Introduction
Inflammatory bowel disease (IBD) refers to chronic
inflammatory disorders of the digestive tract, primarily including
Crohn's disease (CD) and ulcerative colitis (1). CD is characterized by patchy mucosal
and sub-mucosal inflammation, the presence of granulomas in 20% of
patients, and the formation of fissures, fistulae and strictures
that occur throughout the gastrointestinal tract (2,3). CD
has been estimated to affect 322 per 100,000 individuals in Europe
(2,4), and the prevalence of CD in Asia has
been steadily increasing (5).
Despite being widely studied, CD has no cure and its etiology
remains largely unknown (6).
Numerous factors have been suggested to contribute to CD
pathogenesis, such as genetic predisposition, immunological
dysregulation and environmental factors (7). The intestinal barrier is considered
to represent the frontline against adverse challenges, such as
smoking, infection or a high sugar diet (7). The balance between cellular apoptosis
and proliferation is important for maintaining the integrity of
intestinal epithelial cells (IECs). Excessive epithelial apoptosis
disrupts the barrier functions of the intestinal epithelium,
allowing the invasion of bacteria into the sub-mucosa and
subsequently activating host inflammatory responses (7,8).
Increased apoptosis of IECs resulting in epithelial barrier defects
has recently been suggested to directly contribute to CD
development (9). However, the
molecular mechanisms underlying IEC apoptosis in CD remain
unclear.
In eukaryotic cells, the accumulation of unfolded
and misfolded proteins in the endoplasmic reticulum (ER) lumen,
known as ER stress, triggers the unfolded protein response (UPR),
which aims to resolve the protein folding defect and restore ER
homeostasis (10). Three classical
ER sensor proteins, inositol-requiring kinase 1α (IRE1α),
pancreatic ER eIF2α kinase (PERK) and activating transcription
factor 6α (ATF6α), are involved in three main UPR pathways
(11). If ER stress is severe or
chronic, or the UPR is compromised and unable to restore ER
protein-folding homeostasis, numerous apoptotic signaling pathways
are activated (12,13). Several intestinal cell populations,
including Paneth and goblet cells, require robust ER function for
protein folding, maturation and secretion (6). Accumulating preclinical and clinical
studies suggest that ER stress and the UPR have a significant
impact on the pathogenesis of IBD (9,14).
For example, increased expression of ER stress markers has been
observed in the ileal and colonic mucosa of patients with IBD
(15). Furthermore, genetic
deletion of X-box-binding protein 1in IECs, an important
transcription factor of ER stress, has been revealed to result in
spontaneous enteritis in mice (14). In addition, genetic deletion of
IRE1αin IECs has been demonstrated to lead to spontaneous colitis
and increased sensitivity to chemical reagent-induced colitis in
mice (16). Furthermore,
interleukin-10, an anti-inflammatory cytokine, maintains intestinal
homeostasis by promoting correct protein folding under adverse
conditions in goblet cells (17,18).
FK506 binding protein (FKBP11) belongs to the FK506
binding protein family, which possess peptidyl-prolyl cis-trans
isomerase (PPIase) activity and are notable for their capacity to
bind immunosuppressive drugs, including FK506 and rapamycin
(19). FKBP11 mRNA is abundant in
numerous secretory tissues, including the pancreas, stomach and
salivary glands (19). FKBP11
protein contains a cleavable N-terminal signal sequence followed by
a putative PPIase domain with homology to FKBP12 (19). PPIases catalyze the slow cis-trans
isomerization of peptidyl-prolyl bonds to facilitate the protein
folding process, and FKBP11 has been suggested to be involved in ER
stress and UPR (19,20). Highly expressed FKBP11 is involved
in the pathogenesis of numerous ER stress-associated inflammatory
diseases, including type 2 diabetes (21), systemic lupus erythematosus (SLE)
(22) and hepatitis (23). Furthermore, a progressively
elevated expression of FKBP11 has been previously detected during
the development of hepatocellular carcinoma (HCC), which suggests
that FKBP11 may be a potential early marker for HCC (23). In ER stress induced hepatic
steatosis in mice, FKBP11 is a downstream molecule of the classical
UPR transducer IRE1α that promotes viable protein folding (24). Notably, previous bioinformatics
analysis investigating IBD suggested that the FKBP11 gene
might be exclusively highly expressed in human CD, thus suggesting
that FKPB11 may be involved in the pathogenesis of CD (25). However, the exact expression
pattern and biological role of FKBP11 in CD remains unclear.
In the present study, the protein expression of
FKBP11 in human CD colon tissues and a 2, 4,
6-trinitrobenzenesulphonic acid (TNBS)-induced mice colitis model
was investigated. Using interferon-γ (IFN-γ)/tumor necrosis
factor-α (TNF-α)-treated IEC models, the effect of FKBP11 on IECs
apoptosis and its potential association with the ER
stress-associated c-Jun-N-terminal kinase (JNK)-caspase apoptotic
pathway was investigated in inflammation-injured IECs.
Materials and methods
Human tissues
Following approval from the Ethics Committee of the
Affiliated Hospital of Nantong University (Nantong, China), samples
were selected between September 2013 and June 2015. Prior to sample
collection, all patients provided written informed consent.
Patients with CD had been diagnosed according to standard clinical
manifestations, endoscopy examination and histological criteria
(26). The inclusion criteria
included focal inflammation, irregular crypts and granuloma
(27). Inflamed intestinal biopsy
samples were obtained from patients with CD (n=20; 11 males and 9
females; age, 24–45). Control intestinal biopsy samples were
obtained from noninflamed areas of patients with CD (n=20; 10 males
and 10 females; age, 28–44).
Animals and induction of colitis
All animal care and surgical procedures were
performed based on the Guide for the Care and Use of Laboratory
Animals produced by the National Research Council in 1996 (28), and supported by the Chinese
National Committee to Use of Experimental Animals for Medical
Purposes, Jiangsu Branch (Nantong, China). Following the approval
from the Ethics Committee of the Affiliated Hospital of Nantong
University (permit no. 2014-L087), female BALB/c mice (aged 8–10
weeks; weight, 18–20 g; n=60) were obtained from the Animal Center
of Nantong University (Nantong, China), randomized into 2 groups
and kept in the laboratory of Animal Center (22–24°C; humidity,
40±5% and a 12 h light/dark cycle) with free access to food and
water. Mice were fasted for 24 h prior to further
experimentation.
The animal colitis model was established using TNBS
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany). Based on a
previously published method (29),
mice were injected intraperitoneally with pentobarbital sodium to
induce anesthesia (0.3% solution; 75 mg/kg). In the experimental
group, mice were administered 0.1 ml TNBS (2.5% (weight/volume)
TNBS solution in 50% ethanol). In the control group, mice were
administered 0.1 ml of 50% ethanol alone (ETOH group) (30). A 1 ml syringe containing a 3.5 F
catheter was used to administer either TNBS or ethanol via gentle
insertion from the anus into the colon. The depth of the catheter
was ~4 cm from the anus. The solution was slowly delivered into the
intestine, and mice were held vertically for 1 min to improve the
success rate of the enema.
Evaluation of TNBS-induced
colitis
To investigate the severity of colitis, changes in
body weight, piloerection, fecal traits and bloody stools were
recorded daily, and mice colon tissues were obtained for histology
and protein analyses. The mice from the experimental and control
group were sacrificed by cervical dislocation at 0, 1, 2, 3, 4 and
5 days' time intervals (n=5 per time interval). The mice colonic
tissues were removed quickly and washed gently in PBS. Following
this, colonic tissues were fixed in 4% paraformaldehyde (4°C for 48
h), embedded in paraffin, sectioned (4 µm thickness) and stained
with 5% hematoxylin (10 min at room temperature) and 0.5% eosin (5
min at room temperature). The pathological slices were observed
under a light microscope at a magnification of ×200. According to a
well-established scoring system (31), the degree of inflammation on
microscopic colon sections was graded from 0 to 4 (0, no signs of
inflammation; 1, very low levels of inflammation; 2, low levels of
leukocytic infiltration; 3, high levels of thickening of the colon
wall, high vascular density and leukocytic infiltration; 4,
transmural infiltration, high vascular density, loss of goblet
cells and thickening of the colon wall).
Cell culture and stimulation
HT-29 cells (a human colon epithelial cell line)
were purchased from the Cell Library, China Academy of Science
(Shanghai, China). HT-29 cells were cultured in complete medium
consisting of RPMI-1640 (Invitrogen; Thermo Fisher Scientific,
Inc., Waltham, MA, USA), containing 10% fetal bovine serum (FBS;
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) with 100 U/ml
penicillin and 100 µg/ml streptomycin at 37°C in a 95% air and 5%
CO2 atmosphere. The medium was replaced with fresh
medium every 2 days. The cells were passaged every 3–4 days. Prior
to subsequent experiments, HT-29 cells were incubated with IFN-γ
(2.5 ng/ml) and TNF-α (50 ng/ml; Sigma-Aldrich; Merck KGaA) for 24
h.
Western blot analysis
In order to perform western blot analysis, colon
tissues were washed with PBS and then frozen at −80°C. The colon
tissues were cut using scissors on ice to prepare lysates, and then
homogenized in lysis buffer (1% NP-40, 50 mmol/l Tris, pH=7.5, 5
mmol/l EDTA, 1% sodium deoxycholate, 1% sodium dodecyl sulfate
(SDS), 1% Triton X-100, 1 mmol/l PMSF, 1 µg/ml leupeptin and 10
µg/ml aprotinin) and then centrifuged at 4°C for 20 min at 48,000 ×
g to collect the supernatant. Cell cultures were lysed with sodium
lauryl sulfate loading buffer and then stored at −80°C until
further use. Following this, the protein concentration within the
samples was determined using a Bradford assay (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). Proteins from cell lysates
(30 µg/lane) were separated by 10% SDS-PAGE analysis and then
transferred to polyvinylidene difluoride membranes (EMD Millipore,
Billerica, MA, USA). The membranes were blocked with 5% non-fat
milk for 1 h at room temperature and incubated overnight at 4°C
with the following primary antibodies: FK506 binding protein 11
(FKBP11, goat; 1:500; cat. no. AP6790a; Abgent, Inc., San Diego,
CA, USA), 78 kDa glucose-regulated protein (GRP78, mouse; 1:500;
cat. no. sc-13539), Bcl2 associated X apoptosis regulator (Bax,
rabbit; 1:500; cat. no. sc-20067), proliferating cell nuclear
antigen (PCNA, mouse; 1:500; cat. no. sc-25280; all Santa Cruz
Biotechnology, Inc., Dallas, TX, USA), active caspase-3 (rabbit;
1:500; cat. no. AP3725a; Abgent, Inc.), c-Jun N-terminal kinase
(JNK, mouse; 1:500; cat. no. sc-7345), phosphorylated JNK (p-JNK,
mouse; 1:500), caspase-4 (human; 1:500; cat. no. sc-56056),
caspase-12 (human; 1:500; sc56056), and glyceraldehyde-3-phosphate
dehydrogenase (GAPDH, rabbit; 1:1,000; cat. no. sc-32233; all Santa
Cruz Biotechnology, Inc.). Finally, the membranes were incubated
with horseradish peroxidase-conjugated anti-goat, anti-mouse,
anti-rabbit (1:5,000; cat. nos. sc-2347, sc-516102, sc-2357; Santa
Cruz Biotechnology, Inc.) and anti-human (1:5,000; cat. no.
ab200699; Abcam, Cambridge, MA, USA) secondary antibodies for 2 h
at room temperature and visualized using an enhanced
chemiluminescence system (Pierce; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA).
Immunohistochemistry studies
Mice colon tissues were fixed in 10% formalin (4°C
for 24 h) and then incubated with 20% sucrose (4°C, 2–3 days),
followed by 30% sucrose (4°C, 2–3 days). The samples were embedded
in optimal cutting temperature compound (Taihe Huamei
Pharmaceutical Technology Co., Ltd., Shanghai, China) and then cut
into 4 µm sections using a cryostat. Specimens from patients with
CD were fixed in formalin (4°C for 24 h), dehydrated, embedded in
paraffin and cut into 5 µm sections, deparaffinized and rehydrated
via descending alcohol series. All sections were processed in 10 mM
citrate buffer (pH 6.0) and heated to 121°C in an autoclave for 20
min to retrieve the antigen. Following this, the sections were
soaked in 3% hydrogen peroxide to block the endogenous peroxidase
activity for 40 min at room temperature. Following rinsing with PBS
(pH=7.2), the sections were incubated with antibodies against
FKBP11 (goat; 1:500; cat. no. AP6790a; Abgent, Inc.), GRP78 (mouse,
1:500; cat. no. sc-13539; Santa Cruz Biotechnology, Inc.) for 2 h
at room temperature. All slides were processed using the
peroxidase-anti-peroxidase method for secondary antibodies
(anti-goat and anti-mouse; both 1:5,000; cat. nos. sc-2347 and
sc-516102; Santa Cruz Biotechnology, Inc.) at room temperature for
30 min (Dako; Agilent Technologies, Inc., Santa Clara, CA, USA).
Following rinsing with PBS, sections were incubated with
diaminobenzidine mixture (0.1% PBS, 0.02% diaminobenzidine
tetrahydrochloride and 3% H2O2) for 5 min at
room temperature to investigate the peroxidase reaction. Following
rinsing with water, sections were counterstained with hematoxylin
and subsequently dehydrated. Images were captured using a light
microscope at a magnification of ×50 and analyzed using Image-Pro
Plus 7.0 (Media Cybernetics, Inc., Rockville, MD, USA) with the
Olympus microscope (BX53; Olympus Corporation, Tokyo, Japan).
Cell apoptosis
A flow cytometry assay was performed to investigate
the cell apoptosis and necrosis rates using an ApoScreen Annexin V
kit (Southern Biotech, Birmingham, AL, USA), according to the
manufacturer's protocol. Briefly, HT-29 cells were digested using
0.1% trypsin and then resuspended in cold binding buffer (10 mM
HEPES, pH 7.4, 140 mM NaCl, 2.5 mM CaCl2 and 0.1% BSA)
at concentrations between 105 and 106
cells/ml. Following this, labeled Annexin V (10 µl) was added to
100 µl of the cell suspension. Propidium iodide (PI) solution (10
µl) and binding buffer (380 µl) were then added to the cell
suspension following incubation for 15 min on ice. Subsequently,
the number of stained cells was investigated using a BD FACSAriaII
flow cytometer (CXP software, version 2.2; FC500 flow cytometer;
Beckman Coulter, Inc., Brea, CA, USA) (32).
FKBP11 and FKBP11 small interfering
RNA (siRNA) transfection
FKBP11siRNA and control siRNA were obtained from
Biomics Biopharma Limited (Nantong, China). The sequences of
FKBP11siRNA used were as follows: siRNA#1:
5′-CACUAAUCCGAGCCAACUA-3′, siRNA#2: 5′-UGGAGCUGAUUGCACUAAU-3′,
siRNA#3: 5′-UGGUAGAUGGACGUAUUAU-3′ and siRNA#4:
5′-GAGCCAACUACUGGCUAAA-3′. The control siRNA sequence was
5′-UUCUCCGAACGUGUCACGU-3′. The pCMV-HA-FKBP11 plasmid with
hemagglutinin (HA) tag and control plasmid (pCMV-HA) were obtained
from the Plasmid Library of Jikai (Shanghai GeneChem Co., Ltd.,
Shanghai, China). HT-29 cells were seeded 1 day prior to
transfection using RPMI-1640 medium containing 10% FBS. Transient
transfection was performed using Lipofectamine 2000 (Thermo Fisher
Scientific, Inc.), according to the manufacturer's instructions
(33), 2 ml Lipofectamine 2000 and
1 µg FKBP11 plasmid were added individually into the 1.5 ml tubes
and mixed with OPTI-MEM medium for 20 min. Then cells were
incubated for 6 h at 37°C in RPMI-1640 medium in the absence of
serum and antibiotics. Following this, the transfection mixtures
were replaced with 10% FBS containing RPMI-1640 medium, and cells
were cultured for a further 48 h prior to further
experimentation.
Statistical analysis
SPSS software (version 19.0; IBM Corp., Armonk, NY,
USA) was used for data analysis. Data are expressed as the mean ±
standard error of the mean. Statistical analyses between two groups
were performed using the Student's t test, and statistical analyses
between multiple groups were performed using one-way analysis of
variance. One-way analysis of variance followed by Dunnett's post
hoc test was performed for multiple comparisons. Each experiment
consisted of a minimum of three replicates per condition. P<0.05
was considered to indicate a statistically significant result.
Results
FKBP11 and GRP78 expression levels are
increased in the inflamed intestinal mucosal tissues of patients
with CD
To investigate the potential contribution of FKBP11
in CD, western blot analysis was performed to determine the
expression levels of FKBP11 in the colon biopsies from patients
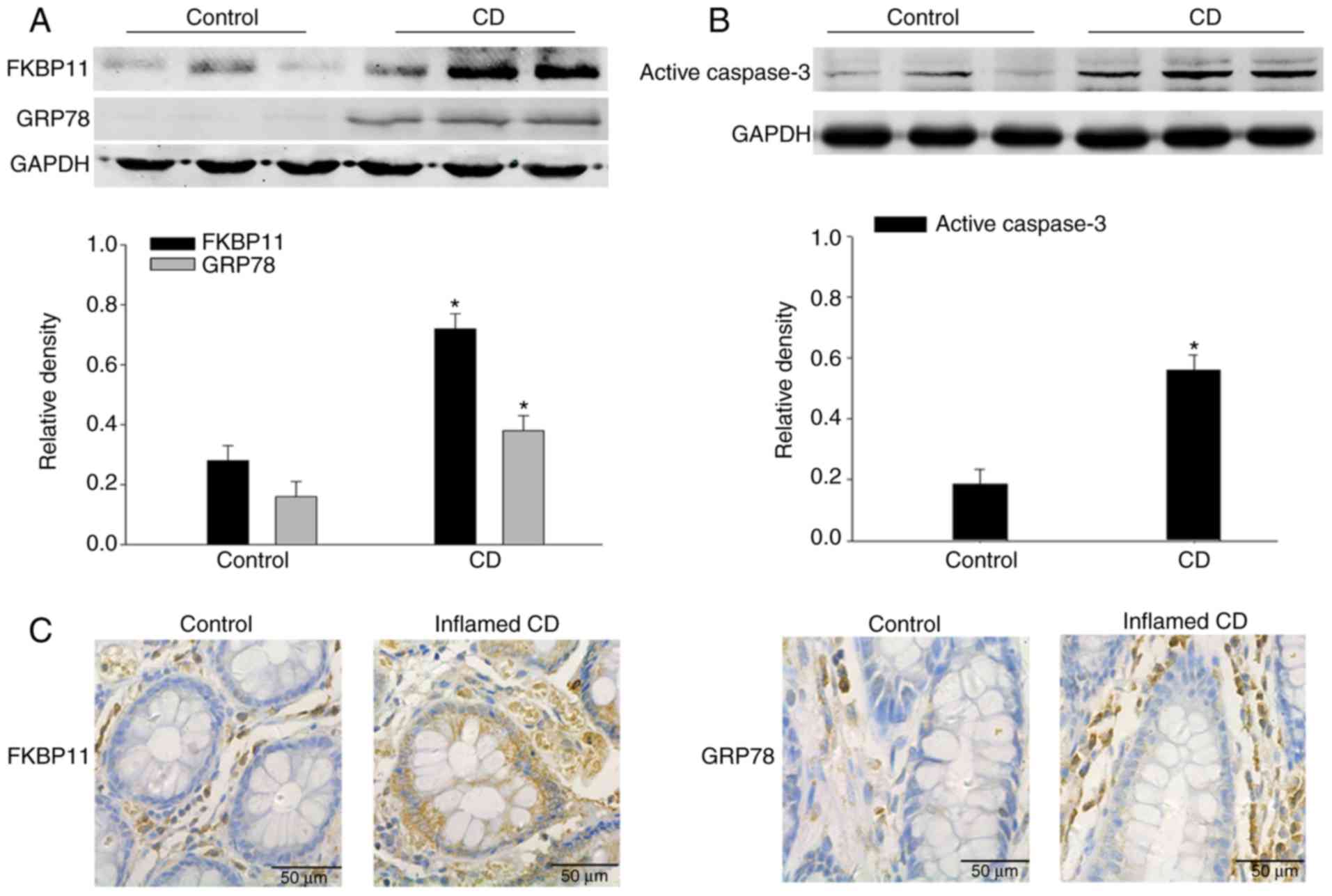
with CD. As shown in Fig. 1A,
FKBP11 and GRP78 protein expression levels were significantly
increased in the inflamed colon tissues of patients with active CD
compared with the control. In addition, the protein expression
level of active caspase-3 was significantly increased in the
inflamed colon tissues of patients with active CD compared with the
control (Fig. 1B).
Immunohistochemistry analysis further verified the upregulated
expression of FKBP11 and GRP78 in the inflamed intestinal mucosal
tissues of patients with CD compared with the control (Fig. 1C and D). These results suggested
that FKBP11 may be involved in the process of human CD.
TNBS-induced experimental colitis in
mice
A TNBS induced mice colitis model was established to
further investigate the role of FKBP11 in CD. This is a
well-established model of colonic inflammation, resembling numerous
primary clinical and morphological features of human CD (34). Compared with the ETOH control
group, the body weight of the TNBS group began to decrease by day 1
of the model and reached a minimum body weight at day 3 (Fig. 2A). The histological scores
reflecting inflammation levels of mice were significantly increased
in the TNBS group compared with the control group, and reached a
maximum score on day 3 (Fig. 2B).
H&E staining revealed some CD-like pathological changes in
TNBS-treated colons, including inflammatory cell infiltration
within the lamina propria, formation of ulceration, depletion of
epithelial cells, edema and thickened colon wall (Fig. 2C), which was consistent with
previous studies (31,35). These results suggest that the
TNBS-induced colitis model was well established in the present
study. Furthermore, the results of the present study demonstrated
that 3 days post-treatment with TNBS was the optimum time interval
of acute inflammation, and therefore this tissue was retained for
use in subsequent experiments.
 | Figure 2.Indicators confirm success of the
TNBS-induced colitis model. (A) Mice body weight changes following
administration of TNBS or ETOH at 0, 1, 2, 3, 4 and 5 days time
intervals post-treatment. (B) Histological scoring of the H&E
staining results at 0, 1, 2, 3, 4 and 5 days time intervals
post-treatment with either TNBS or ETOH. (C) Representative light
microscopy images of H&E stained colonic tissues from mice
following administration of TNBS or ETOH (scale bar, 200 µm).
Arrows indicate areas of inflammatory cell infiltration and
epithelial cell depletion. Data are presented as mean ± standard
error (n=3). *P<0.05 vs. ETOH. H&E, hematoxylin and eosin;
ETOH, ethanol; TNBS, 2, 4, 6-trinitrobenzenesulphonic acid. |
Expression levels of FKBP11 and GRP78
are increased in TNBS-induced colitis in mice
Western blot analysis demonstrated that FKBP11,
GRP78 and active caspase-3 protein expression levels were
significantly increased in the mice colon tissues of the TNBS group
compared with the ETOH group (Fig. 3A
and B), which was consistent with the results obtained from the
biopsies of patients with CD. Furthermore, immunohistochemistry
analysis was performed to investigate the expression levels and
distributions of FKBP11 and GRP78 proteins in the TNBS group. The
TNBS group revealed markedly increased levels of FKBP11 and GRP78
expression, predominantly in the cytoplasm of IECs, compared with
the ETOH group (Fig. 3C).
FKBP11 regulates ER stress in
IFN-γ/TNF-α treated IECs
Previous studies have revealed that ER stress is
involved in the pathogenesis and progression of IBD (36,37).
The expression levels of ER stress markers, such as GRP78 and DNA
damage-inducible transcript 3 protein (CHOP), have been previously
demonstrated to be upregulated in inflamed intestines of IBD
(38). To investigate the
association between FKBP11 and ER stress in CD, HT-29 cells were
treated with IFN-γ (2.5 ng/ml) and TNF-α (50 ng/ml). FKBP11 protein
levels significantly increased in a time-dependent manner following
IFN-γ/TNF-α stimulation compared with the control, which suggests
that an IFN-γ/TNF-α induced ER stress cell model had been
successfully established (Fig.
4A). Following this, cells were transfected with control siRNA
and FKBP11siRNAs, and the expression of FKBP11 was subsequently
determined. Western blot analysis revealed that FKBP11-siRNA#3
demonstrated the most efficient knockdown of FKBP11 expression
(Fig. 4B), and therefore was
subsequently chosen for use in further experiments. To investigate
the association between FKBP11 and ER stress, the protein
expression of FKBP11 was knocked down via RNA interference.
Compared with the control siRNA group, suppression of FKBP11
expression with siRNA significantly enhanced the expression of
GRP78 in IFN-γ/TNF-α-induced HT-29 cells (Fig. 4C). These results suggest that
FKBP11 may regulate IFN-γ/TNF-α-induced ER stress in HT-29
cells.
 | Figure 4.Association of FKBP11 expression
levels with ER stress in IFN-γ/TNF-α-treated HT-29 cells. (A) HT-29
cells were treated with IFN-γ (2.5 ng/ml) and TNF-α (50 ng/ml) for
different time intervals (0, 6, 12 and 24 h) to construct a cell
model of ER stress. Western blot analyses revealed a significant
upregulation of FKBP11 and GRP78 protein levels in
IFN-γ/TNF-α-treated cells in a time-dependent manner. (B) FKBP11
expression following transfection with FKBP11 siRNA in HT-29 cells
was revealed by western blot analyses, and transfection with
FKBP11siRNA#3 was revealed to exhibit the most significant
downregulation of FKBP11 expression. (C) HT-29 cells were
transfected with control siRNA or FKBP11siRNA for 48 h, and then
treated with IFN-γ/TNF-α for 24 h. Western blot analyses revealed
that FKBP11 and GRP78 expression levels increased following
IFN-γ/TNF-α treated cells compared with the untreated control, and
GRP78 expression was significantly increased following transfection
of FKBP11siRNA compared with control siRNA. Bar graphs reveal the
densities of FKBP11 or GRP78 protein levels vs. GAPDH. Data are
presented as mean ± standard error (n=3). *P<0.05 vs. control
siRNA; #P<0.05 vs. IFN-γ/TNF-α untreated control.
IFN-γ, interferon-γ; TNF-α, tumor necrosis factor-α; FKBP11, FK506
binding protein 11; GRP78, 78 kDa glucose-regulated protein; siRNA,
small interfering RNA. |
FKBP11 protects HT-29 cells against
IFN-γ/TNF-α-induced apoptosis
It has been previously reported that increased
apoptosis of IECs disrupts the integrity of the epithelial barrier
and contributes to CD development (8). In the present study, it was
demonstrated that combined IFN-γ/TNF-α stimulation significantly
enhanced the expression levels of pro-apoptotic proteins BAX and
active caspase-3 compared with the control, which suggests that an
inflammation-triggered IEC apoptosis model had been successfully
established (Fig. 5A). To
investigate the function of FKBP11 in IFN-γ/TNF-α-induced IEC
apoptosis, the pCMV-HA-FKBP11 plasmid was transfected into HT-29
cells to overexpress FKBP11 and the cells were then treated with
IFN-γ/TNF-α. The results of the western blot analyses suggested
that the expression of the pro-apoptotic markers BAX and active
caspase-3 were significantly decreased when FKBP11 was
overexpressed compared with the empty control plasmid group
(Fig. 5A). In addition, in cell
treated with IFN-γ/TNF-α, knockdown of FKBP11 using siRNA
significantly increased the expression levels of BAX and active
caspase-3 compared with the control siRNA group (Fig. 5B). These results suggested that
FKBP11 may have an important role in IFN-γ/TNF-α stimulated IEC
apoptosis.
 | Figure 5.FKBP11 is involved in
IFN-γ/TNF-α-induced apoptosis in HT-29 cells. (A) HT-29 cells were
transfected with either the FKBP11 overexpressing plasmid
pCMV-HA-FKBP11, blank control plasmid pCMV-HA, controls siRNA or
FKBP11siRNA for 48 h and then treated with IFN-γ (2.5 ng/ml) and
TNF-α (50 ng/ml) for 24 h. Western blot analyses detected FKBP11,
BAX and active caspase-3 expression levels following treatment with
IFN-γ/TNF-α compared with the untreated control and control plasmid
(HA). #P<0.05 vs. IFN-γ/TNF-α untreated control;
$P<0.05 vs. control plasmid pCMV-HA. (B) Western blot
analyses detected FKBP11, BAX, active caspase-3 and PCNA following
transfection with FKBP11 siRNA and treatment with IFN-γ (2.5 ng/ml)
and TNF-α (50 ng/ml) for 24 h. Data are presented as mean ±
standard error (n=3). *P<0.05 vs. control siRNA. HA,
hemagglutinin; FKBP11, FK506 binding protein 11; siRNA, small
interfering RNA; GRP78, 78 kDa glucose-regulated protein; IFN-γ,
interferon-γ; TNF-α, tumor necrosis factor-α; BAX, Bcl2 associated
X apoptosis regulator; PCNA, proliferating cell nuclear
antigen. |
FKBP11 suppresses IFN-γ/TNF-α-induced
IEC apoptosis by inhibiting the JNK-caspase signaling pathway
To further investigate the importance of FKBP11 in
apoptosis, the effect of FKBP11 overexpression in IECs following
IFN-γ/TNF-α-induced cellular apoptosis was investigated using an
Annexin V/PI staining assay. As shown in Fig. 6A, following IFN-γ/TNF-α
administration, the Annexin V+/PI+ double
positive cell ratio was notably increased compared with the
control, suggesting that a cellular apoptosis model had been
successfully established. Furthermore, FKBP11 overexpression
markedly attenuated the IFN-γ/TNF-α induced apoptosis of cells
compared with the control plasmid group (Fig. 6A). A previous study demonstrated
that excessive or prolonged ER stress leads to the induction of
apoptotic processes via CHOP, JNK and/or caspase dependent pathways
(39). Human caspase-12 has no
role in apoptotic pathways (40)
and it is still controversial that human caspase-12 acts as a
functional counterpart of mouse caspase-12. Of note, the amino acid
sequence of mouse caspase-12 has a 61% identity with human
caspase-4, which is involved in apoptosis induced by ER stress
(41). Therefore, the
phosphorylation levels of JNK and expression levels of caspase-4 in
IECs subjected to IFN-γ and TNF-α administration were investigated.
As shown in Fig. 6B, HT-29 cells
exhibited significantly increased levels of phosphorylated (p)-JNK
following IFN-γ/TNF-α administration compared with the control,
thus suggesting activation of the JNK signaling pathway.
Overexpression of FKBP11 significantly suppressed the levels of
phospho-JNK in IFN-γ/TNF-α-treated cells compared with control
cells treated with IFN-γ/TNF-α (Fig.
6B). In addition, cells treated with IFN-γ/TNF-α and
overexpressing FKBP11 significantly suppressed caspase-4 expression
compared with control cells treated with IFN-γ/TNF-α. In
conclusion, these results suggest that FKBP11 may protect IECs
against IFN-γ/TNF-α-induced apoptosis via negative regulation of
the JNK-caspase signaling pathway.
Discussion
Accumulating evidence has demonstrated that
disrupted ER homeostasis in IECs contributes to the pathogenesis of
IBD. Prolonged ER stress and impaired UPR signaling results in
excessive IEC apoptosis, defects in intestinal mucosal barrier
function and augmentation of the intestinal inflammatory reaction,
which together promote IBD progression (11,42,43).
Previous bioinformatics analysis investigating IBD revealed that
FKBP11, a novel UPR protein, is highly expressed exclusively in the
intestinal tissues of human CD (25). Thus, the present study aimed to
investigate the possible role of FKBP11 in the intestinal
epithelium homeostasis of CD. In the present study, increased
expression of FKBP11 protein was detected in inflamed intestinal
tissues from patients with active CD and mice with TNBS-induced
colitis. Using an ER stress and apoptosis cellular model in IECs
in vitro, the results of the present study suggested that
highly expressed FKBP11 attenuates IFN-γ/TNF-α-induced apoptosis
via regulation of ER stress and inhibiting the detrimental
JNK-caspase apoptosis signaling pathway in IECs.
FKBP11 is a member of the PPIase family, which
catalyzes the folding of proline-containing polypeptides, and thus,
is important for the regulation of UPR (19). Previous studies have suggested that
FKBP11 is closely associated with numerous inflammatory disorders
and digestive tumors. For example, a quantitative proteomics
analysis revealed a close association between highly expressed
FKBP11 and insulin resistance, as well as type 2 diabetes (21). The expression of FKBP11 and other
ER stress genes (activating transcription factor 4 and endoplasmin)
were significantly increased by treatment with palmitate in
pancreatic islet β-cells (20).
Thus, FKBP11 may regulate UPR in β-cells when exposed to abnormally
high levels of circulating free fatty acids, and thus participate
in the pathogenesis of type 2 diabetes. In addition, enhanced
expression of FKBP11 was demonstrated in the B cell
transcriptome of patients with quiescent SLE (44). Furthermore, overexpression of
FKBP11 was revealed to disrupt B cell tolerance against DNA and
initiate plasma cell differentiation by acting upstream of the
paired box 5 master regulator gene, which may synergistically
contribute to B cell gene abnormalities in SLE immunopathology
(22,44). In addition, progressively elevated
expression of FKBP11 during the development of HCC has been
previously demonstrated, which suggests that FKBP11 may represent a
novel early marker for HCC (23).
In the present study, western blot analyses and
immunohistochemistry assays revealed that the expression of FKBP11
is significantly enhanced in the inflamed intestinal tissues of
patients with active CD compared with healthy controls.
Furthermore, using a murine model of TNBS-induced colitis, which
mimics human CD, it was revealed that the expression of FKBP11 and
GRP78 were significantly upregulated in IECs compared with the
control group. The results of the present study supported the
previous findings of a bioinformatics analysis investigating human
IBD (25), and suggested that
FKBP11 may be involved in CD pathogenesis. Following this, an
IFN-γ/TNF-α induced ER stress cell model was established to induce
ER stress in human colon epithelial cell line HT-29, and it was
demonstrated that FKBP11 and GRP78 protein expression levels were
increased in IFN-γ/TNF-α induced HT-29 cells. Furthermore,
knockdown of FKBP11 using siRNA significantly enhanced the
upregulation of GRP78, suggesting that FKBP11 is important for
inflammation-induced ER stress in IECs. The results of the present
study did not determine the exact molecular mechanisms underlying
the effect of FKBP11 upregulation in CD, however, previous studies
may provide some potential suggestions. In a genetic mouse model of
ER stress-induced hepatic steatosis, FKBP11 gene expression
was revealed to be regulated by IRE1α, a conserved UPR sensor in
hepatocytes (24). Notably, as a
major sensor of ER stress, genetic ablation of IRE1α in IECs
has been demonstrated to result in a spontaneous colitis in mice,
thus suggesting that IRE1α functions as an important defense
molecule against IBD (16). In
conclusion, it can be suggested that IRE1α positively regulates
FKBP11 expression in IECs under adverse inflammatory conditions,
and that the IRE1α-FKBP11 UPR mechanism may promote proper protein
folding, attenuate ER stress and regulate intestinal epithelial
homeostasis.
Three UPR pathways (IRE1α, PERK and ATF6α) promote
cell survival by reducing misfolded protein levels, however, UPR
signaling also promotes apoptotic cell death if ER stress is not
attenuated (1). For example, under
persistent ER stress, PERK signaling induces the expression of
CHOP, a key regulator of ER stress associated apoptosis (45). In addition, decreased IRE1α levels
have been revealed to induce IEC apoptosis via activation of the
PERK-CHOP pro-apoptotic pathway, which contributes to spontaneous
colitis in IRE1α knockout mice (16). Furthermore, the pro-apoptotic
IRE1-TNF receptor associated factor 2 (TRAF2)-JNK pathway can be
activated by prolonged ER stress (46). Signal transduction between
IRE1-TRAF2 and phosphorylation of JNK may be regulated in certain
contexts by MAP kinase kinase kinase apoptotic signal-regulating
kinase 1 and its activator kinase (46). JNK-induced apoptosis may involve
pro-apoptotic Bcl-2 family members, BAX and Bcl-2 antagonist/killer
1, which can amplify the IRE1 signal (46). Cysteine proteases (or caspases) are
important mediators of apoptosis. Different from other caspase
members that are activated by membrane- or mitochondrial-targeted
apoptotic signals, caspase-12 is localized to the ER and is
activated by ER stress via disruption of ER calcium homeostasis and
accumulation of excess proteins in the ER (13). Caspase-12 activity can result in
selective apoptosis in response to ER stress via cleavage of
pro-caspase-3 into activated caspase-3 (13). Caspase-12, a member of the cysteine
protease family, serves a role in promoting apoptosis in mice,
whereas caspase-12 in humans had lost its apoptotic regulatory
function due to genetic mutations, and the homeotic caspase-4 may
have the same function (47). In
the present study, caspase-4 was used instead of caspase-12, as
human colon cancer cells HT-29 were used. Two major pro-apoptotic
pathways were revealed to be associated with ER stress: The
caspase-12/caspase-3 pathway and the JNK/BAX pathway, which were
significantly activated in IECs following treatment with
IFN-γ/TNF-α. Notably, the results of the present study demonstrated
that FKBP11 overexpression attenuates ER stress-related
pro-apoptotic signal transduction, which suppresses IFN-γ/TNF-α
induced apoptosis in IECs.
In conclusion, the results of the present study
demonstrated that FKBP11 and GRP78 expression levels are
significantly upregulated in the inflamed intestinal mucosa of CD.
Furthermore, the results revealed that overexpression of FKBP11
could attenuate IFN-γ/TNF-α-induced cellular apoptosis in IECs.
These results are compatible with the hypothesis that increased
FKBP11 expression attenuates IFN-γ/TNF-α induced ER stress
associated with the JNK-caspase apoptotic pathway in IECs. However,
further studies are required to further validate the results of the
present study and to investigate the exact molecular mechanisms of
FKBP11 in intestinal epithelial homeostasis and CD development.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Scientific Foundation of China (grant no. 81572397),
Jiangsu Province's Outstanding Medical Academic Leader program
(grant no. LJ101135) and Infectious Disease Clinical Medicine
Center Program of Suzhou (grant. no. SZZX201508).
Availability of data and materials
All data generated or analyzed during this study are
included in this article.
Authors' contributions
XW, XC, DZ and GZ conceived and designed the study.
XW, XC, JZ, ZS, XS and LW performed the experiments. HW, YS and YN
established and monitored the animal colitis model. XW wrote the
paper. CZ and ML analyzed the cellular apoptosis rates and designed
the chart accordingly. DZ and GZ reviewed and edited the
manuscript. All authors read and approved the manuscript.
Ethics approval and consent to
participate
This experiment was performed with the approval of
the Ethics Committee of the Affiliated Hospital of Nantong
University (permit no. 2014-L087).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Cao SS: Endoplasmic reticulum stress and
unfolded protein response in inflammatory bowel disease. Inflamm
Bowel Dis. 21:636–644. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tawfik A, Flanagan PK and Campbell BJ:
Escherichia coli-host macrophage interactions in the pathogenesis
of inflammatory bowel disease. World J Gastroenterol. 20:8751–8763.
2014.PubMed/NCBI
|
|
3
|
Cheng L, Huang MF, Mei PF, Bo WH and Deng
CS: The clinical, endoscopic and pathologic features of Crohn's
disease in the differentiation from intestinal tuberculosis.
Zhonghua Nei Ke Za Zhi. 52:940–944. 2013.(In Chinese). PubMed/NCBI
|
|
4
|
Galeone C, Pelucchi C, Barbera G, Citterio
C, La Vecchia C and Franchi A: Crohn's disease in Italy: A critical
review of the literature using different data sources. Dig Liver
Dis. 49:459–466. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kim ES, Chen M, Lee J, Lee CK and Kim YS:
Diagnosis of inflammatory bowel disease in Asia: The results of a
multinational web-based survey in the 2(nd) Asian Organization for
Crohn's and Colitis (AOCC) meeting in Seoul. Intest Res.
14:224–230. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Guo B and Li Z: Endoplasmic reticulum
stress in hepatic steatosis and inflammatory bowel diseases. Front
Genet. 5:2422014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hisamatsu T, Kanai T, Mikami Y, Yoneno K,
Matsuoka K and Hibi T: Immune aspects of the pathogenesis of
inflammatory bowel disease. Pharmacol Ther. 137:283–297. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Salim SY and Söderholm JD: Importance of
disrupted intestinal barrier in inflammatory bowel diseases.
Inflamm Bowel Dis. 17:362–381. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cao SS: Epithelial ER stress in Crohn's
disease and ulcerative colitis. Inflamm Bowel Dis. 22:984–993.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kaser A, Adolph TE and Blumberg RS: The
unfolded protein response and gastrointestinal disease. Semin
Immunopathol. 35:307–319. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Luo K and Cao SS: Endoplasmic reticulum
stress in intestinal epithelial cell function and inflammatory
bowel disease. Gastroenterol Res Pract. 2015:3287912015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Urano F, Wang X, Bertolotti A, Zhang Y,
Chung P, Harding HP and Ron D: Coupling of stress in the ER to
activation of JNK protein kinases by transmembrane protein kinase
IRE1. Science. 287:664–666. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nakagawa T, Zhu H, Morishima N, Li E, Xu
J, Yankner BA and Yuan J: Caspase-12 mediates
endoplasmic-reticulum-specific apoptosis and cytotoxicity by
amyloid-beta. Nature. 403:98–103. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kaser A, Lee AH, Franke A, Glickman JN,
Zeissig S, Tilg H, Nieuwenhuis EE, Higgins DE, Schreiber S,
Glimcher LH and Blumberg RS: XBP1 links ER stress to intestinal
inflammation and confers genetic risk for human inflammatory bowel
disease. Cell. 134:743–756. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li M, Zhang S, Qiu Y, He Y, Chen B, Mao R,
Cui Y, Zeng Z and Chen M: Upregulation of miR-665 promotes
apoptosis and colitis in inflammatory bowel disease by repressing
the endoplasmic reticulum stress components XBP1 and ORMDL3. Cell
Death Dis. 8:e26992017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang HS, Chen Y, Fan L, Xi QL, Wu GH, Li
XX, Yuan TL, He SQ, Yu Y, Shao ML, et al: The endoplasmic reticulum
stress sensor IRE1α in intestinal epithelial cells is essential for
protecting against colitis. J Biol Chem. 290:15327–15336. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hasnain SZ, Tauro S, Das I, Tong H, Chen
AC, Jeffery PL, McDonald V, Florin TH and McGuckin MA: IL-10
promotes production of intestinal mucus by suppressing protein
misfolding and endoplasmic reticulum stress in goblet cells.
Gastroenterology. 144:357–368.e9. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shkoda A, Ruiz PA, Daniel H, Kim SC,
Rogler G, Sartor RB and Haller D: Interleukin-10 blocked
endoplasmic reticulum stress in intestinal epithelial cells: Impact
on chronic inflammation. Gastroenterology. 132:190–207. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rulten SL, Kinloch RA, Tateossian H,
Robinson C, Gettins L and Kay JE: The human FK506-binding proteins:
Characterization of human FKBP19. Mamm Genome. 17:322–331. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kim M, Lee JS, Oh JE, Nan J, Lee H, Jung
HS, Chung SS and Park KS: SIRT3 overexpression attenuates
palmitate-induced pancreatic β-cell dysfunction. PLoS One.
10:e01247442015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lu H, Yang Y, Allister EM, Wijesekara N
and Wheeler MB: The identification of potential factors associated
with the development of type 2 diabetes: A quantitative proteomics
approach. Mol Cell Proteomics. 7:1434–1451. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ruer-Laventie J, Simoni L, Schickel JN,
Soley A, Duval M, Knapp AM, Marcellin L, Lamon D, Korganow AS,
Martin T, et al: Overexpression of Fkbp11, a feature of lupus B
cells, leads to B cell tolerance breakdown and initiates plasma
cell differentiation. Immun Inflamm Dis. 3:265–279. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lin IY, Yen CH, Liao YJ, Lin SE, Ma HP,
Chan YJ and Chen YM: Identification of FKBP11 as a biomarker for
hepatocellular carcinoma. Anticancer Res. 33:2763–2769.
2013.PubMed/NCBI
|
|
24
|
Zhang K, Wang S, Malhotra J, Hassler JR,
Back SH, Wang G, Chang L, Xu W, Miao H, Leonardi R, et al: The
unfolded protein response transducer IRE1α prevents ER
stress-induced hepatic steatosis. EMBO J. 30:1357–1375. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Clark PM, Dawany N, Dampier W, Byers SW,
Pestell RG and Tozeren A: Bioinformatics analysis reveals
transcriptome and microRNA signatures and drug repositioning
targets for IBD and other autoimmune diseases. Inflamm Bowel Dis.
18:2315–2333. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ingle SB, Adgaonkar BD, Jamadar NP,
Siddiqui S and Hinge CR: Crohn's disease with gastroduodenal
involvement: Diagnostic approach. World J Clin Cases. 3:479–483.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yu H, Liu Y, Wang Y, Peng L, Li A and
Zhang Y: Clinical, endoscopic and histological differentiations
between Crohn's disease and intestinal tuberculosis. Digestion.
85:202–209. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
National Research Council (US) Institute
for Laboratory Animal Research, . Guide for the care and use of
laboratory animals. Washington (DC): National Academies Press (US);
1996
|
|
29
|
Hollenbach E, Vieth M, Roessner A, Neumann
M, Malfertheiner P and Naumann M: Inhibition of RICK/nuclear
factor-kappaB and p38 signaling attenuates the inflammatory
response in a murine model of Crohn disease. J Biol Chem.
280:14981–14988. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang D, Wang L, Yan L, Miao X, Gong C,
Xiao M, Ni R and Tang Q: Vacuolar protein sorting 4B regulates
apoptosis of intestinal epithelial cells via p38 MAPK in Crohn's
disease. Exp Mol Pathol. 98:55–64. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lin H, Chen R, Jiang X, Wu X, Huang X,
Dong X, Yang X, Lin X, Chen X, Chen X and Huang Z: Elevated
fibrinogen-like protein 2 in TNBS-induced colitis mice: Association
with Th17 and regulatory T cells. Mol Med Rep. 16:3445–3454. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li L, Miao X, Ni R, Miao X, Wang L, Gu X,
Yan L, Tang Q and Zhang D: Epithelial-specific ETS-1 (ESE1/ELF3)
regulates apoptosis of intestinal epithelial cells in ulcerative
colitis via accelerating NF-κB activation. Immunol Res. 62:198–212.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tao T, Cheng C, Ji Y, Xu G, Zhang J, Zhang
L and Shen A: Numbl inhibits glioma cell migration and invasion by
suppressing TRAF5-mediated NF-κB activation. Mol Biol Cell.
23:2635–2644. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Elson CO, Sartor RB, Tennyson GS and
Riddell RH: Experimental models of inflammatory bowel disease.
Gastroenterology. 109:1344–1367. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cui X, Shan X, Qian J, Ji Q, Wang L, Wang
X, Li M, Ding H, Liu Q, Chen L, et al: The suppressor of cytokine
signaling SOCS1 promotes apoptosis of intestinal epithelial cells
via p53 signaling in Crohn's disease. Exp Mol Pathol. 101:1–11.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hosomi S, Kaser A and Blumberg RS: Role of
endoplasmic reticulum stress and autophagy as interlinking pathways
in the pathogenesis of inflammatory bowel disease. Curr Opin
Gastroenterol. 31:81–88. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zeng LX, Tao J, Liu HL, Tan SW, Yang YD,
Peng XJ, Liu ZH, Jiang J and Wu B: β-Arrestin2 encourages
inflammation-induced epithelial apoptosis through ER stress/PUMA in
colitis. Mucosal Immunol. 8:683–695. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kunde DA, Chong WC, Nerurkar PV, Ahuja KD,
Just J, Smith JA, Guven N and Eri RD: Bitter melon protects against
ER stress in LS174T colonic epithelial cells. BMC Complement Altern
Med. 17:22017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hao X, Yao A, Gong J, Zhu W, Li N and Li
J: Berberine ameliorates pro-inflammatory cytokine-induced
endoplasmic reticulum stress in human intestinal epithelial cells
in vitro. Inflammation. 35:841–849. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Saleh M, Vaillancourt JP, Graham RK, Huyck
M, Srinivasula SM, Alnemri ES, Steinberg MH, Nolan V, Baldwin CT,
Hotchkiss RS, et al: Differential modulation of endotoxin
responsiveness by human caspase-12 polymorphisms. Nature.
429:75–79. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hitomi J, Katayama T, Eguchi Y, Kudo T,
Taniguchi M, Koyama Y, Manabe T, Yamagishi S, Bando Y, Imaizumi K,
et al: Involvement of caspase-4 in endoplasmic reticulum
stress-induced apoptosis and Abeta-induced cell death. J Cell Biol.
165:347–356. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Koh SJ, Kim JW, Kim BG, Lee KL, Chun J and
Kim JS: Fexofenadine regulates nuclear factor-κB signaling and
endoplasmic reticulum stress in intestinal epithelial cells and
ameliorates acute and chronic colitis in mice. J Pharmacol Exp
Ther. 352:455–461. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Das I, Png CW, Oancea I, Hasnain SZ,
Lourie R, Proctor M, Eri RD, Sheng Y, Crane DI, Florin TH and
McGuckin MA: Glucocorticoids alleviate intestinal ER stress by
enhancing protein folding and degradation of misfolded proteins. J
Exp Med. 210:1201–1216. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Garaud JC, Schickel JN, Blaison G, Knapp
AM, Dembele D, Ruer-Laventie J, Korganow AS, Martin T,
Soulas-Sprauel P and Pasquali JL: B cell signature during inactive
systemic lupus is heterogeneous: Toward a biological dissection of
lupus. PLoS One. 6:e239002011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lin JH, Li H, Yasumura D, Cohen HR, Zhang
C, Panning B, Shokat KM, Lavail MM and Walter P: IRE1 signaling
affects cell fate during the unfolded protein response. Science.
318:944–949. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Tabas I and Ron D: Integrating the
mechanisms of apoptosis induced by endoplasmic reticulum stress.
Nat Cell Biol. 13:184–190. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Song S, Lee H, Kam TI, Tai ML, Lee JY, Noh
JY, Shim SM, Seo SJ, Kong YY, Nakagawa T, et al: E2-25K/Hip-2
regulates caspase-12 in ER stress-mediated Abeta neurotoxicity. J
Cell Biol. 182:675–684. 2008. View Article : Google Scholar : PubMed/NCBI
|