Introduction
Familial adenomatous polyposis (FAP) is an autosomal
dominant-inherited colorectal cancer syndrome with poor prognosis
(1,2). It is well established that FAP is
linked to germline mutations of the adenomatous polyposis coli
(APC; transcript ID, ENST00000257430.8; https://www.ensembl.org/Homo_sapiens/Transcript/Summary?g=ENSG00000134982;
r=5:112737888-112846239;t=ENST00000257430) gene (3,4), and
screening for APC germline mutations in families with FAP is a
standard and effective tool for the predictive testing of at-risk
individuals (3–6). However, through conventional
screening methods, a number of patients with FAP have no detectable
APC mutations, which is defined as APC mutation-negative FAP
(7,8). Certain patients with FAP without
detectable germline APC mutations were revealed to carry biallelic
mutations in the base-excision-repair gene mutY DNA glycosylase
(MUTYH; transcript ID, ENST00000354383.10), and investigators
demonstrated that mutations in MUTYH may explain up to 25% of APC
mutation-negative FAP cases (8,9).
However, a large subset of families with a history of FAP have
pathogenic alterations that are undetectable, and this is termed
APC/MUTYH mutation-negative FAP (8–10).
In the present study, in order to investigate the
germline mutations and the potential causes of APC/MUTYH
mutation-negative FAP, a cohort of 13 patients with FAP were
enrolled. First, whole-gene sequencing of APC and MUTYH was applied
to determine the pathogenic alterations predisposing patients to
FAP (11–13), and the results identified four
known pathogenic mutations in APC and two novel disease-associated
pathogenic mutations in six individuals. For the APC/MUTYH
mutation-negative FAP cases, missense, synonymous and intronic
mutations in APC and MUTYH were analyzed using bioinformatics
assessment tools and databases; notably, a synonymous mutation of
Tyr486Tyr in APC (APC∆486s) was predicted to be a
disease-causing polymorphism. In-depth analysis of the results
suggested that this mutation may have caused exon skipping in APC
and that the truncated APC protein resulted in FAP.
It has been established that the majority of
synonymous and intronic mutations are silent mutations; however,
previous investigations have also revealed that certain synonymous
substitutions are associated with a risk of disease and other
complex traits including susceptibility to cancer (14,15).
Increasing amounts of evidence indicate that synonymous mutations
may induce major splicing defects at the mRNA level and
consequently result in disease (16).
Combined with the above conclusions and the findings
of the present study, it was hypothesized that APC∆486s
which was previously considered to be non-pathogenic polymorphism
was a potential pathogenic mutation for APC/MUTYH mutation-negative
FAP. To confirm the splicing regulation of the synonymous mutation
APC∆486s in vitro, an exon skipping assay was
performed to verify whether the synonymous mutation
APC∆486s leads to APC protein truncation. It is hoped
that following further studies on the mechanism of exon skipping of
the synonymous mutation APC∆486s, it could be applied as a
potential pathogenic alteration in the diagnostic application for
those APC/MUTYH mutation-negative patients to FAP.
Materials and methods
Ethics statement
The present study was approved by and performed in
accordance with the Research Ethics Board of Kunming Medical
University (Kunming, China). All participants provided written
informed consent, and the ethics committees approved the consent
procedure.
Patients
Between January 2013 and December 2017, 13 patients
[male:female 8:5, mean age 31 years (range, 16–41] clinically
diagnosed with FAP were recruited from the First Affiliated
Hospital of Kunming Medical University (Kunming, China). The
diagnosis of all patients referred for the present study was
confirmed by surgery, and all patients had a family history
suggestive of FAP. Blood samples were collected from all patients
prior to surgery.
Gene-sequencing of APC and MUTYH
Blood samples from all patients with FAP underwent
exon-specific DNA sequencing for APC and DNA was purified following
the manufacturer's protocols of the DNA extraction kit (QIAamp DNA
blood mini kit; Qiagen, Valencia, CA, USA). The extracted DNA was
quantified by a Pearl nanophotometer (Implen, Munich, Germany). The
promoter region, exons 1-14 and 21 segments of exon 15 in the APC
gene were separately amplified by polymerase chain reaction (PCR)
(11,13,17).
Following the DNA sequencing of APC, whole-gene sequencing of MUTYH
was performed for the APC mutation-negative FAP samples. The primer
pairs used for MUTYH sequencing in the present study were designed
as previously described (12,13).
In silico analysis and functional
single nucleotide polymorphism (SNP) assessment
The sequencing data for each gene was initially
analyzed using Mutation Surveyor (18) (www.softgenetics.com). Human Gene Mutation Database
(19) (www.hgmd.org), International HapMap Project (20) (ftp.ncbi.nlm.nih.gov/hapmap/), dbSNP database
(21) (www.ncbi.nlm.nih.gov/SNP), 1000 Genomes (http://www.internationalgenome.org/), and Ensembl
(22) (www.ensembl.org) were also used.
For APC gene mutations, the Leiden Open Variation
Database (LOVD; databases.lovd.nl/shared/genes/APC), BIPMed SNP
Array (bipmed.iqm.unicamp.br/snparray/genes/APC), ClinVar at the
National Center for Biotechnology Information [NCBI; www.ncbi.nlm.nih.gov/clinvar/?term=APC(gene)],
the Universal Mutation Database (UMD)-APC mutations database
(www.umd.be/APC/W_APC), Iran Variation
Database (genet.ir/variome/genes/APC), International Society for
Gastrointestinal Hereditary Tumours Database (www.insight-database.org/genes/APC),
LOVD (proteomics.bio21.unimelb.edu.au/lovd/genes/APC),
Spain Mutation Database (lovd3.isciii.es/genes/APC) and the
Zhejiang University Center for Genetic and Genomic Medicine
(23) (www.genomed.org/lovd2/home.php?select_db=APC) were
applied.
For the mutation screening of the MUTYH gene, the
LOVD (proteomics.bio21.unimelb.edu.au/lovd/genes/MUTYH),
Shared database (databases.lovd.nl/shared/genes/MUTYH), UMD-MUTYH
mutations database (www.umd.be/MUTYH/), Iran Variation Database
(genet.ir/variome/genes/MUTYH),
ClinVar at NCBI [www.ncbi.nlm.nih.gov/clinvar/?term=MUTYH(gene)],
International Society for Gastrointestinal Hereditary Tumours
Database (www.insight-database.org/genes/MUTYH) and PharmGKB
(https://www.pharmgkb.org/gene/PA31328/variantAnnotation)
were used.
Bioinformatics predictions of the
missense, synonymous and intronic substitutions in APC/MUTYH
mutation-negative FAP
To investigate the possible genetic causes in
patients with APC/MUTYH mutation-negative FAP, bioinformatics
prediction tools were used to study the function of any identified
missense, synonymous and intron substitutions. Polymorphism
phenotype version 2 (24), Sorting
Intolerant From Tolerant (SIFT) (25) and SNPs&Gene Ontology (GO)
(26) were used to predict
deleterious missense substitutions. Exonic splicing enhancer (ESE)
finder 3.0 (27) (krainer01.cshl.edu/cgi-bin/tools/ESE3/esefinder.cgi?process=home),
RESCUE-ESE (28) (genes.mit.edu/burgelab/rescue-ese),
exonic-splicing regulatory (ESR) search (26) and putative exonic splicing
enhancers (PESX) (29) were used
to determine the corresponding functional impacts of the synonymous
and intronic substitutions.
Hybrid mini-gene assay of synonymous SNP
APC∆486s
Generation of mini-gene constructs of
synonymous SNP APC∆486s
Through bioinformatics analysis, the synonymous and
intronic variants in MUTYH were predicted to have no effect on the
disease. However, five synonymous SNPs in APC were predicted to be
disease-associated mutations (Fig.
1), and among them, APC∆486s was predicted to be
associated with exon skipping in APC. To confirm the splicing
regulation of the synonymous mutation APC∆486s in
vitro, a mini-gene system of APC∆486s was
constructed to investigate its splicing impact on the APC gene. The
nucleotide sequence of exon 12 with wild-type and mutant
APC∆486s was synthesized, together with 264 bp of the
12th flanking intron, carrying the XhoI restriction site,
and 210 bp of the 13th flanking intron, carrying the HindIII
restriction site (Fig. 2), as
described previously (30). The
wild-type and mutant mini-gene were inserted into pEGFP-N1 to
construct the recombinant plasmids pEGFP-N1-wt-mini-gene and
pEGFP-N1-mt-mini-gene, respectively (Takara Bio USA, Inc., CA,
USA).
 | Figure 1.Synonymous substitutions in the APC
gene and the corresponding bioinformatics prediction of each mutant
polymorphism. I indicates the synonymous substitution c.1458T>C,
located in the first armadillo repeat of APC, which is in the
Asef1, Asef2 and IQGAP binding domain. The splicing impact of
c.1458T>C may have caused the skipping of exon 12 in APC. II
indicates the synonymous substitution c.4425G>A located in the
third 20 amino acid repeat of APC, which is located in the
β-catenin binding/degradation domain. The splicing impact of
c.4425G>A may have caused a frameshift mutation in APC. III
indicates the synonymous substitution c.5034 G>A is located
behind the fourth 20 amino acid repeat of APC, which is in the
β-catenin binding/degradation domain. The splicing impact of c.5034
G>A may have resulted in a frameshift mutation. IV indicates the
synonymous substitution c.5268T>G located behind the second SAMP
repeat of APC. The splicing impact of c.5268T>G may also result
in a frameshift mutation. V indicates the synonymous substitution
c.5880G>A, located in the sixth 20 amino acid repeat of APC,
which is in the β-catenin binding/degradation domain. The splicing
impact of c. 5880G>A may have caused a frameshift mutation in
APC. APC, adenomatous polyposis coli; Asef, APC-stimulated guanine
nucleotide exchange factor; IQGAP1, IQ motif containing GTPase
activating protein 1; EB1, microtubule associated protein RP/EB
family member 1; DLG, discs large MAGUK scaffold protein 1. |
Transfection and reverse transcription
(RT)-PCR
Following identification by plasmid sequencing
(30), the wild-type and mutant
mini-gene constructs were transiently transfected into HeLa cells
using Lipofectamine® 2000 transfection reagent (Thermo
Fisher Scientific, Inc., Waltham, MA, USA), according to
manufacturer's protocol: 1×105 HeLa cells were seeded in
each 12-well plate and then cultured in DMEM (Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany) containing 100 ml/l fetal bovine serum
(Gibco, Thermo Fisher Scientific, Inc.) and 50 ml/l CO2
at 37°C. 24 h following transfection, the cells were collected.
Total RNA was extracted using the TriPure Isolation reagent
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) and analyzed by
RT-PCR (2XTSINGKE Master Mix, TSINGKE Biological Technology,
Beijing, China). The resulting cDNA was amplified by PCR (30
cycles) using the following primers: APC forward,
5′-ATTATTTCGCTCAGCAAGATAAG-3′ and reverse,
5′-TTCCATCTGTAGATGTACCTTTGC-3′. GAPDH was selected as reference and
the corresponding premiers were: GAPDH forward,
5′-TGAAGGTCGGAGTCAACGG-3′ and reverse, 5′-TCCTGGAAGATGGTGATGGG-3′.
The PCR thermocycling conditions included an initial denaturation
step at 95°C for 5 min, followed by 30 cycles of 95°C for 30 sec,
59°C for 30 sec and 72°C for 30 sec, and a final 5 min extension
step at 72°C. PCR products were separated by electrophoresis on
2.5% agarose gels containing ethidium bromide, and visualized by
exposure to ultraviolet light. The experiments were repeated 3
times.
Results
Micromutations of APC and MUTYH
Following DNA sequencing of APC and MUTYH,
micromutations were analyzed using Mutation Surveyor (Table I). For APC, four disease-associated
pathogenic alterations (Lys1061LysfsTer2, Glu1309AspfsTer4,
Arg283Ter and Ser1196Ter) were identified, which had been
previously reported to be pathogenic mutations (31–33,13).
In addition to the pathogenic mutations mentioned above, seven
missense mutations (Ser784Thr, Ile880Leu, Arg1171Gly, Ile1259Leu,
Asp1519Glu, Glu1552Val and Pro2216Thr), five synonymous
substitutions (Tyr486Tyr, Tyr1493Tyr, Gly1678Gly, Ser1756Ser and
Pro1960Pro) and three intronic mutations (c.220+35T>A,
c.645+129A>C and c.1548+133C>A) were identified.
 | Table I.Variants identified in the patient
cohort by Mutation Surveyor. |
Table I.
Variants identified in the patient
cohort by Mutation Surveyor.
| A, APC |
|---|
|
|---|
| Variant | Functional
class |
|---|
| c.3183_3187delACAAA
(p.Lys1061LysfsTer2) | Frameshift
mutations |
| c.3927_3931delAAAGA
(p.Glu1309AspfsX4) | Frameshift
mutations |
| c.847C>T
(p.Arg283Ter) | Nonsense
mutation |
| c.3587C>A
(p.Ser1196Ter) | Nonsense
mutation |
| c.2350T>A
(p.Ser784Thr) | Missense
mutation |
| c.2638A>C
(p.Ile880Leu) | Missense
mutation |
| c.3511C>G
(p.Arg1171Gly) | Missense
mutation |
| c.3775A>C
(p.Ile1259Leu) | Missense
mutation |
| c.4557T>A
(p.Asp1519Glu) | Missense
mutation |
| c.4655A>T
(p.Glu1552Val) | Missense
mutation |
| c.6646C>A
(p.Pro2216Thr) | Missense
mutation |
| c.220+35T>A | Intronic
mutation |
|
c.645+129A>C | Intronic
mutation |
|
c.1548+133C>A | Intronic
mutation |
| c.1458T>C
(p.Tyr486Tyr) | Synonymous
mutation |
| c.4425G>A
(p.Tyr1493Tyr) | Synonymous
mutation |
| c.5034 G>A
(p.Gly1678Gly) | Synonymous
mutation |
| c.5268T>G
(p.Ser1756Ser) | Synonymous
mutation |
| c. 5880G>A
(p.Pro1960Pro) | Synonymous
mutation |
|
| B,
MUTYH |
|
| Variant | Functional
class |
|
| c.456T>A
(p.Tyr152Ter) | Nonsense
mutation |
| c.1564T>G
(p.Ter522Gly) | Nonstop
mutation |
| c.553G>C
(p.Glu185Gln) | Missense
mutation |
| c.1232A>T
(p.Gln411Leu) | Missense
mutation |
| c.1268G>A
(p.Arg423His) | Missense
mutation |
| c.1483C>G
(p.Gln495Glu) | Missense
mutation |
| c.264+11G>A | Intronic
mutation |
| c.304+23G>A | Intronic
mutation |
| c.420+35A>G | Intronic
mutation |
|
c.1566+33_1566+35het_delTGT | Intronic
mutation |
| c.423G>A
(p.Glu141Glu) | Synonymous
mutation |
| c.450C>A
(p.Glu150Glu) | Synonymous
mutation |
| c.1347G>C
(p.Tyr449Tyr) | Synonymous
mutation |
For MUTYH, one novel nonsense mutation (Tyr152Ter)
and one nonstop mutation (Ter522Gly) were identified as pathogenic
mutations and were predicted to cause premature protein truncation
and the continued translation of an mRNA, respectively. In
addition, four missense mutations (Glu185Gln, Gln411Leu, Arg423His
and Gln495Glu), three synonymous substitutions (Glu141Glu,
Glu150Glu and Tyr449Tyr) and four intronic mutations
(c.264+11G>A, c.304+23G>A, c.420+35A>G and
c.1566+33_1566+35het_delTGT) were identified.
Computational prediction of missense
substitutions and ‘silent’ mutations
Computational prediction suggested that no missense
substitutions in APC or MUTYH in the cohort of the present study
were disease-associated variants (Table II). The synonymous and intronic
substitutions of APC and MUTYH were further analyzed, and the
in-silico analysis results indicated that the synonymous
substitutions (Tyr486Tyr, Tyr1493Tyr, Gly1678Gly, Ser1756Ser and
Pro1960Pro) of APC may have affected splicing regulation by
creating or removing ESEs or exon splicing silencers. Through
bioinformatics prediction, the synonymous mutations Tyr1493Tyr,
Gly1678Gly, Ser1756Ser and Pro1960Pro were predicted to cause a
frame shift in APC, and the synonymous mutation Tyr486Tyr
(APC∆486s) was predicted to induce exon skipping in APC
(Table III). No intron
substitutions in APC or MUTYH were predicted to be
disease-associated polymorphisms.
| Table II.Computational predictions of
deleterious missense mutations. |
Table II.
Computational predictions of
deleterious missense mutations.
| A, APC |
|---|
|
|---|
| Variant | Functional
class | PolyPhen-2 | SIFT | SNPs&GO |
|---|
|
| c.2350T>A
(p.Ser784Thr) | Missense
mutation | BENIGN (0.017) | Tolerated
(0.41) | Neutral (RI:
5) |
| c.2638A>C
(p.Ile880Leu) | Missense
mutation | BENIGN (0.376) | Tolerated
(0.92) | Disease (RI:
6) |
| c.3511C>G
(p.Arg1171Gly) | Missense
mutation | BENIGN (0.000) | Tolerated
(0.11) | Disease (RI:
1) |
| c.3775A>C
(p.Ile1259Leu) | Missense
mutation | BENIGN (0.008) | Tolerated
(0.58) | Disease (RI:
7) |
| c.4557T>A
(p.Asp1519Glu) | Missense
mutation | N/A | Tolerated
(0.16) | Disease (RI:
6) |
| c.4655A>T
(p.Glu1552Val) | Missense
mutation | BENIGN (0.104) | Tolerated
(0.24) | Neutral (RI:
1) |
| c.6646C>A
(p.Pro2216Thr) | Missense
mutation | BENIGN (0.000) | Tolerated
(0.12) | Neutral (RI:
0) |
| B,
MUTYH |
|
| Variant | Functional
class |
PolyPhen-2 | SIFT |
SNPs&GO |
|
| c.553G>C
(p.Glu185Gln) | Missense
mutation | BENIGN (0.020) | Tolerated
(0.08) | Neutral (RI:
8) |
| c.1232A>T
(p.Gln411Leu) | Missense
mutation | BENIGN (0.000) | Tolerated
(0.10) | Neutral (RI:
9) |
| c.1268G>A
(p.Arg423His) | Missense
mutation | BENIGN (0.001) | Tolerated
(0.12) | Neutral (RI:
4) |
| c.1483C>G
(p.Gln495Glu) | Missense
mutation | BENIGN (0.146) | Tolerated
(1.00) | Neutral (RI:
9) |
| Table III.Computational predictions of the
effect on splicing of variants in each gene. |
Table III.
Computational predictions of the
effect on splicing of variants in each gene.
| A, APC |
|---|
|
|---|
| Variant |
Functionalclass | ESE Finder | ESR Search | PESX | RESCUE_ESE | Splicingeffect |
|---|
|
c.1458T>C(Tyr486Tyr) |
Synonymousmutation | Changed | Changed | Changed | Notchanged | Exonskipping |
|
c.4425G>A(Tyr1493Tyr) |
Synonymousmutation | Changed | Changed | Changed | Notchanged | Frameshifting |
|
c.5034G>A(Gly1678Gly) |
Synonymousmutation | Changed | Changed | Changed | Changed | Frameshifting |
|
c.5268T>G(Ser1756Ser) |
Synonymousmutation | Changed | Changed | Changed | Changed | Frameshifting |
|
c.5880G>A(Pro1960Pro) |
Synonymousmutation | Changed | Changed | Changed | Changed | Frameshifting |
| c.220+35T>A | Intronmutation | N/A | N/A | N/A | N/A | Noeffect |
|
c.645+129A>C | Intronmutation | N/A | N/A | N/A | N/A | Noeffect |
|
c.1548+133C>A | Intronmutation | N/A | N/A | N/A | N/A | Noeffect |
|
c.8532+99T>A | Intronmutation | N/A | N/A | N/A | N/A | Noeffect |
|
| B,
MUTYH |
|
| Variant |
Functionalclass | ESE
Finder | ESR
Search | PESX |
RESCUE_ESE |
Splicingeffect |
|
|
c.423G>A(Glu141Glu) |
Synonymousmutation | N/A | N/A | N/A | N/A | Noeffect |
|
c.450C>A(Glu150Glu) |
Synonymousmutation | N/A | N/A | N/A | N/A | Noeffect |
|
c.1347G>C(Tyr449Tyr) |
Synonymousmutation | N/A | N/A | N/A | N/A | Noeffect |
| c.264+11G>A | Intronmutation | N/A | N/A | N/A | N/A | Noeffect |
| c.304+23G>A | Intronmutation | N/A | N/A | N/A | N/A | Noeffect |
| c.420+35A>G | Intronmutation | N/A | N/A | N/A | N/A | Noeffect |
|
c.1566+33_1566+35het_delTGT | Intronmutation | N/A | N/A | N/A | N/A | Noeffect |
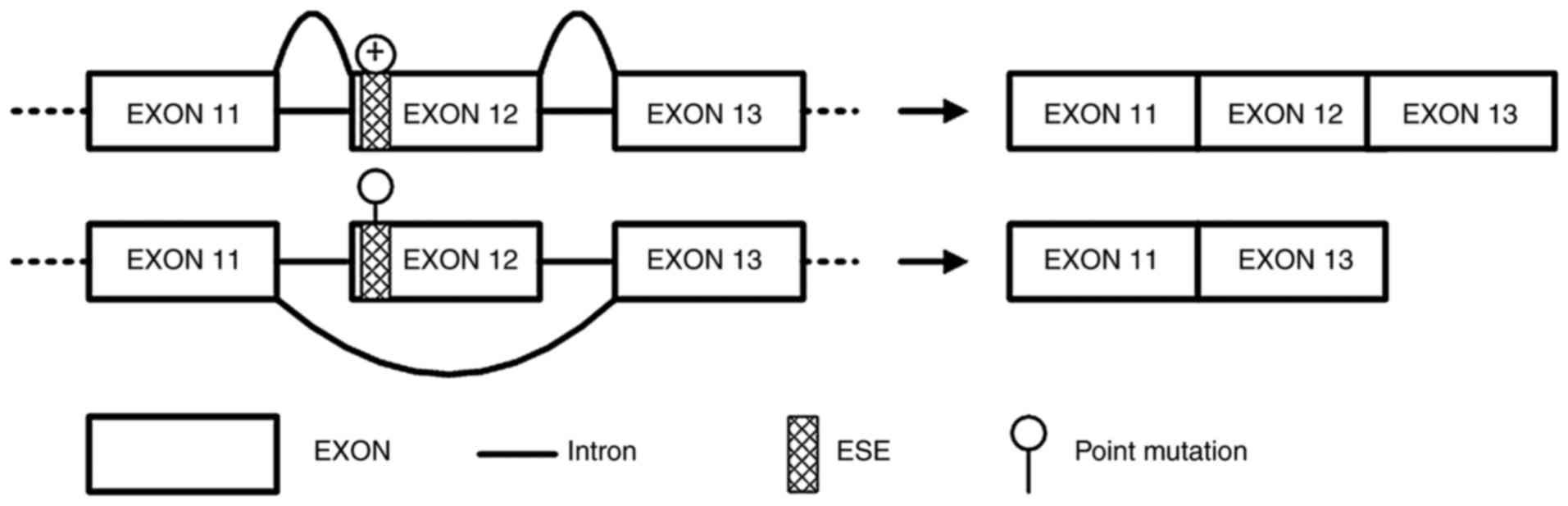
Synonymous variant APC∆486s
induces APC exon 12 skipping in a mini-gene splicing assay
Due to the splicing impact predicted by
bioinformatics analysis, APC∆486s was further studied to
verify its exon splicing impact on the APC gene. Mini-gene systems
encoding wild-type and mutant APC∆486s in APC exon 12
were synthesized and inserted into the pEGFP-N1 plasmid. The
recombined constructs were subsequently transfected into HeLa
cells, and the corresponding PCR products were separated by
electrophoresis. The results revealed that the PCR product from the
mutant mini-gene was smaller than that from the wild-type mini-gene
(Fig. 3). Sequencing results of
the two PCR products further confirmed the skipping of exon 12 in
APC (data not shown). These results indicated that the mini-gene
system encoding mutant APC∆486s induced the skipping of
exon 12 in APC.
Discussion
FAP has been linked to germline mutations in the APC
gene. However, a number of patients with FAP have no APC mutations
(1–3). Among the known genes, evidence has
also implicated MUTYH mutations in the pathogenesis of FAP, which
may explain up to 25% of all APC mutation-negative FAP cases
(7–10). However, a large subset of families
with FAP have undetectable pathogenic alterations in APC and MUTYH,
and this condition is designated as APC/MUTYH mutation-negative
FAP.
This apparent mutation negativity may suggest that
APC has alterations that escape detection by routine techniques,
and that other known or unknown genes are involved in FAP
predisposition (8). Genetic
counselling for these cases of FAP without an identified pathogenic
alteration is frequently limited by a lack of knowledge about the
pathogenic role of a large fraction of germline sequence variations
(34).
In the present study, in order to investigate the
germline mutations in Chinese patients with FAP, 13 patients with a
classical clinical phenotype were examined by exon-specific DNA
sequencing of APC and MUTYH to determine their micromutation types.
Following the use of conventional analysis methods, only six types
of clearly pathogenic mutations were identified in six individuals.
Among the APC/MUTYH mutation-negative cases, seven missense
mutations, five synonymous substitutions and three intron mutations
were identified in APC, and four missense mutations, three
synonymous substitutions and four intron mutations were identified
in MUTYH.
Previous reports have indicated that certain
missense mutations are deleterious (35). However, examining candidate
missense mutations that may cause pathogenic alterations and
identifying their relative effects on function is a time-consuming
and labor-wasting process. Recently, computational prediction has
been proposed as an efficient and economical strategy to screen for
potential pathogenic missense mutations (36). By applying bioinformatics tools and
databases in the present study, it was predicted that no identified
missense mutations in APC or MUTYH were deleterious.
It was previously thought that all synonymous
mutations were silent. However, further research into these
mutations has demonstrated that silent mutations may alter protein
expression, conformation and function (14). In 2007, it was demonstrated that
synonymous mutations in transporter ATP binding cassette subfamily
B member 1 may be implicated in drug resistance to chemotherapeutic
agents, and it was further confirmed that synonymous SNPs (sSNPs)
affect protein conformation and have functional and clinical
consequences (37). Since then, a
number of other synonymous mutations associated with human disease
have been reported. For example, polymorphisms rs709816 and
rs1061302 in gene NBN may be linked to smoking-associated cancer
(38), and polymorphism rs11615 in
ERCC1 aids in clinical outcome prediction following
oxaliplatin-based chemotherapy in metastatic colorectal cancer
(39). In addition, sSNPs
rs2229069 and rs2227985 of the gene ABL1in the fusion protein BCR,
RhoGEF and GTPase activating protein/ABL proto-oncogene 1,
non-receptor tyrosine kinase, may contribute to primary, although
not secondary, resistance to tyrosine kinase inhibitors (40). Furthermore, rs2293347 in gene EGFR
may be a potential predictor of clinical outcome in patients with
advanced non-small cell lung carcinoma when treated with gefitinib
(41), and rs1045642 in gene ABCB1
has been associated with multidrug resistance to cancer
chemotherapy (42).
Based on the above findings, the identified
synonymous and intronic substitutions of APC and MUTYH were further
analyzed by applying bioinformatics tools and databases, and the
synonymous mutation Tyr486Tyr (APC∆486s) was predicted
to have induced exon skipping in APC, which suggested that it may
have caused pathogenic alterations that lead to FAP predisposition.
Therefore, the synonymous substitution c.1458T>C was further
studied to investigate its effect on exon splicing of the APC gene.
A mini-gene system encoding wild-type or mutant c.1458T>C APC in
exon 12 was synthesized and inserted into pEGFP-N1 plasmids. The
synonymous substitution of APC∆486s was confirmed to
induce a major splicing defect with skipping of exon 12 in APC.
In the present study, DNA sequencing of the APC and
MUTYH genes in 13 patients with FAP was conducted. A total of four
known pathogenic mutations in APC and two novel disease-associated
pathogenic nonsense mutations in MUTYH were identified. For samples
that did not present with obvious pathogenic alterations, the
functional effects of the identified missense, synonymous and
intronic mutations were analyzed with bioinformatics tools and
databases. Bioinformatics analyses predicted that the synonymous
mutation APC∆486s was likely a disease-causing
polymorphism and that it may have induced exon skipping in APC.
Using a hybrid mini-gene assay, synonymous SNP APC∆486s
was demonstrated to induce a major splicing defect with the
skipping of exon 12 in APC (Fig.
4).
Therefore, it was concluded that the synonymous
mutation APC∆486s that affected exon splicing in APC was
a pathogenic alteration that predisposed patients to APC/MUTYH
mutation-negative FAP. However, a limitation to the present study
was that the exon skipping effect of synonymous mutation of
APC∆486s was predicted by bioinformatics analysis. This
was the main focus of the present study; therefore, further
ESE-dependent experiments and protein truncation tests are required
to investigate the mechanism of exon skipping and to verify that
this occurs in patients with FAP presenting with
APC∆486s.
Acknowledgements
Not applicable.
Funding
This work was supported by grants from the National
Natural Science Foundation of China (grant nos. 81660472 and
81760511), the Scientific Research Foundation of Yunnan Province
(grant no. 2015Y177), Yunnan Applied Basic Research Projects-Union
Foundation (grant no. 2015FB035), the Scientific Research Project
of Internal Research Institute in Yunnan Province (grant no.
2016NS009) and the Medical Candidate of Yunnan Province (grant no.
H-201608).
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
WQL, JY and JD conceived the present study. WQL, JY
and YP carried out the bioinformatics prediction work and analyzed
the data. WQL, JY, WLL and JD performed the experiments of hybrid
mini-gene assay. All authors discussed the results and contributed
to the final manuscript.
Ethics approval and consent to
participate
The present study was approved by and performed in
accordance with the Research Ethics Board of Kunming Medical
University (Kunming, China). All participants provided written
informed consent, and the ethics committees approved the consent
procedure.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Gardner EJ, Burt RW and Freston JW:
Gastrointestinal polyposis: Syndromesand genetic mechanisms. West J
Med. 132:488–499. 1980.PubMed/NCBI
|
|
2
|
Kanth P, Grimmett J, Champine M, Burt R
and Samadder NJ: Hereditary colorectal polyposis and cancer
syndromes: A primer on diagnosis and management. Am J
Gastroenterol. 112:1509–1525. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kinzler KW, Nilbert MC, Vogelstein B,
Bryan TM, Levy DB, Smith KJ, Preisinger AC, Hamilton SR, Hedge P,
Markham A, et al: Identification of a gene located at chromosome
5q21 that is mutated in colorectal cancers. Science. 251:1366–1370.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Groden J, Thliveris A, Samowitz W, Carlson
M, Gelbert L, Albertsen H, Joslyn G, Stevens J, Spirio L, Robertson
M, et al: Identification and characterization of the familial
adenomatous polyposis coligene. Cell. 66:589–600. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Syngal S, Brand RE, Church JM, Giardiello
FM, Hampel HL and Burt RW; American College of Gastroenterology, :
ACG clinical guideline: Genetic testing and management of
hereditary gastrointestinal cancer syndromes. Am J Gastroenterol.
110:223–262. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Boman BM and Fields JZ: An APC: WNT
counter-current-like mechanism regulates cell division along the
human colonic crypt Axis: A mechanism that explains how APC
mutations induce proliferative abnormalities that drive colon
cancer development. Front Oncol. 3:2442013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Al-Tassan N, Chmiel NH, Maynard J, Fleming
N, Livingston AL, Williams GT, Hodges AK, Davies DR, David SS,
Sampson JR and Cheadle JP: Inherited variants of MYH associated
with somatic G:C->T: A mutations in colorectal tumors. Nat
Genet. 30:227–232. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Renkonen ET, Nieminen P, Abdel-Rahman WM,
Moisio AL, Järvelä I, Arte S, Järvinen HJ and Peltomäki P:
Adenomatous polyposis families that screen APC mutation-negative by
conventional methods are genetically heterogeneous. J Clin Oncol.
23:5651–5659. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sieber OM, Lipton L, Crabtree M, Heinimann
K, Fidalgo P, Phillips RK, Bisgaard ML, Orntoft TF, Aaltonen LA,
Hodgson SV, et al: Multiple colorectal adenomas, classic
adenomatous polyposis, and germ-line mutations in MYH. N Engl J
Med. 348:791–799. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ricci MT, Salemme M, Villanacci V and
Varesco L: The genetics of inherited predispositions to colorectal
polyps: A quick guide for clinicians. Colorectal Dis. 17 Suppl
1:S3–S9. 2015. View Article : Google Scholar
|
|
11
|
Wei SC, Su YN, Tsai-Wu JJ, Wu CH, Huang
YL, Sheu JC, Wang CY and Wong JM: Genetic analysis of the APC gene
in Taiwanese familial adenomatous polyposis. J Biomed Sci.
11:260–265. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Prior TW and Bridgeman SJ: Identifying
mutations for MYH-associated polyposis. Curr Protoc Hum Genet
Chapter. 10:Unit 10.13. 2010. View Article : Google Scholar
|
|
13
|
Yang J, Liu WQ, Li WL, Chen C, Zhu Z, Hong
M, Wang ZQ and Dong J: Investigating polymorphisms by
bioinformatics is a potential cost-effective method to screen for
germline mutations in Chinese familial adenomatous polyposis
patients. Oncol Lett. 12:421–428. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sauna ZE and Kimchi-Sarfaty C:
Understanding the contribution of synonymous mutations to human
disease. Nat Rev Genet. 12:683–691. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Plotkin JB and Kudla G: Synonymous but not
the same: The causes and consequences of codon bias. Nat Rev Genet.
12:32–42. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Supek F, Miñana B, Valcárcel J, Gabaldón T
and Lehner B: Synonymous mutations frequently act as driver
mutations in human cancers. Cell. 156:1324–1335. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chiang JM, Chen HW, Tang RP, Chen JS,
Changchien CR, Hsieh PS and Wang JY: Mutation analysis of the APC
gene in Taiwanese FAP families: Low incidence of APC germline
mutation in a distinct subgroup of FAP families. Fam Cancer.
9:117–124. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Minton JA, Flanagan SE and Ellard S:
Mutation surveyor: Software for DNA sequence analysis. Methods Mol
Biol. 688:143–153. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Stenson PD, Mort M, Ball EV, Shaw K,
Phillips A and Cooper DN: The human gene mutation database:
Building a comprehensive mutation repository for clinical and
molecular genetics, diagnostic testing and personalized genomic
medicine. Hum Genet. 133:1–9. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Thorisson GA, Smith AV, Krishnan L and
Stein LD: The International HapMap project web site. Genome Res.
15:1592–1593. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sherry ST, Ward MH, Kholodov M, Baker J,
Phan L, Smigielski EM and Sirotkin K: dbSNP: The NCBI database of
genetic variation. Nucleic Acids Res. 29:308–311. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zerbino DR, Achuthan P, Akanni W, Amode
MR, Barrell D, Bhai J, Billis K, Cummins C, Gall A, Girón CG, et
al: Ensembl 2018. Nucleic Acids Res. 46:(Database issue).
D754–D761. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pan M, Cong P, Wang Y, Lin C, Yuan Y, Dong
J, Banerjee S, Zhang T, Chen Y, Zhang T, et al: Novel LOVD
databases for hereditary breast cancer and colorectal cancer genes
in the Chinese population. Hum Mutat. 32:1335–1340. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ramensky V, Bork P and Sunyaev S: Human
non-synonymous SNPs: Server and survey. Nucleic Acids Res.
30:3894–900. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ng PC and Henikoff S: Predicting
deleterious amino acid substitutions. Genome Res. 11:863–874. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Capriotti E, Calabrese R, Fariselli P,
Martelli PL, Altman RB and Casadio R: WS-SNPs&GO: A web server
for predicting the deleterious effect of human protein variants
using functional annotation. BMC Genomics. 14 Suppl 3:S62013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cartegni L, Wang J, Zhu Z, Zhang MQ and
Krainer AR: ESEfinder: A web resource to identify exonic splicing
enhancers. Nucleic Acids Res. 31:3568–3571. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yeo G and Burge CB: Variation in sequence
and organization of splicing regulatory elements in vertebrate
genes. In the Proceeding of Proc. Natl Acad Sci. 101:15700–15705.
2004. View Article : Google Scholar
|
|
29
|
Fairbrother WG, Yeh RF, Sharp PA and Burge
CB: Predictive identification of exonic splicing enhancers in human
genes. Science. 297:1007–1013. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cooper TA: Use of minigene systems to
dissect alternative splicing elements. Methods. 37:331–340. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gómez-Fernández N, Castellví-Bel S,
Fernández-Rozadilla C, Balaguer F, Muñoz J, Madrigal I, Milà M,
Graña B, Vega A, Castells A, et al: Molecular analysis of the APC
and MUTYH genes in Galician and Catalonian FAP families: A
different spectrum of mutations? BMC Med Genet. 10:572009.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kashfi SM, Farahbakhsh Behboudi F,
Golmohammadi M, Mojarad Nazemalhosseini E, Azimzadeh P and Aghdaie
Asadzadeh H: Frameshift mutations (deletion at codon 1309 and codon
849) in the APC gene in iranian FAP patients: A c series and review
of the literature. Int J Mol Cell Med. 3:196–202. 2014.PubMed/NCBI
|
|
33
|
Mohamed Z, Ahmad R, Yoke NS, Zakaria Z,
Ahmad H and Yew TH: A nonsense mutation in exon 8 of the APC gene
(Arg283Ter) causes clinically variable FAP in a Malaysian Chinese
family. Cancer Sci. 94:725–728. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yang J, Liu QW, Li LW, Wang QZ, Hong M and
Dong J: Familial adenomatous polyposis in China. Oncol Lett.
12:4877–4882. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Menéndez M, González S, Obrador-Hevia A,
Domínguez A, Pujol MJ, Valls J, Canela N, Blanco I, Torres A,
Pineda-Lucena A, et al: Functional characterization of the novel
APC N1026S variant associated with attenuated familial adenomatous
polyposis. Gastroenterology. 134:56–64. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sucularli C and Arslantas M: Computational
prediction and analysis of deleterious cancer associated missense
mutations in DYNC1H1. Mol Cell Probes. 34:21–29. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kimchi-Sarfaty C, Oh JM, Kim IW, Sauna ZE,
Calcagno AM, Ambudkar SV and Gottesman MM: A ‘silent’ polymorphism
in the MDR1 gene changes substrate specificity. Science.
315:525–528. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Park SL, Bastani D, Goldstein BY, Chang
SC, Cozen W, Cai L, Cordon-Cardo C, Ding B, Greenland S, He N, et
al: Associations between NBS1 polymorphisms, haplotypes and
smoking-related cancers. Carcinogenesis. 31:1264–1271. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liang J, Jiang T, Yao RY, Liu ZM, Lv HY
and Qi WW: The combination of ERCC1 and XRCC1 gene polymorphisms
better predicts clinical outcome to oxaliplatin-based chemotherapy
in metastatic colorectal cancer. Cancer Chemother Pharmacol.
66:493–500. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ernst T, Hoffmann J, Erben P, Hanfstein B,
Leitner A, Hehlmann R, Hochhaus A and Müller MC: ABL single
nucleotide polymorphisms may masquerade as BCR-ABL mutations
associated with resistance to tyrosine kinase inhibitors in
patients with chronic myeloid leukemia. Haematologica.
93:1389–1393. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ma F, Sun T, Shi Y, Yu D, Tan W, Yang M,
Wu C, Chu D, Sun Y, Xu B and Lin D: Polymorphisms of EGFR predict
clinical outcome in advanced non-small-cell lung cancer patients
treated with Gefitinib. Lung Cancer. 66:114–119. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Fung KL and Gottesman MM: A synonymous
polymorphism in a common MDR1 (ABCB1) haplotype shapes protein
function. Biochim Biophys Acta. 1794:860–871. 2009. View Article : Google Scholar : PubMed/NCBI
|