but had
no personal involvement in the reviewing process, or any influence
in terms of adjudicating on the final decision, for this
article.
Glossary
Abbreviations
Abbreviations:
|
IPF
|
idiopathic pulmonary fibrosis
|
|
RA
|
rheumatoid arthritis
|
|
RA-ILD
|
rheumatoid arthritis-interstitial lung
disease
|
|
HRCT
|
high resolution computed
tomography
|
|
COPD
|
chronic obstructive pulmonary
disease
|
|
BALF
|
bronchoalveolar lavage fluid
|
|
AECs
|
alveolar epithelial cells
|
|
RA-FLS
|
rheumatoid arthritis fibroblast-like
synoviocytes
|
References
|
1
|
Cuervo AM: Autophagy and aging: Keeping
that old broom working. Trends Genet. 24:604–612. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Deretic V, Saitoh T and Akira S: Autophagy
in infection, inflammation and immunity. Nat Rev Immunol.
13:722–737. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nakahira K, Pabon Porras MA and Choi AM:
Autophagy in pulmonary diseases. Am J Respir Crit Care Med.
194:1196–1207. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hwang JW, Chung S, Sundar IK, Yao H,
Arunachalam G, McBurney MW and Rahman I: Cigarette smoke-induced
autophagy is regulated by SIRT1-PARP-1-dependent mechanism:
Implication in pathogenesis of COPD. Arch Biochem Biophys.
500:203–209. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen ZH, Kim HP, Sciurba FC, Lee SJ,
Feghali-Bostwick C, Stolz DB, Dhir R, Landreneau RJ, Schuchert MJ,
Yousem SA, et al: Egr-1 regulates autophagy in cigarette
smoke-induced chronic obstructive pulmonary disease. PLoS One.
3:e33162008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Monick MM, Powers LS, Walters K, Lovan N,
Zhang M, Gerke A, Hansdottir S and Hunninghake GW: Identification
of an autophagy defect in smokers' alveolar macrophages. J Immunol.
185:5425–5435. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wolters PJ, Collard HR and Jones KD:
Pathogenesis of idiopathic pulmonary fibrosis. Annu Rev Pathol.
9:157–179. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Johnson C, Giles JT, Bathon J, Lederer D,
Hoffman EA, Barr RG and Danoff SK: Smoking and Subclinical ILD in
RA versus the Multi-Ethnic Study of Atherosclerosis. PLoS One.
11:e01530242016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bernstein EJ, Barr RG, Austin JHM, Kawut
SM, Raghu G, Sell JL, Hoffman EA, Newell JD Jr, Watts JR Jr, Nath
PH, et al: Rheumatoid arthritis-associated autoantibodies and
subclinical interstitial lung disease: The Multi-Ethnic Study of
Atherosclerosis. Thorax. 71:1082–1090. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Solomon JJ, Chung JH, Cosgrove GP,
Demoruelle MK, Fernandez-Perez ER, Fischer A, Frankel SK, Hobbs SB,
Huie TJ, Ketzer J, et al: Predictors of mortality in rheumatoid
arthritis-associated interstitial lung disease. Eur Respir J.
47:588–596. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Luban S and Li ZG: Citrullinated peptide
and its relevance to rheumatoid arthritis: An update. Int J Rheum
Dis. 13:284–287. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Olson AL, Swigris JJ, Sprunger DB, Fischer
A, Fernandez-Perez ER, Solomon J, Murphy J, Cohen M, Raghu G and
Brown KK: Rheumatoid arthritis-interstitial lung disease-associated
mortality. Am J Respir Crit Care Med. 183:372–378. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kim HP, Wang X, Chen ZH, Lee SJ, Huang MH,
Wang Y, Ryter SW and Choi AM: Autophagic proteins regulate
cigarette smoke-induced apoptosis: Protective role of heme
oxygenase-1. Autophagy. 4:887–895. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cuervo AM, Bergamini E, Brunk UT, Dröge W,
Ffrench M and Terman A: Autophagy and aging: The importance of
maintaining ‘clean’ cells. Autophagy. 1:131–140. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Patel AS, Lin L, Geyer A, Haspel JA, An
CH, Cao J, Rosas IO and Morse D: Autophagy in idiopathic pulmonary
fibrosis. PLoS One. 7:e413942012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rangarajan S, Kurundkar A, Kurundkar D,
Bernard K, Sanders YY, Ding Q, Antony VB, Zhang J, Zmijewski J and
Thannickal VJ: Novel mechanisms for the antifibrotic action of
Nintedanib. Am J Respir Cell Mol Biol. 54:51–59. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shin YJ, Han SH, Kim DS, Lee GH, Yoo WH,
Kang YM, Choi JY, Lee YC, Park SJ, Jeong SK, et al: Autophagy
induction and CHOP under-expression promotes survival of
fibroblasts from rheumatoid arthritis patients under endoplasmic
reticulum stress. Arthritis Res Ther. 12:R192010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ireland JM and Unanue ER: Autophagy in
antigen-presenting cells results in presentation of citrullinated
peptides to CD4 T cells. J Exp Med. 208:2625–2632. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Samara KD, Trachalaki A, Tsitoura E,
Koutsopoulos AV, Lagoudaki ED, Lasithiotaki I, Margaritopoulos G,
Pantelidis P, Bibaki E, Siafakas NM, et al: Upregulation of
citrullination pathway: From Autoimmune to Idiopathic Lung
Fibrosis. Respir Res. 18:2182017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Selman M and Pardo A: Revealing the
pathogenic and aging-related mechanisms of the enigmatic idiopathic
pulmonary fibrosis. an integral model. Am J Respir Crit Care Med.
189:1161–1172. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wynn TA and Vannella KM: Macrophages in
tissue repair, regeneration, and fibrosis. Immunity. 44:450–462.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Drakopanagiotakis F, Xifteri A, Tsiambas
E, Karameris A, Tsakanika K, Karagiannidis N, Mermigkis D,
Polychronopoulos V and Bouros D: Decreased apoptotic rate of
alveolar macrophages of patients with idiopathic pulmonary
fibrosis. Pulm Med. 2012:9817302012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Larson-Casey JL, Deshane JS, Ryan AJ,
Thannickal VJ and Carter AB: Macrophage Akt1 kinase-mediated
mitophagy modulates apoptosis resistance and pulmonary fibrosis.
Immunity. 44:582–596. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Antoniou KM, Margaritopoulos G, Economidou
F and Siafakas NM: Pivotal clinical dilemmas in collagen vascular
diseases associated with interstitial lung involvement. Eur Respir
J. 33:882–896. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lake F and Proudman S: Rheumatoid
arthritis and lung disease: From mechanisms to a practical
approach. Semin Respir Crit Care Med. 35:222–238. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Raghu G: Idiopathic pulmonary fibrosis:
Guidelines for diagnosis and clinical management have advanced from
consensus-based in 2000 to evidence-based in 2011. Eur Respir J.
37:743–746. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Arnett FC, Edworthy SM, Bloch DA, McShane
DJ, Fries JF, Cooper NS, Healey LA, Kaplan SR, Liang MH, Luthra HS,
et al: The American Rheumatism Association 1987 revised criteria
for the classification of rheumatoid arthritis. Arthritis Rheum.
31:315–324. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Barth S, Glick D and Macleod KF:
Autophagy: Assays and artifacts. J Pathol. 221:117–124. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Margaritopoulos GA, Tsitoura E, Tzanakis
N, Spandidos DA, Siafakas NM, Sourvinos G and Antoniou KM:
Self-eating: Friend or foe? The emerging role of autophagy in
idiopathic pulmonary fibrosis. BioMed Res Int. 2013:4204972013.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mora AL, Bueno M and Rojas M: Mitochondria
in the spotlight of aging and idiopathic pulmonary fibrosis. J Clin
Invest. 127:405–414. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bueno M, Lai YC, Romero Y, Brands J, St
Croix CM, Kamga C, Corey C, Herazo-Maya JD, Sembrat J, Lee JS, et
al: PINK1 deficiency impairs mitochondrial homeostasis and promotes
lung fibrosis. J Clin Invest. 125:521–538. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kobayashi K, Araya J, Minagawa S, Hara H,
Saito N, Kadota T, Sato N, Yoshida M, Tsubouchi K, Kurita Y, et al:
Involvement of PARK2-mediated mitophagy in idiopathic pulmonary
fibrosis pathogenesis. J Immunol. 197:504–516. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Park JH, Kim DS, Park IN, Jang SJ,
Kitaichi M, Nicholson AG and Colby TV: Prognosis of fibrotic
interstitial pneumonia: Idiopathic versus collagen vascular
disease-related subtypes. Am J Respir Crit Care Med. 175:705–711.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Antoniou KM, Walsh SL, Hansell DM, Rubens
MR, Marten K, Tennant R, Hansel T, Desai SR, Siafakas NM, du Bois
RM, et al: Smoking-related emphysema is associated with idiopathic
pulmonary fibrosis and rheumatoid lung. Respirology. 18:1191–1196.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Juge PA, Borie R, Kannengiesser C, Gazal
S, Revy P, Wemeau-Stervinou L, Debray MP, Ottaviani S,
Marchand-Adam S, Nathan N, et al: FREX consortium: Shared genetic
predisposition in rheumatoid arthritis-interstitial lung disease
and familial pulmonary fibrosis. Eur Respir J. 49:492017.
View Article : Google Scholar
|
|
36
|
Araya J, Kojima J, Takasaka N, Ito S,
Fujii S, Hara H, Yanagisawa H, Kobayashi K, Tsurushige C, Kawaishi
M, et al: Insufficient autophagy in idiopathic pulmonary fibrosis.
Am J Physiol Lung Cell Mol Physiol. 304:L56–L69. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kato M, Ospelt C, Gay RE, Gay S and Klein
K: Dual role of autophagy in stress-induced cell death in
rheumatoid arthritis synovial fibroblasts. Arthritis Rheumatol.
66:40–48. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sorice M, Iannuccelli C, Manganelli V,
Capozzi A, Alessandri C, Lococo E, Garofalo T, Di Franco M,
Bombardieri M, Nerviani A, et al: Autophagy generates citrullinated
peptides in human synoviocytes: A possible trigger for
anti-citrullinated peptide antibodies. Rheumatology (Oxford).
55:1374–1385. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lee J, Hong EC, Jeong H, Hwang JW, Kim H,
Bae EK, Ahn JK, Choi YL, Han J, Cha HS, et al: A novel histone
deacetylase 6-selective inhibitor suppresses synovial inflammation
and joint destruction in a collagen antibody-induced arthritis
mouse model. Int J Rheum Dis. 18:514–523. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Rockel JS and Kapoor M: Autophagy:
Controlling cell fate in rheumatic diseases. Nat Rev Rheumatol.
12:517–531. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Liang XH, Jackson S, Seaman M, Brown K,
Kempkes B, Hibshoosh H and Levine B: Induction of autophagy and
inhibition of tumorigenesis by beclin 1. Nature. 402:672–676. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Semenza GL: Involvement of
hypoxia-inducible factor 1 in pulmonary pathophysiology. Chest. 128
Suppl 6:592S–594S. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Bellot G, Garcia-Medina R, Gounon P,
Chiche J, Roux D, Pouysségur J and Mazure NM: Hypoxia-induced
autophagy is mediated through hypoxia-inducible factor induction of
BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol.
29:2570–2581. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang H, Bosch-Marce M, Shimoda LA, Tan
YS, Baek JH, Wesley JB, Gonzalez FJ and Semenza GL: Mitochondrial
autophagy is an HIF-1-dependent adaptive metabolic response to
hypoxia. J Biol Chem. 283:10892–10903. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ricci A, Cherubini E, Scozzi D,
Pietrangeli V, Tabbì L, Raffa S, Leone L, Visco V, Torrisi MR,
Bruno P, et al: Decreased expression of autophagic beclin 1 protein
in idiopathic pulmonary fibrosis fibroblasts. J Cell Physiol.
228:1516–1524. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sosulski ML, Gongora R, Danchuk S, Dong C,
Luo F and Sanchez CG: Deregulation of selective autophagy during
aging and pulmonary fibrosis: The role of TGFβ1. Aging Cell.
14:774–783. 2015. View Article : Google Scholar : PubMed/NCBI
|