Introduction
Pyramidal neurons in the hippocampal cornu ammonis 1
(CA1) area are killed four to five days after brief (5 to 10 min)
transient global cerebral ischemia in gerbils (1). A complex series of molecular
mechanisms of ischemia-induced neuronal degeneration/death is
related to increased glutamate excitotoxicity, oxidative stress,
and inflammation (2,3). Many researchers have been struggling
to find relevant molecular targets from those mechanisms to protect
neurons against ischemic damage for developing therapeutics. For
example, protective effects of antioxidants, superoxide dismutases
(SODs), and their mimetics, have been demonstrated (4,5).
Intermittent fasting (IF) is defined as a severe
dietary energy restriction during a certain period of normal energy
intake (6). The duration of IF is
variable, and previous studies have typically used alternate-day
fasting or a daily time-restricted (4 to12 h) food-deprivation
regimen in rodents (rats or mice) (7). It has been reported that the liver
and adipose tissue act as energy storage, allowing fasting for
various periods in mammals (8).
Additionally, during fasting, body systems, including the
metabolic, endocrine, and nervous systems, adjust to enable a high
level of physical and mental activities (8).
Beneficial effects of dietary restriction are
increased resistance to aging and degenerative diseases, and their
associated mechanisms have been demonstrated in previous studies.
IF activates the sirtuin 1 signaling pathway, which plays a major
role in life span and cellular health, and decreases apoptotic
pathways in the brain of senescence-accelerated mice p8 (SAMP8)
(9). In addition, IF-related
insulin-like signaling and FoxO transcription factors are known to
stimulate antioxidant enzymes to help cells resist stress (10).
For the brain, many researchers have studied whether
an IF regimen provides neuroprotection or not. Zhu et al
(11), have reporetd that IF
protects hippocampal neurons against kainate excitotoxicity in a
mouse model of Alzheimer's disease (presenilin1 mutant knockin
mice) by reducing oxidative stress. In ischemia, it has been
demonstrated that dietary restriction or an IF regimen reduces
infarct volume in rodent models of cerebral focal cerebral ischemia
by inhibiting the accumulation of autophagosomes in neurons
(12), by suppressing inflammasome
activity (13), and by increasing
a preconditioning stress response (14). The above-mentioned studies
attenuate or protect ischemic damage in focal cerebral ischemia
models; however, the possibility that IF protects neurons from
transient global cerebral ischemia (tGCI) has not been examined.
Therefore, in this study, we investigated effects of IF on
expressions of endogenous antioxidant enzymes, and then examined
the effect of IF on expressions of antioxidant enzymes, neuronal
damage/degeneration, and reactive glia cells following tGCI in
gerbils, which are a good animal model of tGCI (15,16).
Materials and methods
Experimental animals
Male gerbils were obtained at 6 months of age (B.W.,
70±5.2 g) from the Experimental Animal Center, Kangwon University,
Chuncheon, Gangwon, Republic of Korea, and maintained at a constant
temperature (23°C) and humidity (50%) with a 12-h light/dark cycle.
The process of handling and caring animals conformed to the
guidelines being in compliance with current international laws and
policies (NIH Guide for the Care and Use of Laboratory Animals, The
National Academies Press, 8th ed., 2011). The protocol of this
experiment was approved by the Institutional Animal Care and Use
Committee (IACUC) at Kangwon National University (approval no.
KW-180124-1).
IF and experimental groups
Animals were fed commercially available rodent
normal diet or IF (24 h fasting and 24 h feeding) was applied for 2
months according to method by published methods (9,12,17).
During procedures, food intake of IF group was controlled daily (10
g per day), and body weight of normal diet and IF groups was
monitored every week. After 2 months, animals with normal diet or
IF were randomly assigned to following groups: i) Sham groups
(n=7), which were allowed free access to water and food and
received no ischemia; ii) IF and sham (IF+Sham) group (n=7), which
was subjected to IF and received no ischemia; iii) Ischemia groups
(n=7), which received tGCI without IF and iv) IF+Ischemia groups
(n=7), which were subjected to IF and received tGCI. To investigate
effects of IF on neuronal death (loss), antioxidant enzymes, and
gliosis, all animals were sacrificed at 5 days after ischemia,
because death (loss) of pyramidal neurons in the gerbil hippocampal
CA1 region occurs 5 days follwong transient cerebral ischemia
(1).
Induction of tGCI
As previously described (18), in brief, gerbils in all groups were
anesthetized with a mixture of 2.5% isoflurane (Baxtor, Deerfield,
IL, USA) in 33% oxygen and 67% nitrous oxide. The gerbils received
a midline incision on the ventral surface of the neck, and both
common carotid arteries were occluded for 5 min using non-traumatic
aneurysm clips. We controlled normal body (rectal) temperature
(37±0.5°C) using a thermometric blanket throughout the surgery,
monitoring the temperature with a rectal temperature probe (TR-100;
Fine Science Tools, Foster City, Inc., CA, USA).
Preparation of histological
sections
For histology, as described previously (18), gerbils were anesthetized with 30
mg/kg Zoletil 50 (Virbac, Carros, France) 5 days after tGCI (at
this point in time, pyramidal neurons are dead after tGCI), and
perfused transcardially with 0.1 m phosphate buffered saline (PBS,
pH 7.4) followed by 4% paraformaldehyde in 0.1 m phosphate buffer
(PB, pH 7.4). The brain tissues containing hippocampi were
cryoprotected and serially sectioned into 30-µm coronal sections in
a cryostat (Leica Microsystems GmbH, Wetzlar, Germany).
Immunohistochemistry
In brief, according to our published method
(19), sheep anti-superoxide
dismutase 1 (SOD1) (1:1,000; EMD Millipore, Billerica, MA, USA),
sheep anti-mitochondrial (SOD2) (1:1,000; EMD Millipore), rabbit
anti-catalase (CAT) (1:500; EMD Millipore), mouse anti-glutathione
peroxidase (GPX) (1:500; EMD Millipore), mouse anti-NeuN (a marker
for neuron) (1:1,000; Thermo Fisher Scientific, Inc., Waltham, MA,
USA), mouse anti-GFAP (a marker for astrocyte) (1:800; Abcam,
Cambridge, MA, USA), and rabbit anti-Iba1 (a marker for microglia)
(1:800; Wako Pure Chemical Industries, Ltd., Osaka, Japan) were
used as primary antibodies. The sections were sequentially treated
with 0.3% hydrogen peroxide (H2O2) in PBS for
30 min and 10% normal goat serum in 0.05 M PBS for 30 min. The
treated sections were incubated with the primary antibodies
overnight at 4°C, thereafter, the reacted sections were exposed to
biotinylated goat anti-mouse or goat anti-rabbit IgG (1:200; Vector
Laboratories, Inc., Burlingame, CA, US) and streptavidin peroxidase
complex (1:200; Vector Laboratories, Inc.). Finally, the reacted
sections were visualized by staining with 3, 3′-diaminobenzidine
tetrahydrochloride in 0.1 M Tris-HCl buffer (pH 7.2).
Fluoro-Jade (F-J) B histofluorescence
staining
To investigate neuronal death in the hippocampal CA1
at 5 days after tGCI, F-J B (a fluorescent marker for cell
degeneration) histofluorescence staining was conducted according to
method published by Candelario-Jalil et al (20). In brief, the sections were first
immersed in a solution containing 1% sodium hydroxide in 80%
alcohol and followed in 70% alcohol. They were then transferred to
a solution of 0.06% potassium permanganate, and to a 0.0004% F-J B
(Histochem, Inc., Jefferson, AR, USA) staining solution. After
washing them, they were placed on a slide warmer (approximately
50°C) to be reacted. The stained sections were examined using an
epifluorescent microscope (Carl Zeiss AG, Oberkochen, Germany) with
blue (450–490 nm) excitation light and a barrier filter (Schmued
and Hopkins, 2000).
Data analysis
First, we quantitatively analyzed SOD1, SOD2, GPX,
CAT, GFAP, and Iba-1 immunoreactivities according to our published
method (19). In brief, we
selected six sections from each animal with 120-µm interval
according to AP (Antero-posterior) −1.4 to −2.2 mm of the gerbil
brain atlas and took images of them from the CA1 through an AxioM1
light microscope (Carl Zeiss AG) equipped with a digital camera
(Axiocam; Carl Zeiss AG) connected to a PC monitor. The image of
each immunoreactivity was calibrated into an array of 512×512
pixels corresponding to a tissue area of 250×250 µm2
(20× primary magnification). Each immunoreactivity was measured by
a 0–255 gray scale system and evaluated by optical density (OD),
which was obtained after transformation of the mean gray level
using the formula: OD=log (255/mean gray level). A ratio of the OD
was calibrated as % (relative OD, ROD) using Adobe Photoshop
version 8.0 and analyzed using Image J 1.46 software (National
Institutes of Health, Bethesda, MD, USA). A ratio of the ROD was
calibrated as %, with the Sham group designated as 100%.
Second, we analyzed numbers of NeuN- and F-J
B-positive cells according to our published method (19). In brief, we selected six sections
like the above-mentioned method. Images of NeuN- and F-J B-positive
cells were captured through an AxioM1 light microscope (Carl Zeiss
AG) equipped with a digital camera (Axiocam; Carl Zeiss AG)
connected to a PC monitor. CA1 pyramidal neurons were captured in a
250×250 µm square. Cell counts were obtained by averaging the total
number of NeuN- and F-J B-positive cells from each animal using an
image analyzing system (Optimas v.6.5; CyberMetrics, Scottsdale,
AZ, USA).
Statistical analysis
The data shown here represent the means ± SEM.
Differences of the means among the groups were statistically
analyzed by one-way analysis of variance with Duncan's post hoc
test using SPSS v.17.0 software (SPSS, Inc., Chicago, IL, USA).
P<0.05 was considered to indicate a statistically significant
difference.
Results
Body weight
Normal diet or IF was treated for 2 months. Body
weight in the normal diet animals was slowly increased. Change in
body weight in the IF animals was not significantly different from
that in the normal diet animals (Fig.
1).
Immunoreactivities of antioxidant
enzymes
SOD1 immunoreactivity
When we examined SOD1 immunoreactivity in the Sham
group, SOD1 immunoreactivity was mainly shown in neurons of the
stratum pyramidale in the CA1, which are called CA1 pyramidal
neurons (Fig. 2A). In the IF+Sham
group, SOD1 immunoreactivity in CA1 pyramidal neurons was not
different from that in the Sham group (Fig. 2B).
In the Ischemia group, SOD1 immunoreactivity was
hardly found in CA1 pyramidal neurons, but increased in many
non-pyramidal cells in stratum oriens and radiatum of the CA1, and
the SOD1 immunoreactivity was increased by about 43% compared to
the Sham group (Fig. 2C). In the
IF+Ischemia group, the pattern and immunoreactivity of SOD1 in the
CA1 was similar to that in the Ischemia group (Fig. 2D).
Each immunoreactivity of SOD1 in the CA1 region at 5
days after tGCI in the Sham, IF+Sham, Ischemia, and IF+Ischemia
groups was shown in Fig. 2E.
SOD2 immunoreactivity
In the Sham group, very weak SOD2 immunoreactivity
was detected in CA1 pyramidal neurons (Fig. 3A). In the IF+Sham group, SOD2
immunoreactivity in CA1 pyramidal neurons was significantly
increased by about 40% compared to the Sham group (Fig. 3B).
In the Ischemia group, SOD2 immunoreactivity was
rarely shown in CA1 pyramidal neurons, instead, SOD2
immunoreactivity was increased in non-pyramidal cells (Fig. 3C). In the IF+Ischemia group, the
pattern of SOD2 expression in the CA1 was not different from the
Ischemia group; however, SOD2 immunoreactivity in non-pyramidal
cells was increased by about 41% compared to the Ischemia group
(Fig. 3D).
Each immunoreactivity of SOD2 in the CA1 region at 5
days after tGCI in the Sham, IF+Sham, Ischemia, and IF+Ischemia
groups was shown in Fig. 3E.
CAT immunoreactivity
CAT immunoreactivity was observed in CA1 pyramidal
neurons in the Sham group (Fig.
4A). In the IF+Sham group, CAT immunoreactivity in CA1
pyramidal neurons was increased by about 102% compared to the Sham
group (Fig. 4B).
 | Figure 4.CAT immunohistochemistry.
Immunoreactivities of CAT in the CA1 of (A) Sham, (B) IF+Sham, (C)
Ischemia, and (D) IF+Ischemia groups 5 days after tGCI. In the
IF+Sham group, CAT immunoreactivity is significantly increased in
pyramidal neurons (star in B) compared to the Sham group. In the
IF+Ischemia group, CAT immunoreactivity is significantly increased
in non-pyramidal cells (arrow in D) compared to the Ischemia group.
Scale bar=50 µm. SO, stratum oriens; SP, stratum pyramidale; SR,
stratum radiatum. (E) CAT immunoreactivity as percent values in
pyramidal and non-pyramidal cells (n=7 in each group, *P<0.05
vs. Sham group, †P<0.05 vs. Ischemia group). Bars
indicate the means ± SEM. |
In the Ischemia group, CAT immunoreactivity was
hardly found in CA1 pyramidal neurons, instead, weak CAT
immunoreactivity was shown in non-pyramidal cells (Fig. 4C). In the IF+Ischemia group, CAT
immunoreactivity was strong in many non-pyramidal cells, and the
immunoreactivity was increased by about 85% compared to the
Ischemia group (Fig. 4D).
Each immunoreactivity of CAT in the CA1 region at 5
days after tGCI in the Sham, IF+Sham, Ischemia, and IF+Ischemia
groups was shown in Fig. 4E.
GPX immunoreactivity
Strong GPX immunoreactivity was found in CA1
pyramidal neurons in the Sham group (Fig. 5A). In the IF+Sham group, no
significant difference in GPX immunoreactivity in CA1 pyramidal
neurons was observed compared to the Sham group (Fig. 5B).
 | Figure 5.GPX immunohistochemistry.
Immunoreactivities of GPX in the CA1 of (A) Sham, (B) IF+Sham, (C)
Ischemia, and (D) IF+Ischemia groups 5 days after tGCI. GPX
immunoreactivity in the IF+Sham group is shown in pyramidal
neurons, and the immunoreactivity is not changed compared to the
Sham group. In the IF+Ischemia, GPX immunoreactivity is shown in
non-pyramidal cells, and the immunoreactivity is not different from
the Ischemia group. Scale bar=50 µm. SO, stratum oriens; SP,
stratum pyramidale; SR, stratum radiatum. (E) GPX immunoreactivity
as percent values in pyramidal and non-pyramidal cells (n=7 in each
group, *P<0.05 vs. Sham group). Bars indicate the means ±
SEM. |
In the Ischemia group, GPX immunoreactivity in CA1
pyramidal neurons was hardly identified, instead, strong GPX
immunoreactivity was observed in many non-pyramidal cells (Fig. 5C). In the IF+Ischemia group, GPX
immunoreactivity in non-pyramidal cells was not different from that
in the Ischemia group (Fig.
5D).
Each immunoreactivity of GPX in the CA1 region at 5
days after tGCI in the Sham, IF+Sham, Ischemia, and IF+Ischemia
groups was shown in Fig. 5E.
Neuroprotection
NeuN-immunoreactive neurons
In the Sham group, NeuN-immunoreactive neurons, as
pyramidal neurons, were predominantly distributed in the stratum
pyramidale (Fig. 6A). In the
IF+Sham group, NeuN-immunoreactive pyramidal neurons were not
different in their distribution compared with the Sham group
(Fig. 6B).
 | Figure 6.NeuN immunohistochemistry. NeuN
immunoreactive neurons in the CA1 of (A) Sham, (B) IF+Sham, (C)
Ischemia, and (D) IF+Ischemia groups 5 days after tGCI. A few NeuN
immunoreactive pyramidal neurons are shown in the stratum
pyramidale (SP, star in C) in the Ischemia group. In the
IF+Ischemia group, numbers of NeuN-immunoreactive pyramidal neurons
(star in D) are similar to the Ischemia group. The scale bar
represents 400 µm for the top row and 50 µm for the bottom row. (E)
Number of NeuN-immunoreactive neurons per 250×250 µm2 in
the CA1 (n=7 in each group, *P<0.05 vs. Sham group). Bars
indicate the means ± SEM. CA, cornu ammonis; DG, dentate gyrus; SO,
stratum oriens; SR, stratum radiatum. |
In the Ischemia group, numbers of
NeuN-immunoreactive neurons was significantly decreased only in the
stratum pyramidale of the CA1 (Fig.
6C). In the IF+Ischemia group, the distribution and numbers of
NeuN-immunoreactive neurons were similar to the Ischemia group
(Fig. 6D and E).
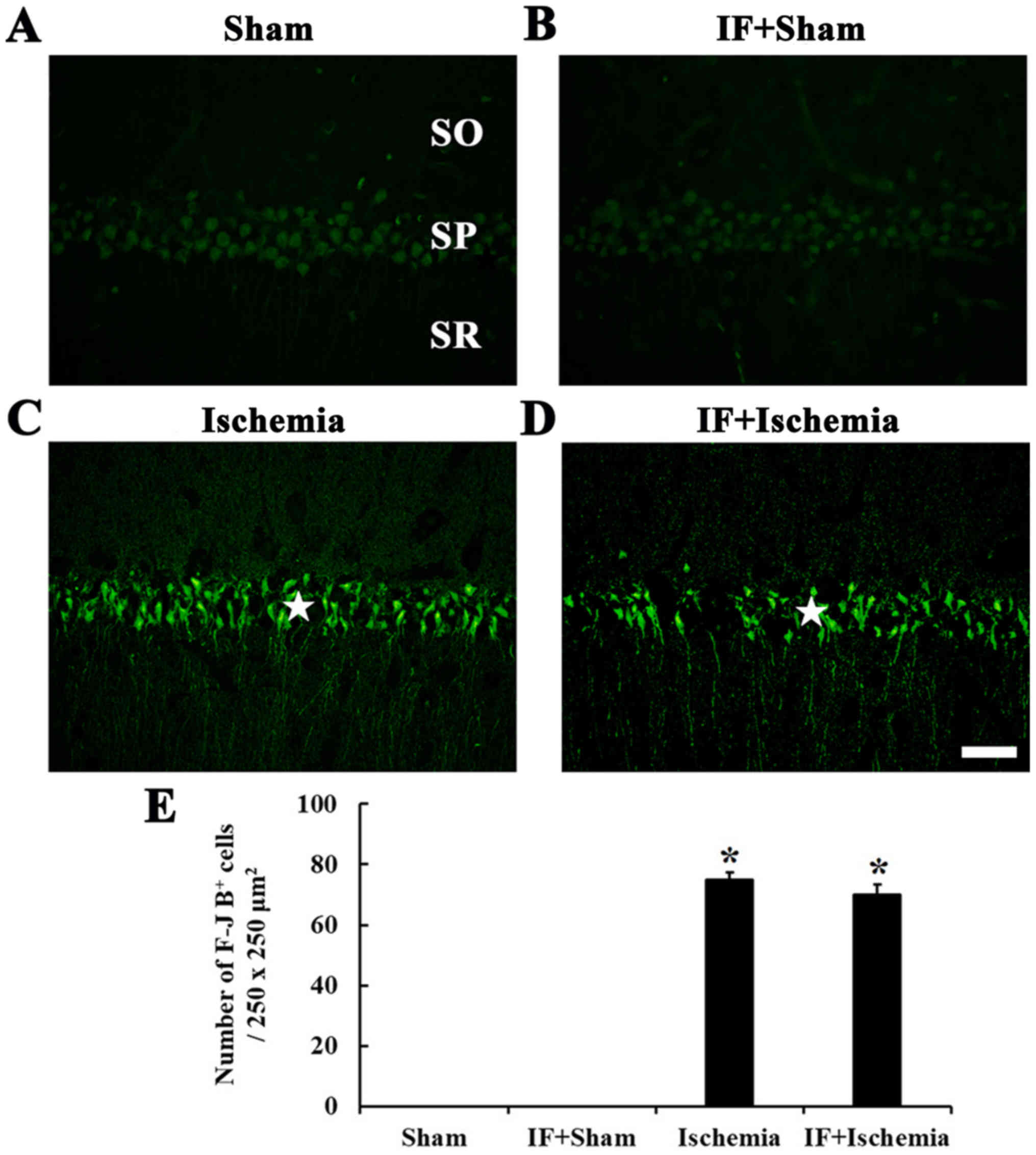
F-J B-positive cells
F-J B-positive cells, which are dead/degenerated
cells, were not detected in the stratum pyramidale of the CA1 in
the Sham group (Fig. 7A). In the
IF+Sham group, F-J B-positive cells were not shown like the Sham
group (Fig. 7B).
In the Ischemia group, many F-J B positive cells
were shown in the stratum pyramidale of the CA1 (Fig. 7C). In the IF+Ischemia group, the
distribution and numbers of F-J B-positive cells were similar to
the Ischemia group (Fig. 7D and
E).
Glial activation and Inflammation
GFAP immunoreactivity
In the Sham group, GFAP immunoreactive cells, which
were astrocytes, had small cytoplasm and fine processes, and
scattered throughout in all layers (Fig. 8A). In the IF+Sham group, the
morphology and immunoreactivity of GFAP was similar to that in the
Sham group (Fig. 8B).
In the Ischemia group, GFAP-immunoreactive cells
displayed thick processes, and GFAP immunoreactivity was increased
by about 279% compared to the Sham group (Fig. 8C). In the IF+Ischemia group, the
morphology of GFAP-immunoreactive cells and GFAP immunoreactivity
was similar to those in the Ischemia group (Fig. 8D).
Each immunoreactivity of GFAP in the CA1 region at 5
days after tGCI in the Sham, IF+Sham, Ischemia, and IF+Ischemia
groups was shown in Fig. 8E.
Iba-1 immunoreactivity
In the Sham group, Iba-1-immunoreactive cells, which
were microglia, had small cell body and distributed in all layers
(Fig. 9A). In the IF+Sham group,
the morphology and immunoreactivity of Iba-1-immunoreactive cells
was not different from the Sham group (Fig. 9B).
In the Ischemia group, Iba-1-immunoreactive cells
showed activated form with hypertrophied cell bodies and branched
processes, and many of them were aggregated into and near the
stratum pyramidale, showing that Iba-1 immunoreactivity was by
about 226% compared to the Sham group (Fig. 9C). In the IF+Ischemia group, the
distribution of Iba-1-immunoreactive cells and their Iba-1
immunoreactivity was not different from the Ischemia group
(Fig. 9D).
Each immunoreactivity of Iba-1 in the CA1 region at
5 days after tGCI in the Sham, IF+Sham, Ischemia, and IF+Ischemia
groups was shown in Fig. 9E.
Discussion
This study was examined effects of IF on endogenous
antioxidant enzymes, SOD1, SOD2, GPX and CAT, in the hippocampal
CA1 of the gerbil, and investigated effects of IF on tGCI-induced
antioxidant enzymes, neuronal damage/degeneration, and reactive
glia cells.
We found that weight gain in the IF-sham group for
two months was similar to that of the Sham group. In line with our
results, it was reported that C57BL6 mice lost little or no weight,
although other mice or rats lost weight after IF (21). These results indicate that weight
loss after IF is different according to kinds or species of
experimental animals. We need to study exact changes in weight gain
or loss induced by IF in various kinds of experimental animals and
its causes and mechanisms.
It is well known that SODs convert superoxide to
hydrogen peroxide (H2O2), and that
H2O2 is converted to H2O by
scavenger enzymes, such as CAT and GPX, to detoxify harmful
radicals and reactive oxidative stress (22). It has been reported that SOD2
(mitochondrial enzyme) is a more important enzyme, since SOD2
knockout mice die earlier after birth or suffer from severe
neurodegeneration (23), but SOD1
(cytosolic enzyme) knockout mice, which are phenotypically normal
with only reproductive problems, can survive (24). Furthermore, it has been
demonstrated that CAT removes peroxides more effectively than does
GPX in neurons (22,25). In a previous study, alternate-day
fasting for four or five months has shown increased levels of heme
oxygenase (HO)-1, an antioxidant enzyme in the mouse brain
(26). Similar to the previous
study, our current study showed that IF for two months
significantly increased SOD2 and CAT immunoreactivities, but not
SOD1 and GPX immunoreactivities in CA1 pyramidal neurons. These
findings indicate that IF could induce increases of basal
antioxidant expressions, especially SOD2 and CAT, in hippocampal
CA1 pyramidal neurons in gerbils, which have been used for an
animal model of tGCI (15,16).
We have demonstrated that neuronal protection or
improvement of neuronal survival (after drug treatment) is closely
related to maintenance or increase of SOD1, SOD2, CAT, and GPX
expressions in the gerbil hippocampus after 5 min of tGCI (19,27,28).
In addition, Walsh et al (2014) extensively reviewed
previous studies and summarized that IF was effective in increasing
antioxidant enzymes, in particular, glutathione activity (29). Furthermore, Arumugam et al
(2010) showed that IF increased the HO-1 level in vulnerable brain
regions, and the increased HO-1 level was correlated with
decreasing infarct volume and neurological deficit following focal
cerebral ischemic stroke in mice (26). Recently, Hu et al (2017)
reported that postoperative IF for a week after chronic cerebral
hypoperfusion in rats significantly decreased malondialdehyde (MDA)
activity, maintained glutathione, SOD1, and SOD2 levels in the
hippocampus, and improved memory deficit induced by the
hypoperfusion (30). In our
present study, we found that SOD2 and CAT immunoreactivities were
hardly shown in CA pyramidal cells in the IF+Ischemia group;
instead, the immunoreactivities were increased in non-pyramidal
cells, which were found to be astrocytes (31,32),
compared to the Ischemia group. Nevertheless, there was no
IF-mediated neuronal protection in the hippocampal CA1 following
tGCI. Our present study indicates that IF could increase SOD2 and
CAT expressions in CA1 pyramidal cells in the IF+Sham groups and in
non-pyramidal cells in the IF+Ischemia group but does not protect
neurons from ischemic injury in a gerbil model of tGCI, which is
different from transient focal cerebral ischemia.
Activations of microglia and astrocytes in the acute
phase of post-ischemia are increased in response to
ischemia-induced neuronal damage in the gerbil hippocampal CA1
(18,33). The activations (gliosis) are one of
the main reasons for the secondary damage that increases cytokine
production during neuronal degeneration after ischemia (34,35).
Our previous studies have shown that the attenuation of glial
activation is strongly correlated with the protection of
hippocampal CA1 neurons from tGCI (36,37).
It was reported that three months of IF could decrease
seizure-induced microgliosis in the lesioned hippocampus (38). It was reported that postoperative
IF for a week after chronic cerebral hypoperfusion in rats
significantly attenuated microglial activation in the hippocampus
induced by chronic cerebral hypoperfusion (30). Although postoperative IF reduces
injury-induced microglial activation in the hippocampus, as shown
in our present study, preoperative IF prior to tGCI does not
inhibit activations of microglia and astrocytes in the hippocampus
induced by tGCI.
In summary, our results showed that preoperative IF
increased immunoreactivities of SOD2 and CAT in CA1 pyramidal cells
before tGCI and in non-pyramidal cells after tGCI. However, the IF
did not protect death of CA1 pyramidal neurons following tGCI in
gerbils. In this regard, we need to study the causes of the failure
in protecting CA1 pyramidal neurons after 5 min of tGCI.
Acknowledgements
Not applicable.
Funding
The present study was supported by Basic Science
Research Program through the National Research Foundation of Korea
(NRF) funded by the Ministry of Education (grant no.
NRF-2015R1D1A1A01059728), by the Bio & Medical Technology
Development Program of the NRF funded by the Korean government,
MSIP (grant no. NRF-2015M3A9B6066968), by Basic Science Research
Program through the National Research Foundation of Korea (NRF)
funded by the Ministry of Science, ICT &Future Planning (grant
no. NRF-2017R1A2B4009079), and by Korea Institute of Planning and
Evaluation for Technology in Food, Agriculture, Forestry (IPET)
through High Value-added Food Technology Development Program,
funded by Ministry of Agriculture, Food and Rural Affairs (MAFRA)
(grant no. 117055-3).
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
YN, BS, TL, MS and HK performed the measurements.
SK, JP, JL, JY, IK and YL analyzed and interpreted data. JA, MW and
JK made substantial contributions to conception and design, and
were involved in drafting, revising the manuscript and interpreting
all data. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The protocol of this experiment was approved by the
Institutional Animal Care and Use Committee (IACUC) at Kangwon
National University (approval no. KW-180124-1).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kirino T and Sano K: Selective
vulnerability in the gerbil hippocampus following transient
ischemia. Acta Neuropathol. 62:201–208. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hou ST and MacManus JP: Molecular
mechanisms of cerebral ischemia-induced neuronal death. Int Rev
Cytol. 221:93–148. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mehta SL, Manhas N and Raghubir R:
Molecular targets in cerebral ischemia for developing novel
therapeutics. Brain Res Rev. 54:34–66. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Warner DS, Sheng H and Batinic-Haberle I:
Oxidants, antioxidants and the ischemic brain. J Exp Biol.
207:3221–3231. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gilgun-Sherki Y, Rosenbaum Z, Melamed E
and Offen D: Antioxidant therapy in acute central nervous system
injury: Current state. Pharmacol Rev. 54:271–284. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Brown JE, Mosley M and Aldred S:
Intermittent fasting: A dietary intervention for prevention of
diabetes and cardiovascular disease? Br J Diabetes Vasc Dis.
13:68–72. 2013. View Article : Google Scholar
|
|
7
|
Kim J, Kang SW, Mallilankaraman K, Baik
SH, Lim JC, Balaganapathy P, She DT, Lok KZ, Fann DY, Thambiayah U,
et al: Transcriptome analysis reveals intermittent fasting-induced
genetic changes in ischemic stroke. Hum Mol Genet. 27:1497–1513.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mattson MP, Longo VD and Harvie M: Impact
of intermittent fasting on health and disease processes. Ageing Res
Rev. 39:46–58. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tajes M, Gutierrez-Cuesta J, Folch J,
Ortuño-Sahagun D, Verdaguer E, Jiménez A, bJunyent F, Lau A, Camins
A and Pallàs M: Neuroprotective role of intermittent fasting in
senescence-accelerated mice P8 (SAMP8). Exp Gerontol. 45:702–710.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Martin B, Mattson MP and Maudsley S:
Caloric restriction and intermittent fasting: Two potential diets
for successful brain aging. Ageing Res Rev. 5:332–353. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhu H, Guo Q and Mattson MP: Dietary
restriction protects hippocampal neurons against the
death-promoting action of a presenilin-1 mutation. Brain Res.
842:224–229. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jeong JH, Yu KS, Bak DH, Lee JH1, Lee NS,
Jeong YG, Kim DK, Kim JJ3 and Han SY: Intermittent fasting is
neuroprotective in focal cerebral ischemia by minimizing autophagic
flux disturbance and inhibiting apoptosis. Exp Ther Med.
12:3021–3028. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fann DY, Santro T, Manzanero S,
Widiapradja A, Cheng YL, Lee SY, Chunduri P, Jo DG, Stranahan AM,
Mattson MP and Arumugam TV: Intermittent fasting attenuates
inflammasome activity in ischemic stroke. Exp Neurol. 257:114–119.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yu ZF and Mattson MP: Dietary restriction
and 2-deoxyglucose administration reduce focal ischemic brain
damage and improve behavioral outcome: Evidence for a
preconditioning mechanism. J Neurosci Res. 57:830–839. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kirino T: Delayed neuronal death in the
gerbil hippocampus following ischemia. Brain Res. 239:57–69. 1982.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ginsberg MD and Busto R: Rodent models of
cerebral ischemia. Stroke. 20:1627–1642. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li L, Wang Z and Zuo Z: Chronic
intermittent fasting improves cognitive functions and brain
structures in mice. PLoS One. 8:e660692013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lee CH, Yoo KY, Choi JH, Park OK, Hwang
IK, Kim SK, Kang IJ, Kim YM and Won MH: Neuronal damage is much
delayed and microgliosis is more severe in the aged hippocampus
induced by transient cerebral ischemia compared to the adult
hippocampus. J Neurol Sci. 294:1–6. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lee TK, Park JH, Ahn JH, Shin MC, Cho JH,
Bae EJ, Kim YM, Won MH and Lee CH: Pretreated duloxetine protects
hippocampal CA1 pyramidal neurons from ischemia-reperfusion injury
through decreases of glial activation and oxidative stress. J
Neurol Sci. 370:229–236. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Candelario-Jalil E, Alvarez D, Merino N
and León OS: Delayed treatment with nimesulide reduces measures of
oxidative stress following global ischemic brain injury in gerbils.
Neurosci Res. 47:245–253. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Goodrick CL, Ingram DK, Reynolds MA,
Freeman JR and Cider N: Effects of intermittent feeding upon body
weight and lifespan in inbred mice: Interaction of genotype and
age. Mech Ageing Dev. 55:69–87. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Armogida M, Nistico R and Mercuri NB:
Therapeutic potential of targeting hydrogen peroxide metabolism in
the treatment of brain ischaemia. Br J Pharmacol. 166:1211–1224.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lebovitz RM, Zhang H, Vogel H, Cartwright
J Jr, Dionne L, Lu N, Huang S and Matzuk MM: Neurodegeneration,
myocardial injury, and perinatal death in mitochondrial superoxide
dismutase-deficient mice. Proc Natl Acad Sci USA. 93:9782–9787.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ho YS, Gargano M, Cao J, Bronson RT,
Heimler I and Hutz RJ: Reduced fertility in female mice lacking
copper-zinc superoxide dismutase. J Biol Chem. 273:7765–7769. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dringen R, Pawlowski PG and Hirrlinger J:
Peroxide detoxification by brain cells. J Neurosci Res. 79:157–165.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Arumugam TV, Phillips TM, Cheng A, Morrell
CH, Mattson MP and Wan R: Age and energy intake interact to modify
cell stress pathways and stroke outcome. Ann Neurol. 67:41–52.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yan BC, Park JH, Shin BN, Ahn JH, Kim IH,
Lee JC, Yoo KY, Hwang IK, Choi JH, Park JH, et al: Neuroprotective
effect of a new synthetic aspirin-decursinol adduct in experimental
animal models of ischemic stroke. PLoS One. 8:e748862013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yan BC, Park JH, Ahn JH, Kim IH, Park OK,
Lee JC, Yoo KY, Choi JH, Lee CH, Hwang IK, et al: Neuroprotection
of posttreatment with risperidone, an atypical antipsychotic drug,
in rat and gerbil models of ischemic stroke and the maintenance of
antioxidants in a gerbil model of ischemic stroke. J Neurosci Res.
92:795–807. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Walsh ME, Shi Y and Van Remmen H: The
effects of dietary restriction on oxidative stress in rodents. Free
Radic Biol Med. 66:88–99. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hu Y, Zhang M, Chen Y, Yang Y and Zhang
JJ: Postoperative intermittent fasting prevents hippocampal
oxidative stress and memory deficits in a rat model of chronic
cerebral hypoperfusion. Eur J Nutr. Jan 11–2018.(Epub ahead of
print). View Article : Google Scholar
|
|
31
|
Chen KY, Chiu CH and Wang LC:
Anti-apoptotic effects of Sonic hedgehog signalling through
oxidative stress reduction in astrocytes co-cultured with
excretory-secretory products of larval Angiostrongylus cantonensis.
Sci Rep. 7:415742017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lee JC, Park JH, Kim IH, Cho GS, Ahn JH,
Tae HJ, Choi SY, Cho JH, Kim DW, Kwon YG, et al: Neuroprotection of
ischemic preconditioning is mediated by thioredoxin 2 in the
hippocampal CA1 region following a subsequent transient cerebral
ischemia. Brain Pathol. 27:276–291. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yan BC, Park JH, Ahn JH, Choi JH, Yoo KY,
Lee CH, Cho JH, Kim SK, Lee YL, Shin HC and Won MH: Comparison of
glial activation in the hippocampal CA1 region between the young
and adult gerbils after transient cerebral ischemia. Cell Mol
Neurobiol. 32:1127–1138. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Patel AR, Ritzel R, McCullough LD and Liu
F: Microglia and ischemic stroke: A double-edged sword. Int J
Physiol Pathophysiol Pharmacol. 5:73–90. 2013.PubMed/NCBI
|
|
35
|
Kawabori M and Yenari MA: Inflammatory
responses in brain ischemia. Curr Med Chem. 22:1258–1277. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Park JH, Park CW, Ahn JH, Choi SY, Shin
MC, Cho JH, Lee TK, Kim IH, Cho JH, Lee JC, et al: Neuroprotection
and reduced gliosis by pre- and post-treatments of hydroquinone in
a gerbil model of transient cerebral ischemia. Chem Biol Interact.
278:230–238. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Park CW, Lee TK, Cho JH, Kim IH, Lee JC,
Shin BN, Ahn JH, Kim SK, Shin MC, Ohk TG, et al: Rufinamide
pretreatment attenuates ischemia-reperfusion injury in the gerbil
hippocampus. Neurol Res. 39:941–952. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lee J, Auyeung WW and Mattson MP:
Interactive effects of excitotoxic injury and dietary restriction
on microgliosis and neurogenesis in the hippocampus of adult mice.
Neuromolecular Med. 4:179–196. 2003. View Article : Google Scholar : PubMed/NCBI
|