Introduction
With heart failure becoming increasingly prevalent,
it threatens the health of many individuals (1,2). As
an common health problem, myocardial infarction (MI) is one of the
primary causes of heart failure. The fibers of the myocardial
tissue will be elongated under normal conditions. However, if the
myocardial tissue works overload, the fibers may not properly
shorten, therefore increasing the size of the myocardial fibers.
This eventually leads to the fact that the blood pumping cannot be
effectively exerted and the oxygen cannot be effectively delivered
to the working tissues in the body (3). The risk factors for myocardial
hypertrophy may be coronary artery disease or high blood pressure
(4). As a compensatory response of
the heart to pathological stimuli, myocardial hypertrophy is often
accompanied with MI that is characterized by an enlarged heart
(5).
From the molecular perspective, the myocardial
hypertrophy is a consequence of the reprograming of gene expression
and the alteration of signaling pathways (6,7).
Brain natriuretic factor (BNP) and β-myosin heavy chain (β-MHC) are
two typical markers to indicate the myocardial hypertrophy
(8). In addition, the activity of
intracellular signaling pathway may produce even more profound
effect on the biological events. Moreover, emerging evidences have
demonstrated that reactive oxygen species (ROS) are critical in
triggering the hypertrophic responses to various stimulus, for
example, stretch (9) or Ang II
(10–12). In addition, nuclear factor
(erythroid-derived 2)-like 2 (Nrf2) is a transcription factor that
regulates the expressions of target genes, for instance,
NAD(P)H:quinone oxidoreductase 1 (NQO1) and heme oxygenase-1 (HO-1)
(13), through antioxidant
response element (ARE). The protective role of Nrf2 has been
discovered in tissue injuries (14). However, the role of Nrf2 remains
less known in hypertrophy after MI. Hence, investigating the
effects of Nrf2 is of significance for the purpose of understanding
the molecular event in heart failure.
The antihypertensive therapy has been proved to be
increasingly effective (15);
however, the prognosis of the patients suffering from acute MI
remains unsatisfactory Therefore, it is necessary to develop new
options that are helpful to the treatment of myocardial hypertrophy
induced by MI. Increasing attention has recently been paid to the
traditional Chinese medicine, which has been demonstrated to have
valuable therapeutic effects by modern medicines (16). Hydroxysafflor yellow A (HSYA), a
compound isolated from Carthamus tinctorius L., has its main
activity in the treatment of cardiovascular diseases (17). HSYA is able to promote the blood
circulation. Previous studies have proved its role in myocardial
I/R injury (18). However, the
effect of HSYA and the association between HSYA and Nrf2 in
myocardial hypertrophy after MI remain unclear.
Thus, this study aimed to estimate the role of HSYA
in myocardial hypertrophy induced by MI as well as its role in Nrf2
signaling pathway both in vivo and in vitro. Our
results would not only provide a promising agent for the management
of myocardial hypertrophy after MI, but also the possible molecular
target in the prevention of heart failure.
Materials and methods
Animals and MI in vivo model
This study concerned animals trials and was approved
by Animal Subjects Committee of the Affiliated Hospital of Hangzhou
Normal University. All the clinical and surgical procedures were
conducted under the Institutional Animal Care guidelines. Male
Sprague-Dawley rats (275–300 g) were purchased from Guangdong
Medical Laboratory Animal Center. The animals were randomly grouped
into six groups: control group, sham surgery group (sham), model
group, captopril (Ka) group, HSYA low concentration group (HSYA-L)
and HSYA high concentration group (HSYA-H). The rats had access to
standard rat chow and water ad libitum and they were fed in
the Affiliated Hospital of Hangzhou Normal University. Permanent
ligation of the left anterior descending coronary artery was
performed to set up the MI model in vivo. The sham group was
only performed with open-heart surgery. The treatment groups were
intraperitoneally injected with captopril (10 mg/kg/day; Tianjin
Lisheng Pharmaceutical Co., Ltd., Tianjin, China) or HSYA (2 or 5
mg/kg/day; Shanghai Yuanye Biotechnology Co. Ltd., Shanghai,
China). The rats were anesthetized with 1.5% isoflurane at four
weeks after MI. The heart were excised, rinsed in ice-cold saline
and weighted. The doses of each agent were adopted as previously
described (19,20).
Cell culture and grouping
H9c2, derived from the embryonic rat ventricle, was
purchased from ATCC. Cells were maintained in Dulbecco's modified
Eagle's medium (DMEM) supplemented with 1% penicillin/streptomycin,
10% fetal bovine serum (FBS) at 37°C with 5% CO2 in a
humidified incubator. The cell grouping was as follows: in control
group: The normal cells; in Ang II group: The cells were treated
with 1 µM Ang II for 12 h at 37°C; in Ang II/mock group (A/mock):
The cells were transfected with si-negative control and treated
with 1 µM Ang II; in Ang II/siNrf2 group (A/siNrf2): The cells were
transfected with si-Nrf2 and treated with 1 µM Ang II; in Ang
II/HSYA group (A/H): The cells were pretreated with 80 µM HSYA for
8 h and then incubated with 1 µM Ang II for 12 h; in Ang
II/HSYA/mock group (A/H/mock): The cells were transfected with
si-negative control and then treated with 1 µM Ang II for 12 h
following 8 h incubation of 80 µM HSYA; in Ang II/HSYA/siNrf2 group
(A/H/siNrf2): The cells were transfected with si-Nrf2 control and
then treated with 1 µM Ang II for 12 h following 8 h incubation of
80 µM HSYA.
Hematoxylin and eosin (H&E)
staining
The myocardial tissue was fixed 4% formaldehyde at
4°C for 72 h and was embedded in paraffin. The paraffin embedded
tissue was sliced into 3–4 µm. Then, the slides were subject to the
treatment as follows: Dewaxing with xylene for 15 min, incubation
in gradient alcohol, hematoxylin staining for 15 min, incubation in
1% Hydrochloric acid alcohol for 15 sec, 1% eosin staining for 1
min. The images for H&E staining were captured under an
inverted microscope (Olympus, Tokyo, Japan). The slides were sealed
and the cell area was measured by Image-Pro Plus 6.0 software
(Media Cybernetics, Inc., Rockville, MD, USA). Five random fields
were selected.
Downregulation of Nrf2 by siRNA
Cells were transfected with siRNA using the
Lipofectamine® 3000 reagent (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA). Gene specific siRNA for Nrf2
and negative control siRNA were purchased from GeneCopoeia, Inc.
(Rockville, MD, USA). The transfection efficiency of RNA knockdown
was estimated at 48 h after post-transfection.
Cell viability and 3H
Leucine incorporation assay
Cells were seeded at a density of 3×104
cells in 96-well plate. After being treated as designed for each
group, the cells were prepared for conducting the cell viability
assay. To explain further, first, the Cell Counting Kit-8 (CCK-8)
solution (10 µl) was added into each well. After 4 h of incubation,
the plate was removed from the incubator. The absorbance was read
at 450 nm with a microplate reader (BioTek Instruments Inc.,
Winooski, VT, USA). For detecting protein synthesis rate,
3H Leucine incorporation assay was applied as previously
described (21). To be more
specific, the cells were pulsed with 1 µCi/ml [3H]
leucine (PerkinElmer, Inc., Waltham, MA, USA) in PBS at 37°C for 2
h. After being washed by PBS buffer, the cells were incubated with
10% trichloroacetic acid. The glass micro-fiber filter under vacuum
was used to collect the precipitate. The precipitate was then
suspended in scintillation fluid. The incorporation of 3H-Leu
[counts per minute (CPM)] was counted on the iquid scintillation
counter (GE Healthcare Bio-Sciences, Pittsburgh, PA, USA).
Determination of ROS, malondialdehyde
(MDA) and superoxide dismutase (SOD)
The method of ROS detection and the cell treatment
with Ang II was referred to previous studies (22,23).
The ROS generation was assessed by a fluorescent probe
dihydrodichlorofluorescein diacetate (DCFH-DA; Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany). The cells were treated as described in
cell grouping. Then, the cells were detached with 0.25%
trypsin-EDTA and washed with PBS buffer. Then, cells in each group
were incubated with 10 µM DCFH-DA for 30 min at 37°C. Next, the
cells were washed with serum-free DMEM. And then centrifugated for
5 min at 1,000 × g at room temperature. The supernatants were
collected and prepared for the fluorescence determination. With the
excitation wavelength of 480 nm and the emission wavelength of 525
nm, the fluorescent signal was determined using a FLUOstarOPTIMA
fluorescence spectrophotometer (BMG Labtech, Ortenberg, Germany).
The mean fluorescence intensity (MFI) was considered as the index
for the production of ROS. MFIs from five random fields were
detected using Image J 1.41 software. The assessment of the
activity of SOD and MDA was conducted following the protocls
provided by each commercial kit (Beyotime Institute of
Biotechnology, Haimen, China). The measurement was read adopting
spectrophotometric methods with a plate reader (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). Absorbance at 412 nm was
estimated for SOD. The result was expressed as SOD U/ml. The MDA
level was expressed as MDA nmol/mg protein.
Reverse transcription-quantitative polymerase chain
reaction (RT-qPCR). The total RNA was lysed with TRIzol reagent
(Thermo Fisher Scientific, Inc., Waltham, MA, USA). The transparent
RNA phase was taken by RNAse free chloroform and transfered into a
new RNAse free tube containing 70% alcohol. The integrity of RNA
was determined by gel electrophoresis. The QIAGEN RNA Cleanup kit
was used to purify RNA. Invitrogen cDNA synthesis kit was used to
synthesis cDNA. SYBR-Green Real Time PCR detection system was
adopted to detect the gene expression on ABI 7500 instrument. The
amplification conditions was as follows: At 50°C, 2 min, at 95°C,
10 min, and then 32 cycles at 95°C for 15 sec and at 60°C for 1
min. The expression levels were normalized to β-actin expression.
The specific primers were as follows: Nrf2 Forward (F):
5′-TTCCTCTGCTGCCATTAGTCAGTC-3′, and reverse (R):
5′-GCTCTTCCATTTCCGAGTCACTG-3′; NQO-1 F:
5′-GCAGTTTCTAAGAGCAGAACG-3′, and R: 5′-GTAGATTAGTCCTCACTCAGCCG-3′;
HO-1 F: 5′-CTGGAAGAGGAGATAGAGCGAA-3′, and R:
5′-TCTTAGCCTCTTCTGTCACCCT-3′; BNP F: 5′-TCGGCGCAGTCAGTCGCTTG-3′,
and R: 5′-CGCAGGCAGAGTCAGAAGCCG-3′; β-MHC F:
5′-TGCAAAGGCTCCAGGTCTGAT-3′, and R: 5′-GCCAACACCAACCTGTCAAG-3′;
β-actin F: 5′-GCCATGTACGTAGCCATCCA-3′, and R:
5′-GAACCGCTCATTGCCGATAG-3′.
Western blot analysis
Protein was isolated from cultured cells using lysis
buffer that contained protease/phosphatase inhibitors. The
quantification was determiend by the BCA Protein Assay kit (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). Subsequently, after 5 min
of denaturation at 95°C, samples in each group were separated by
sodium dodecyl sulfate polyacrylamide (SDS-PAGE) gel. The PVDF
membranes (EMD Millipore, Billerica, MA, USA) were used for the
tank blot system. Next, the membrane was incubated with 5% skimmed
milk containing 0.05% Tween-20 (TBST) at room temperature for 2 h.
The primary antibodies were incubated at 4°C overnight: anti-Nrf2
(1:1,000, ab62352; Abcam, Cambridge, UK), anti-NQO-1 (1:800,
ab34173), anti-HO-1 (1:1,000, ab137749), anti-BNP (1:500, ab19645),
anti-β-MHC (1:500, ab23990), anti-GAPDH (ab9385, 1:5,000). The
horseradish-peroxidase-coupled secondary IgG antibodies were
incubated at room temperature for 1 h. ECL Western Blotting
Substrate (Amersham; GE Healthcare, Chicago, IL, USA) was adopted
to develop chemiluminescence signals using the ChemiDoc XRS Imaging
System (Bio-Rad Laboratories, Inc.). Finally, the expression level
was calculated by the densitometric analysis (Quantity One; Bio-Rad
Laboratories, Inc.).
Statistics
GraphPad Prism 6.0 (GraphPad Software, Inc., La
Jolla, CA, USA) was used to perform statistical analyses. The data
were shown as mean ± standard deviation. One-way ANOVA with
Dunnet's multiple comparison test was used to compare the
difference the multiple groups (>2 groups). P<0.05 was
considered to indicate a statistically significant difference.
Results
HSYA reduced MI-induced hypertrophy in
rats
As shown in Fig.
1A, animals in model group showed an obvious increase in the
heart size, compared to sham and control group. By contrast, the
treatment with captopril (Ka) or HSYA alleviated the cardiac
hypertrophy. Moreover, there was a blunted (in HSYA-L group) or
significant decrease (in Ka and HSYA-H group) in the heart
weight/body weight ratio compared to model group (Fig. 1B). Furthermore, the H&E
staining showed that the enlarged cell area of myocytes induced by
MI was attenuated by the treatment with Ka or HSYA. And this
reduction was more obvious in HSYA-H group than in HSYA-L group
(Fig. 1C).
HSYA rescued the expression of Nrf2 in
rats
Research has reported that the abrogation of Nrf2
may enhance atrial hypertrophy in response to exercise stress
(24). Thus, we detected the
expression of Nrf2 in myocardial tissue. The results showed that
the Nrf2 expression was depressed sharply in model group both in
mRNA and protein levels. The treatment with Ka or HSYA recovered
its expression and the recovery effect was most obvious in HSYA-H
group (Fig. 2A-C). There was no
significant difference observed in Nrf2 protein levels in both
HSYA-H and HSYA-L group.
HSYA rescued the expression of Nrf2 in
H9c2 treated with Ang II
To further confirm the effect of HSYA on hypertrophy
after MI and the role of Nrf2 in hypertrophy, we performed
subsequent experiments in H9c2 cardiomyocytes. The transfection
efficiency of Nrf2 was presented in Fig. 3A-C. Ang II was used to set up the
hypertrophy model in the study. We observed that the Nrf2
expression was mitigated by about 50% in Ang II group both in terms
of the transcriptional and translational levels. Noticeably, the
treatment with HSYA inhibited this decrease in A/H group. By
contrast, the combination use of Ang II and siNrf2 aggravated the
reduced expression of Nrf2, however, the effect of HSYA was
reversed by the siNrf2 treatment (Fig.
3D and E).
Downregulation of Nrf2 reversed the
protective effect of HSYA in H9c2 treated with Ang II
CCK-8 results showed that the Ang II-induced
decreased cell viability was deteriorated in A/siNrf2 group but
improved in A/H group. And the cell viability was smaller in
A/H/siNrf2 group than that in A/H group (Fig. 4A). Moreover, while the leucine
incorporation was induced by Ang II, it was reduced by the
treatment of HSYA. The treatment with siNrf2 enhanced the leucine
incorporation both in A/siNrf2 group and A/H/siNrf2 group, compared
to Ang II and A/H group (Fig. 4B).
In addition, the expression of hypertrophy-related genes was
examined. As shown in Fig. 5A and
B, the increased mRNA expressions of BNP and β-MHC caused by
Ang II was depressed by HSYA, and the effect of HSYA was mitigated
by the downregulation of Nrf2. Consistently, the protein level of
BNP and β-MHC showed a similar trend to mRNA level (Fig. 5C and D), suggesting that the
protective effect delivered by HSYA was reversed by the
downregulation of Nrf2.
HSYA altered the oxidative stress and
the activation of NQO-1/HO-1 signaling pathway
As shown in Fig.
6A-C, the treatment of Ang II caused the oxidative stress in
H9c2, and this was achieved by triggering ROS accumulation,
increasing the MDA content and inhibiting the SOD activity. The
oxidative stress was increased and decreased by the down-regulation
of Nrf2 and the treatment of HSYA, respectively. By contrast, the
treatment of siNrf2 partly reversed the effect of HSYA. NQO-1/HO-1
are the typical downstream targets of Nrf2 through ARE. Thus, we
detected the expressions of these two genes. Our results revealed
that the treatment of Ang II inhibited the expressions of NQO-1 and
HO-1, and combined treatment with Ang II and siNrf2 presented even
more obvious effects than treatment with Ang II alone. Therefore,
while HSYA apparently abolished the effects of Ang II on the
expressions of NQO-1 and HO-1, the down-regulation of Nrf2 partly
blocked the effect produced by HSYA (Fig. 6D-G).
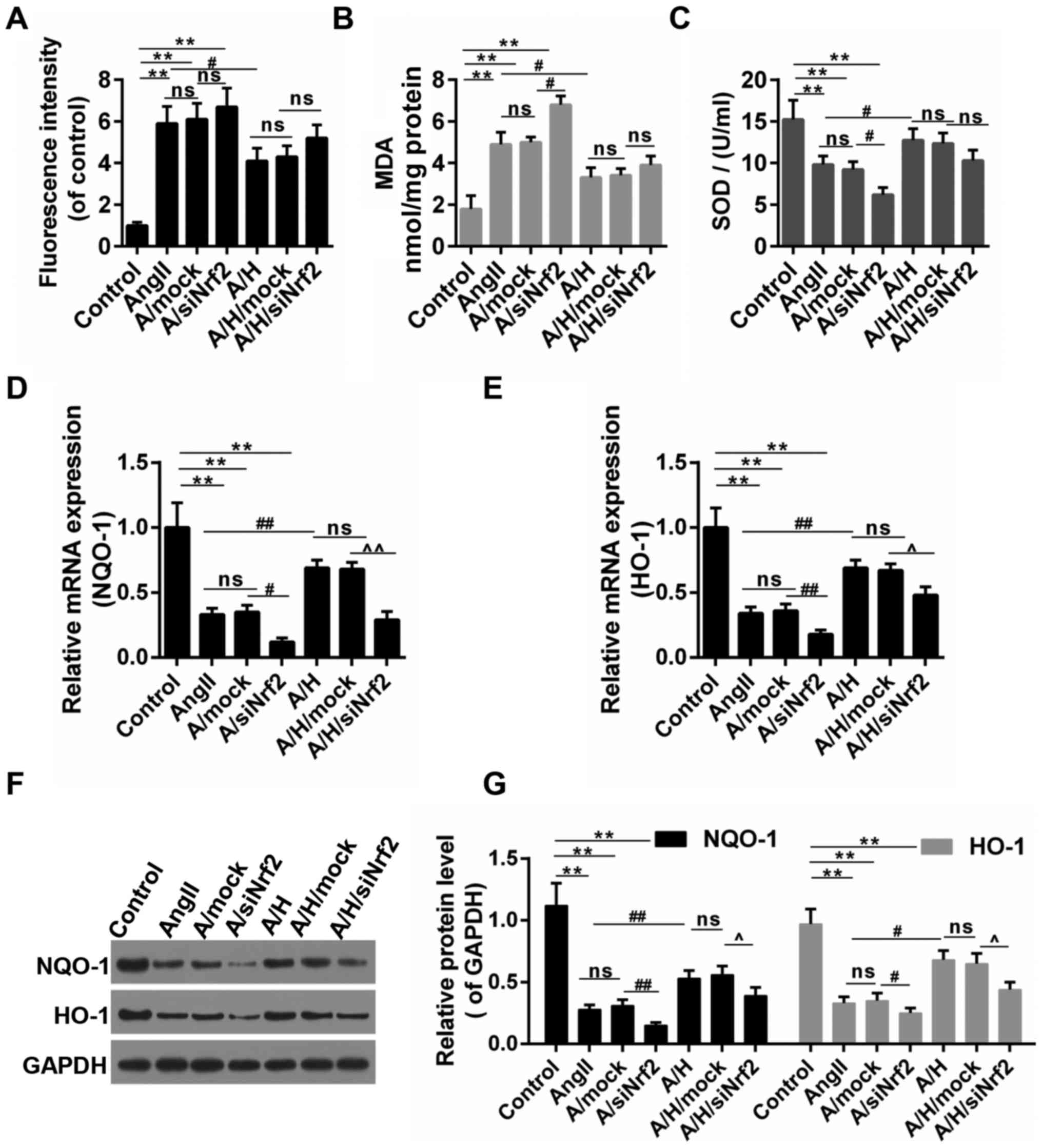 | Figure 6.HSYA ameliorated oxidative stress and
increased the expression of NQO-1 and HO-1. (A) ROS production was
examined by DCHF-DA; (B) The content of MDA; (C) The activity of
SOD; (D-G) The expression NQO-1 and HO-1 by RT-qPCR [(D) for NQO-1,
(E) for HO-1] and (F and G) Western blot assays. **P<0.01,
#P<0.05, ##P<0.01,
^P<0.05 and ^^P<0.01 as indicated. ns,
no significant difference. HSYA, Hydroxysafflor yellow A; RT-qPCR,
reverse transcription-quantitative polymerase chain reaction;
NQO-1, NAD(P)H:quinone oxidoreductase 1 (NQO1) HO-1 heme
oxygenase-1; MDA, malondialdehyde; SOD, superoxide dismutase; ROS,
reactive oxygen species. |
Discussion
The onset of MI would happen when the coronary
artery occlude, and this could induce cardiac hypertrophy (25,26).
This pathological hypertrophy in the heart will cause many
ailments, leading to morbidity and mortality (27,28).
This study uncovered a protective effect of HSYA on cardiac
hypertrophy after MI in vivo and in vitro. Animals
and/or H9c2 cells treated with HSYA exhibited reduced heart size,
declined hypertrophic area and oxidative stress, which were closely
related to Nrf2 signaling pathway.
In this work, HSYA alleviated the heat size and
heart weight/body weight ratio induced by MI in rats. The
cross-sectional area of myocardial cells, an indicator of
hypertrophy, was reduced by the administration of HSYA in rats.
Importantly, the expression of Nrf2 was depressed in hypertrophic
cardiomyocyte in vivo, which was recovered by HSYA. These
results pointed to the protective role of HSYA, in which the
activation of Nrf2 may be involved.
To confirm the role of HSYA and Nrf2 in hypertrophy
caused by MI, we conducted experiments using H9c2 in vitro.
Consistently, the expression of Nrf2 was decreased in H9c2 under
the stimuli of Ang II that was combated by the treatment of HSYA.
Although in HSYA-L group, the expression of Nrf2 was significantly
rescued, the Hw/BW was not decreased significantly. This
discrepancy may be caused by the complexity of the intracellular
regulation network. The crosstalk between signals is very common in
the living cells. Some other signals (that was not identified in
this study) may also affect the Hw/BW.
The downregulation of Nrf2 blunted the effect of
HSYA. Accompanied by the reduced cell viability, Ang II increased
the [3H] leucine incorporation of cardiomyocytes. This
was in line with the results obtained in a previous study (29). Moreover, the expressions of
hypertrophic markers, BNP and β-MHC, was significantly induced by
Ang II. The actions of Ang II were deteriorated by the decreased
Nrf2. By contrast, the treatment of HSYA attenuated the effect of
Ang II. Nevertheless, the effect of HSYA was partly blocked by the
down-regulation of Nrf2. These results suggested that the
anti-hypertrophic effect of HSYA was largely dependent on the
activation of Nrf2. Consistently, in a previous study, the
over-expression of Nrf2 could significantly inhibit the expression
of the hypertrophic factor (30).
These listed results confirmed that the activation of Nrf2 was
helpful in combating the cardiac hypertrophy.
Researchers proposed that ROS contributed to
cardiomyocyte hypertrophy (11,31).
Moreover, the inhibition of ROS accumulation is helpful to the
amelioration of hypertrophy (32).
In our work, the ROS burst caused by Ang II was alleviated by HSYA.
The intracellular ROS accumulation will lead to the disruption of
redox system, for example, the activity of MDA and SOD (33,34).
In this study, the content of MDA and the activity of SOD were
dysregualted by Ang II, however, the treatment of HSYA recovered
their balance. And the down-regulation of Nrf2 reversed the effect
of HSYA. Taken together, we concluded that anti-hypertrophic effect
produced by HSYA was closely associated with the anti-oxidative
capacity. Coincidentally, Nrf2 is outstanding through its ARE in
the protection against oxidative stress. NQO-1 and HO-1 are two
downstream targets of Nrf2. In addition, the induction of NQO-1 and
HO-1 was believed to have protective effects on oxidative injury
(35–37). Therefore, we speculated that HSYA
may prevent cardiac hypertrophy in relation with the activation of
NQO-1/HO-1. Our results showed that the expressions of NQO-1 and
HO-1 were higher in A/H group than those in Ang II and A/siNrf2
groups in terms of transcriptional and translational levels,
suggesting that the activations of NQO-1 and HO-1 contributed to
the protective role of HSYA. In addition, the results showed that
the down-regulation of Nrf2 reversed the effect of HSYA on the
expressions of NQO-1 and HO-1. These data suggested that Nrf2 was
necessary for the protective effects of HSYA against cardiac
hypertrophy caused by MI. The cardio-protective role HSYA as such
was consistent with previous studies (38–40).
Therefore, the results illustrated that HSYA delivered a positive
effect on the activation of Nrf2/NQO-1/HO-1 pathway, which is a
promising thread for increasing the antioxidant and
anti-hypertrophic effect.
Collectively, we discovered that the protective
effect of HSYA on cardiac hypertrophy after MI. The anti-oxidant
capacity of HSYA may be associated with this work model. Moreover,
some studies have pointed out that HSYA could exert anti-apoptotic
effect on tissue injury (20,41,42).
Thus, to assess other aspects of this protective effect could also
lead to productive outcome. Furthermore, we found that Nrf2
signaling was necessary to the effect produced by HSYA.
Nevertheless, due to the complexity of cellular signal cascades, we
did not exclude the possibility that other signaling pathways may
play a part in the protective effect conferred by HSYA.
In summary, HSYA significantly mitigated cardiac
hypertrophy in vitro and in vivo. This effect may
depend on the antioxidant effects via Nrf2/NQO-1/HO-1 signaling
pathway. Our study inspired insight into the mechanisms that is
responsible for HSYA. Our results may provide effective regimen and
strategies to relive heart failure.
Acknowledgements
Not applicable.
Funding
This work was supported Natural Science Foundation
of Zhejiang Province, China (grant no. LY17H020001).
Availability of data and materials
The analyzed data sets generated during the study
are available from the corresponding author on reasonable
request.
Authors' contributions
BN, SL and DZ provided substantial contributions to
conception and design of the present study. YJ contributed to the
data acquisition, data analysis and interpretation. SL drafted the
article and critically revised it for important intellectual
content. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present approved by Animal Subjects Committee of
the Affiliated Hospital of Hangzhou Normal University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Eriksson H: Heart failure: A growing
public health problem. J Intern Med. 237:135–141. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dickstein K, Cohensolal A, Filippatos G,
McMurray JJ, Ponikowski P, Poole-Wilson PA, Strömberg A, Van
Veldhuisen DJ, Atar D, Hoes AW, et al: ESC Guidelines for the
diagnosis and treatment of acute and chronic heart failure 2008:
The Task Force for the diagnosis and treatment of acute and chronic
heart failure 2008 of the European Society of Cardiology. Developed
in collaboration with the Heart Failure Association of the ESC
(HFA) and endorsed by the European Society of Intensive Care
Medicine (ESICM). Eur Heart J. 29:2388–2442. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Frey N and Olson EN: Cardiac hypertrophy:
The good, the bad and the ugly. Annu Rev Physiol. 65:452003.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
English BA, Appalsamy M, Diedrich A,
Ruggiero AM, Lund D, Wright J, Keller NR, Louderback KM, Robertson
D and Blakely RD: Tachycardia, reduced vagal capacity and
age-dependent ventricular dysfunction arising from diminished
expression of the presynaptic choline transporter. Am J Physiol
Heart Circ Physiol. 299:H799–H810. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Singh MV and Anderson ME: Is CaMKII a link
between inflammation and hypertrophy in heart? J Mol Med.
89:537–543. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Molkentin JD and Dorn GW II: Cytoplasmic
signaling pathways that regulate cardiac hypertrophy. Annu Rev
Physiol. 63:391–426. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nicol RL, Frey N and Olson EN: From the
sarcomere to the nucleus: Role of genetics and signaling in
structural heart disease. Annu Rev Genomics Hum Genet. 1:179–223.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yang K, Xu X, Nie L, Xiao T, Guan X, He T,
Yu Y, Liu L, Huang Y, Zhang J and Zhao J: Indoxyl sulfate induces
oxidative stress and hypertrophy in cardiomyocytes by inhibiting
the AMPK/UCP2 signaling pathway. Toxicol Lett. 234:110–119. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pimentel DR, Amin JK, Xiao L, Miller T,
Viereck J, Oliver-Krasinski J, Baliga R, Wang J, Siwik DA, Singh K,
et al: Reactive oxygen species mediate amplitude-dependent
hypertrophic and apoptotic responses to mechanical stretch in
cardiac myocytes. Circ Res. 89:453–460. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nakamura K, Fushimi K, Kouchi H, Mihara K,
Miyazaki M, Ohe T and Namba M: Inhibitory effects of antioxidants
on neonatal rat cardiac myocyte hypertrophy induced by tumor
necrosis factor-alpha and angiotensin II. Circulation. 98:794–799.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bendall JK, Cave AC, Heymes C, Gall N and
Shah AM: Pivotal role of a gp91(phox)-containing NADPH oxidase in
angiotensin II-induced cardiac hypertrophy in mice. Circulation.
105:293–296. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hingtgen SD, Tian X, Yang J, Dunlay SM,
Peek AS, Wu Y, Sharma RV, Engelhardt JF and Davisson RL:
Nox2-containing NADPH oxidase and Akt activation play a key role in
angiotensin II-induced cardiomyocyte hypertrophy. Physiol Genomics.
26:180–191. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nguyen T, Nioi P and Pickett CB: The
Nrf2-antioxidant response element signaling pathway and its
activation by oxidative stress. J Biol Chem. 284:13291–13295. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Soares MP and Ribeiro AM: Nrf2 as a master
regulator of tissue damage control and disease tolerance to
infection. Biochem Soc Trans. 43:663–668. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Psaty BM, Smith NL, Siscovick DS, Koepsell
TD, Weiss NS, Heckbert SR, Lemaitre RN, Wagner EH and Furberg CD:
Health outcomes associated with antihypertensive therapies used as
first-line agents. A systematic review and meta-analysis. JAMA.
277:739–745. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang L, Zhou GB, Liu P, Song JH, Liang Y,
Yan XJ, Xu F, Wang BS, Mao JH, Shen ZX, et al: Dissection of
mechanisms of Chinese medicinal formula Realgar-Indigo naturalis as
an effective treatment for promyelocytic leukemia. Proc Natl Acad
Sci USA. 105:pp. 4826–4831. 2008; View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang J, Wang Y and Guo ML: Identification
and mapping of a novel hydroxysafflor yellow A (HSYA) biosynthetic
gene in Carthamus tinctorius. Biochem Genet. 49:410–415. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Min J and Wei C: Hydroxysafflor yellow A
cardioprotection in ischemia-reperfusion (I/R) injury mainly via
Akt/hexokinase II independent of ERK/GSK-3β pathway. Biomed
Pharmacother. 87:419–426. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nurzynska D, Meglio FD, Castaldo C,
Miraglia R, Romano V, Sacco AM, Barbato V, Granato G, Belviso I,
Bancone C, et al: Cardiac primitive cells in the adult human heart
are influenced by Angiotensin II in chronic heart failure. Ital J
Anat Embryol. 119:2014.
|
|
20
|
Liu SX, Zhang Y, Wang YF, Li XC, Xiang MX,
Bian C and Chen P: Upregulation of heme oxygenase-1 expression by
hydroxysafflor yellow A conferring protection from
anoxia/reoxygenation-induced apoptosis in H9c2 cardiomyocytes. Int
J Cardiol. 160:95–101. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sahni A, Wang N and Alexis JD: UAP56 is an
important regulator of protein synthesis and growth in
cardiomyocytes. Biochem Biophys Res Commun. 393:106–110. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu Y, Jiao R, Ma ZG, Liu WEI, Wu QQ, Yang
Z, Li FF, Yuan Y, Bian ZY and Tang QZ: Sanguinarine inhibits
angiotensin II-induced apoptosis in H9c2 cardiac cells via
restoring reactive oxygen species-mediated decreases in the
mitochondrial membrane potential. Mol Med Rep. 12:3400–3408. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Prathapan A, Vineetha VP and Raghu KG:
Protective effect of Boerhaavia diffusa L. against mitochondrial
dysfunction in angiotensin II induced hypertrophy in H9c2
cardiomyoblast cells. PLoS One. 9:e962202014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kumar RR, Madhusudhanan N, Shanmugam G,
Hong J, Devarajan A, Palaniappan S, Zhang J, Halade GV,
Darley-Usmar VM, Hoidal JR, et al: Abrogation of Nrf2 impairs
antioxidant signaling and promotes atrial hypertrophy in response
to high-intensity exercise stress. J Transl Med. 14:862016.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Meloni M, Caporali A, Graiani G, Lagrasta
C, Katare R, Van Linthout S, Spillmann F, Campesi I, Madeddu P,
Quaini F and Emanueli C: Nerve growth factor promotes cardiac
repair following myocardial infarction. Circ Res. 106:1275–1284.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Olivetti G, Quaini F, Sala R, Lagrasta C,
Corradi D, Bonacina E, Gambert SR, Cigola E and Anversa P: Acute
myocardial infarction in humans is associated with activation of
programmed myocyte cell death in the surviving portion of the
heart. J Mol Cell Cardiol. 28:2005–2016. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
McMullen JR and Jennings GL: Differences
between pathological and physiological cardiac hypertrophy: Novel
therapeutic strategies to treat heart failure. Clin Exp Pharmacol
Physiol. 34:255–262. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Neeland IJ, Drazner MH, Berry JD, Ayers
CR, de Filippi C, Seliger SL, Nambi V, McGuire DK, Omland T and de
Lemos JA: Biomarkers of chronic cardiac injury and hemodynamic
stress identify a malignant phenotype of left ventricular
hypertrophy in the general population. J Am Coll Cardiol.
61:187–195. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hayashi D, Kudoh S, Shiojima I, Zou Y,
Harada K, Shimoyama M, Imai Y, Monzen K, Yamazaki T, Yazaki Y, et
al: Atrial natriuretic peptide inhibits cardiomyocyte hypertrophy
through mitogen-activated protein kinase phosphatase-1. Biochem
Biophys Res Commun. 322:310–319. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li J, Ichikawa T, Villacorta L, Janicki
JS, Brower GL, Yamamoto M and Cui T: Nrf2 protects against
maladaptive cardiac responses to hemodynamic stress. Arterioscler
Thromb Vasc Biol. 29:1843–1850. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sugden PH and Clerk A: Cellular mechanisms
of cardiac hypertrophy. J Mol Med. 76:725–746. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tanaka K, Honda M and Takabatake T: Redox
regulation of MAPK pathways and cardiac hypertrophy in adult rat
cardiac myocyte. J Am Coll Cardiol. 37:676–685. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Todorova I, Simeonova G, Kyuchukova D,
Dinev D and Gadjeva V: Reference values of oxidative stress
parameters (MDA, SOD, CAT) in dogs and cats. Comp Clin Path.
13:190–194. 2005. View Article : Google Scholar
|
|
34
|
Omidi A, Namazi F, Jabire S, Afsar M,
Honarmand M and Nazifi S: The effects of starvation and refeeding
on oxidative stress parameters (MDA, SOD, GPx), lipid profile,
thyroid hormones and thyroid histopathology in male wistar rats.
Int Arch Med. 9(238)2016.
|
|
35
|
Wu CC, Hsu MC, Hsieh CW, Lin JB, Lai PH
and Wung BS: Upregulation of heme oxygenase-1 by
Epigallocatechin-3-gallate via the phosphatidylinositol
3-kinase/Akt and ERK pathways. Life Sci. 78:2889–2897. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li P, Su L, Li X, Di W, Zhang X, Zhang C,
He T, Zhu X, Zhang Y and Li Y: Remote limb ischemic
postconditioning protects mouse brain against cerebral
ischemia/reperfusion injury via upregulating expression of Nrf2,
HO-1 and NQO-1 in mice. Int J Neurosci. September 17–2015.(Epub
ahead of print). View Article : Google Scholar :
|
|
37
|
Tomita M, Okuyama T, Katsuyama H, Hidaka
K, Otsuki T and Ishikawa T: Gene expression in rat lungs during
early response to paraquat-induced oxidative stress. Int J Mol Med.
17:37–44. 2006.PubMed/NCBI
|
|
38
|
Wang T, Fu FH, Han B, Li GS, Zhang LM and
Liu K: Hydroxysafflor yellow A reduces myocardial infarction size
after coronary artery ligation in rats. Pharm Biol. 47:458–462.
2009. View Article : Google Scholar
|
|
39
|
Wei G, Yin Y, Duan J, Guo C, Zhu Y, Wang
Y, Xi M and Wen A: Hydroxysafflor yellow A promotes
neovascularization and cardiac function recovery through
HO-1/VEGF-A/SDF-1α cascade. Biomed Pharmacother. 88:409–420. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Liu YN, Zhu JH, Xiang WU and Wang ZH:
Mitochondrial mechanism of cardioprotective effect of
hydroxysafflor yellow A against anoxia/reoxygenation injury in
rats. J Jiangsu Univ (Medicine Edition). 23:2013.
|
|
41
|
Chen L, Xiang Y, Kong L, Zhang X, Sun B,
Wei X and Liu H: Hydroxysafflor yellow A protects against cerebral
ischemia-reperfusion injury by anti-apoptotic effect through
PI3K/Akt/GSK3β pathway in rat. Neurochem Res. 38:2268–2275. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhou MX, Fu JH, Zhang Q and Wang JQ:
Effect of hydroxy safflower yellow A on myocardial apoptosis after
acute myocardial infarction in rats. Genet Mol Res. 14:3133–3141.
2015. View Article : Google Scholar : PubMed/NCBI
|



















