Introduction
Age-related macular degeneration (AMD) is one of the
most common causes of irreversible vision loss in the elderly
population of developed countries (1,2). AMD
is characterized by an accumulation of drusen in the macula, the
thickening of Bruch's membrane, the denaturation of retinal pigment
epithelial (RPE) cells, and abnormal angiogenesis, and is further
complicated by choroidal neovascularization (CNV) (3). There are two types of AMD: wet AMD,
which causes permanent vision damage despite therapeutic
intervention, and dry AMD, which causes gradual blindness without
specific treatment (4). In most
cases, degeneration of the retinal pigment epithelium occurs,
leading to death of the secondary photoreceptor and resulting in
vision loss (5). Both genetic and
environmental factors, such as persistent oxidative stress,
smoking, excessive fat intake, and UV exposure, contribute to the
development of AMD (6). In
particular, oxidative stress and senescence are the leading causes
of AMD. The retina experiences oxidative stress as a result of high
oxygen tension, constant exposure to light, and a high proportion
of unsaturated fatty acids contained within the photoreceptors
(6). Although the main
pathogenesis of AMD is unknown, oxidative stress has been reported
as one of the leading causes and is also known to accelerate the
process of aging, which again contributes to the development of AMD
(2,3). In other words, aging and oxidative
stress are anticipated to be critically involved in the
pathogenesis of AMD.
Continuous oxidative stress could induce cellular
senescence in RPE cells (6). It
may also cause progressive cellular damage and inflammation,
contributing to protein misfolding and functional anomalies in RPE
cells during cellular senescence (1). Reactive oxygen species (ROS) are
common free radicals that induce oxidative stress. Hypoxia,
metabolic defects, oncogene activation, and ER stress are all
factors that can contribute to the production of ROS. When ROS is
produced in response to various endogenous or exogenous stressors,
it contributes strongly to DNA damage, cell oxidation and
apoptosis, cellular senescence, and eventually inflammation and
tissue damage. The retina has a high metabolic activity and is
composed of various lipid compounds and high oxygen concentrations.
It is easy to generate ROS when exposed to persistent light and
high oxygen. These ROS damage mitochondria and lipids, leading to
modification of retinal functions (3).
Usually, cells will gradually lose the ability to
divide as cellular proliferation declines, typically, fibroblasts
divided up to 50 times. And cellular senescence is a process that
limits this proliferation in normal cells (7), senescence only affects mitotic cells.
Cellular senescence is characterized as an inability to proliferate
despite the presence of sufficient nutrients but where cell
viability and metabolic activity are still maintained (8). It is also a process through which
cells undergo phenotypic changes, including chromatin alterations,
tumor suppression, and interruption of cell division (9). Senescence is triggered by extreme
cellular stress and may act as a protective mechanism against
malignant cell degeneration. Common triggers for the onset of
senescence are telomere shortening and dysfunction, DNA damage,
oxidative stress, chromatin perturbations, oncogene activity, and
strong mitogenic signals (7,9).
Thus, common traits of senescent cells are altered cell morphology,
cell cycle arrest, DNA damage, tolerance to apoptosis, inflammatory
protein secretion, activation of tumor-suppressing mechanisms, and
changes in gene expression. Short-term cellular senescence is
beneficial for tumor suppression, wound healing, and embryo
development, but long-term cellular senescence promotes tumor
formation and senescence-related diseases. Markers of cellular
senescence include senescence-associated β-galactosidase
(SA-β-gal), cell cycle arrest mechanisms, cell proliferation
deficiency, DNA damage, DNA damage response activity, and various
immune-related genes and cell regulatory factors. SA-β-gal is a
representative senescence biomarker, as SA-β-gal activity is
induced if cellular senescence occurs (7,9).
Lutein is a kind of xanthophyll and one of the most
prevalent carotenoids, which are a group of fat-soluble yellow
pigments abundantly present in fruits and green vegetables
(10). Lutein has protective
effects against photo-oxidation and photo-destruction and is also
known as a potent antioxidant. However, lutein cannot be
synthesized human's body so must be ingested and absorbed. One
unique characteristic of lutein is the fact that it exists in
certain eye tissues. It is highly concentrated in the macula, a
small part of the retina, and is the only carotenoid present in the
tissue (11). Lutein is known as a
powerful antioxidant that suppresses and scavenges ROS, acting as a
filter for high energy blue light. By blocking harmful light, it
prevents sun damage and promotes tumor cell death. Also, it has
also been reported that ingestion of lutein is inversely related to
the risk of developing eye diseases such as AMD and cataracts
(11). In addition to eye health,
recent studies have shown that lutein can help maintain heart
health by reducing the risk of arteriosclerosis.
Given the existing known benefits of lutein, we
sought to determine the anti-senescence efficacy of lutein
treatment, and the mechanism by which such an effect might be
mediated, using a premature cellular senescence model. In this
study, we explored the effect of lutein on
H2O2-induced premature senescence in RPE
cells in order to determine whether it could counteract the induced
oxidative stress and therefore provide a new therapeutic strategy
for the treatment of AMD.
Materials and methods
Reagents
DMEM/F-12 medium (cat. no: LM002-04), was purchased
from Welgene, Inc., (Daegu, Korea), FBS (fetal bovine serum, cat.
no: 16000-044), penicillin-streptomycin (cat. no: 15140-122) and
Trypsin-EDTA (cat. no: 15400-054) were purchased from Gibco; Thermo
Fisher Scientific, Inc., (Waltham, MA, USA). Cell viability assay
kit (D-Plus CCK; cat. no: CCK-1000) was purchased from Donginbio
(Seoul, Korea). LysoTracker Green DND-26 (cat. no: 8783) was
purchased from Cell Signaling Technology, Inc., (Danvers, MA, USA),
and CM-H2DCFDA (chloromethyl derivative of
2′,7′-dichlorodihydrofluorescein diacetate; cat. no: C6827) was
purchased from Invitrogen; Thermo Fisher Scientific, Inc.
Quantitative Cellular Senescence Assay kit (cat. no: CBA 232) was
purchased from Cell Biolabs, Inc., (San Diego, CA), and senescence
β-galactosidase staining kit (cat. no. 9860) was purchased from
Cell Signaling Technology, Inc. PI (Propidium iodide solution; cat.
no: p4864) was supplied from Sigma-Aldrich; Merck KGaA (Darmstadt,
Germany). Bradford reagent (cat. no: 500-0006) was purchased from
Bio-Rad Laboratories Inc., (Hercules, CA, USA), passive lysis
buffer (cat. no: E194A) was acquired Promega Corporation (Madison,
WI, USA). And primary antibodies; p-SIRT1 (cat. no: 2314), SIRT1
(cat. no: 9475), SIRT3 (cat. no: 5490), p-p53 (cat. no: 9286), p53
(cat. no: 2524), and p21 (cat. no: 2947) were obtained from Cell
Signaling Technology, Inc. HO-1 (sc-10789), NQO1 (sc-16464), Nrf2
(sc-722), and α-tubulin (sc-23948) were obtained from Santa Cruz
Biotechnology, Inc., (Dallas, TX, USA). Secondary antibodies;
anti-rabbit IgG, HRP-linked antibody (cat. no: 7074), anti-mouse
IgG, HRP-linked antibody (cat. no: 7076) were obtained from Cell
Signaling Technology, Inc.
Cell culture
The human RPE cell line ARPE-19 was incubated at
37°C with 5% CO2 in DMEM/F-12 medium containing 10%
heat-inactivated FBS and 1% antibiotics. Cells were passaged 2 to 3
times weekly, with experiments being performed on cells between
passages 15 and 20.
Cell viability assay
Cell viability was measured by WST-1 (water-soluble
tetrazolium salt-1) assay. The WST-1 assay was performed the
manufacturer's protocol. It is a colorimetric assay for the
quantification of cell viability and proliferation, using the
cleavage of the WST-1 by mitochondrial dehydrogenases. ARPE-19
cells were seeded in 24 well plates at a density of
4×104 cells/well. The cells were treated with 1, 5, 10,
and 20 µM lutein and incubated at 37°C with 5% CO2 for
24, 48, or 72 h. After that 10 µl CCK-8 solution was add to each
well as and allow to react for 2 h at 37°C with 5% CO2.
The absorbance was measured at 450 nm using a microplate reader
(Wallac 1420; PerkinElmer, Inc., Waltham, MA, USA).
Treatment of cells with
H2O2 and lutein
ARPE-19 cells were seeded in 6 well plates at a
density of 1.5×105 cells/well, grown to 80% confluence,
and treated with 100 µM H2O2 for 2 h. They
were then washed with PBS and incubated in normal growth medium for
3 days. Lutein treatment (indicated concentration) was applied 1 h
prior to H2O2 treatment.
Lysosome contents and ROS
generation
Cells that had been treated with both
H2O2 (100 µM) and Lutein (5, 10, and 20 µM)
were washed with PBS and harvested using Trypsin-EDTA treatment.
Lysosome contents were analyzed using 50 nM LysoTracker Green
DND-26 and ROS generation was analyzed using 2.5 µM CM-H2DCFDA.
CM-H2DCFDA was used to detect H2O2-induced
ROS. Cells were treated with these dyes at room temperature in the
dark for 30 min. Fluorescence intensity was then measured by flow
cytometry at an excitation wavelength of 488 nm and an emission
wavelength of 525 nm. Data analysis was performed using CXP
software 2.0 (Beckman Coulter, Inc., Brea, CA, USA).
SA-β-gal activity and staining
Cells that had been treated with both
H2O2 (100 µM, 2 h) and Lutein (5, 10, and 20
µM) incubated for 3 days at 37°C with 5% CO2. The
SA-β-gal activity of H2O2 and lutein treated
cells was evaluated using the Quantitative Cellular Senescence
Assay Kit according to the manufacturer's protocol. Briefly, the
cells were washed with PBS and harvested using Trypsin-EDTA before
adding the pretreatment solution and incubating at 37°C with 5%
CO2 for 2 h. They were then incubated with SA-β-gal
substrate solution at 37°C with 5% CO2 in the dark for 5
h before being analyzed using flow cytometry. Data analysis was
performed using CXP software 2.0 (Beckman Coulter, Inc.).
The degree of SA-β-gal staining was observed using a
senescence β-galactosidase staining kit. ARPE-19 cells were treated
with lutein (20 µM) for 1 h, followed by 100 µM
H2O2 treatment at 37°C with 5% CO2
for 2 h. The cells were washed with PBS, changed into normal growth
medium, and then incubated with lutein at 37°C with 5%
CO2 for an additional 6 days. Finally, the treated cells
were washed with PBS and fixed using fixative solution at room
temperature for 20 min. The cells were again washed with PBS and
then incubated with β-galactosidase staining solution at 37°C
overnight (no CO2). After incubation, the stained cells
were observed under a microscope.
Cell cycle
ARPE-19 cells were seeded in 60 mm dishes at a
density of 4×105 cells per dish. Cells were treated with
lutein (20 µM) for 1 h, followed by 100 µM
H2O2 treatment for 2 h. After replacing to
normal growth medium, the treated cells were again incubated with
lutein at 37°C with 5% CO2 for 48 h, before being
harvested using Trypsin-EDTA and fixed with 70% cold methanol at
4°C for 1 h. They were then centrifuged (2,000 rpm, 3 min) and
washed with PBS. Thereafter, 30 µg/mL RNase and PI were added to
the cells and the cells were incubated at room temperature in the
dark for 30 min. Cell cycle analysis was performed using flow
cytometry. Data analysis was performed using CXP software v2.0
(Beckman Coulter, Inc.).
Western blot analysis
ARPE-19 cells were seeded in 60 mm dishes at a
density of 4×105 cells per dish. Cells were treated with
lutein (20 µM) for 1 h, followed by 100 µM
H2O2 treatment for 2 h. After replacement
with normal growth medium, the treated cells were incubated again
with lutein at 37°C with 5% CO2. Cells were collected
using a scraper by adding RIPA buffer, and centrifuged (13,000 rpm,
10 min) to lyse the cells, and obtained the protein. The protein
content of cell lysates was determined using the Bradford reagent.
Protein (30 µg) from each sample was electrophoresed on 8.5 or 10%
SDS-polyacrylamide gels, transferred to a PVDF (polyvinylidene
difluoride) membrane. Membranes were incubated overnight at 4°C
with the primary antibodies against p-SIRT1, SIRT1, SIRT3, p-p53,
p53, and p21 (all 1:1,000). Also they were reacted for 4 h at room
temperature with primary antibodies such as HO-1 (1:500), NQO1
(1:5,000), Nrf2 (1:1,000), and α-tubulin (1:5,000). Secondary
antibodies were used HRP-conjugated anti-rabbit, anti-mouse, or
anti-goat antibodies (all 1:1,000) for 2 h at room temperature.
Target bands were visualized using an enhanced chemiluminescence
detection system (GE Healthcare, Chicago, IL, USA). Images were
acquired using an ImageQuant 350 analyzer (GE Healthcare).
Statistical analysis
Statistical analyses were performed using SPSS (v23;
IBM Corp., Armonk, NY, USA) to determine significant differences
based on one-way analyses of variance (followed by Tukey's post-hoc
test). P<0.05 were considered to indicate a statistically
significant difference.
Results
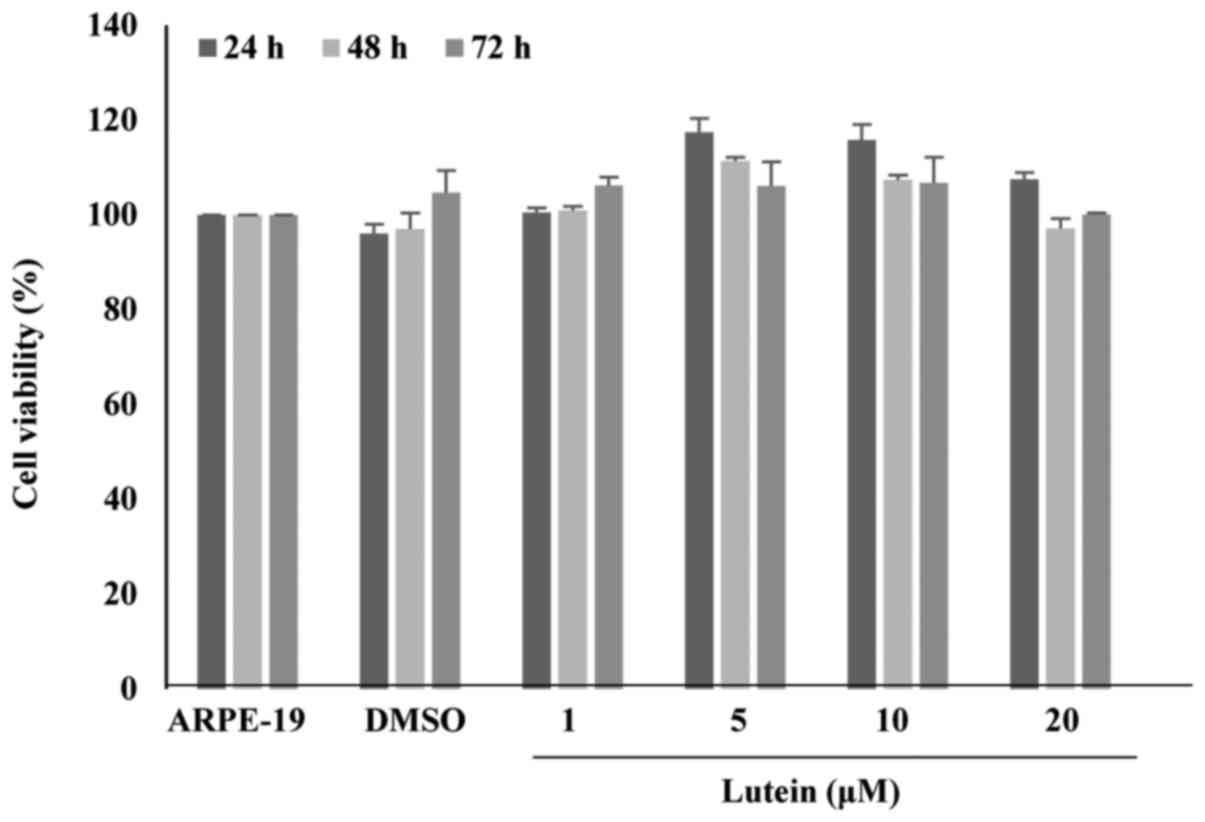
Cell viability
Cell viability of ARPE-19 cells treated with lutein
at concentrations of 1 to 20 µM for 24, 48, or 72 h were examined
(Fig. 1). Cell viability was
performed using the WST-1 assay. The WST results showed that lutein
treatment at 20 µM for 72 h resulted in no significant changes in
viability when compared with the control.
Effects of lutein on lysosome contents
and ROS generation in H2O2-treated ARPE-19
cells
The lysosome contents of ARPE-19 cells treated with
H2O2 alone greatly increased (27.06%) when
compared with the control (10.44%), whereas co-treatment with
lutein resulted in a dose-dependent reduction (22.34, 17.08, and
13.0%) when compared with the cells treated with
H2O2 alone. Lutein treatment alone did not
affect the lysosome contents of the cells (Fig. 2A). Lutein treatment alone did not
affect ROS production (Fig. 2B).
However, H2O2 treatment considerably
increased ROS production (33.57%) when compared to the normal
control (10.42%). Lutein treatment of H2O2
treated cells resulted in a marked decrease in expected ROS levels
(26.03%). These data suggest that oxidative stress is triggered in
ARPE-19 cells by H2O2 and that this induction
is inhibited by lutein. We have demonstrated that lutein suppresses
H2O2-induced oxidative stress through
anti-oxidative effects.
Inhibitory effects of lutein on
SA-β-gal in H2O2-treated ARPE-19 cells
To evaluate the effects of lutein on
H2O2-treated ARPE-19 cells, we employed
SA-β-gal staining. The morphology and degree of staining were
confirmed under a microscope. As indicated in Fig. 3A, normal cells did not show
staining. However, after H2O2 treatment,
changes in cell shape and an increase in SA-β-gal staining were
observed. In cells treated with H2O2 and
lutein, the SA-β-gal staining was considerably decreased, while
lutein treatment alone did not produce SA-β-gal staining. Thus,
H2O2 induced senescence in ARPE-19 cells and
lutein greatly inhibited the production of SA-β-gal in cells
induced towards senescence by H2O2 treatment,
indicating that lutein protected ARPE-19 cells from
H2O2-induced senescence.
To evaluate the effects of lutein on
H2O2-treated ARPE-19 cells, SA-β-gal activity
was measured. As shown in Fig. 3B,
the number of SA-β-gal positive cells increased in cells treated
with H2O2 (41.08%) to a level roughly
four-fold higher than is seen in normal control cells. In cells
treated with both H2O2 and lutein, the number
of SA-β-gal positive cells was effectively decreased, in a
lutein-dose-dependent manner (35.38, 27.39, and 20.41%). In cells
treated with lutein alone, the number of SA-β-gal positive cells
did not increase. These data show that SA-β-gal positive cells are
increased in cells triggered for senescence by
H2O2 and that this effect is markedly
decreased by lutein treatment. Together, these data clearly
indicate the inhibitory effects of lutein treatment on
H2O2-induced senescence.
Cell cycle analysis of lutein and
H2O2-treated ARPE-19 cells
Oxidative stress and the resulting DNA damage can
contribute to cell cycle arrest in senescent cells. Therefore, we
performed cell cycle analysis using PI staining to assess the
effects of lutein on H2O2-treated ARPE-19
cells. We observed G2 arrest after H2O2
treatment. Lutein treatment significantly attenuated the percentage
of G2 arrest observed in these H2O2-treated
cells but did not affect the cell cycle distribution in control
cells (Fig. 4). These data
indicate that G2 arrest is increased in ARPE-19 cells by
H2O2 treatment, however, this is reversed by
lutein treatment. Thus, these results clearly support the
hypothesis that lutein protects ARPE-19 cells from DNA damage
caused by H2O2-induced oxidative stress.
Induction of HO-1, NQO1, and Nrf2 in
ARPE-19 cells
When oxidative stress occurs, Nrf2 is activated,
followed by the expression of antioxidant enzymes such as HO-1 and
NQO1. We observed the induction of HO-1 and NQO1, and the
activation of Nrf2, upon lutein treatment. These effects are most
strongly observed at 8, 4, and 1 h after lutein treatment,
respectively (Fig. 5A and C).
Furthermore, HO-1 and NQO1 were most strongly expressed at lutein
treatment levels of 10 or 20 µM (Fig.
5B). HO-1 and NQO1 are both known to be involved in protecting
cells from various stresses. These results indicate that HO-1
induction via lutein may activate cellular protective and/or
anti-oxidative effects.
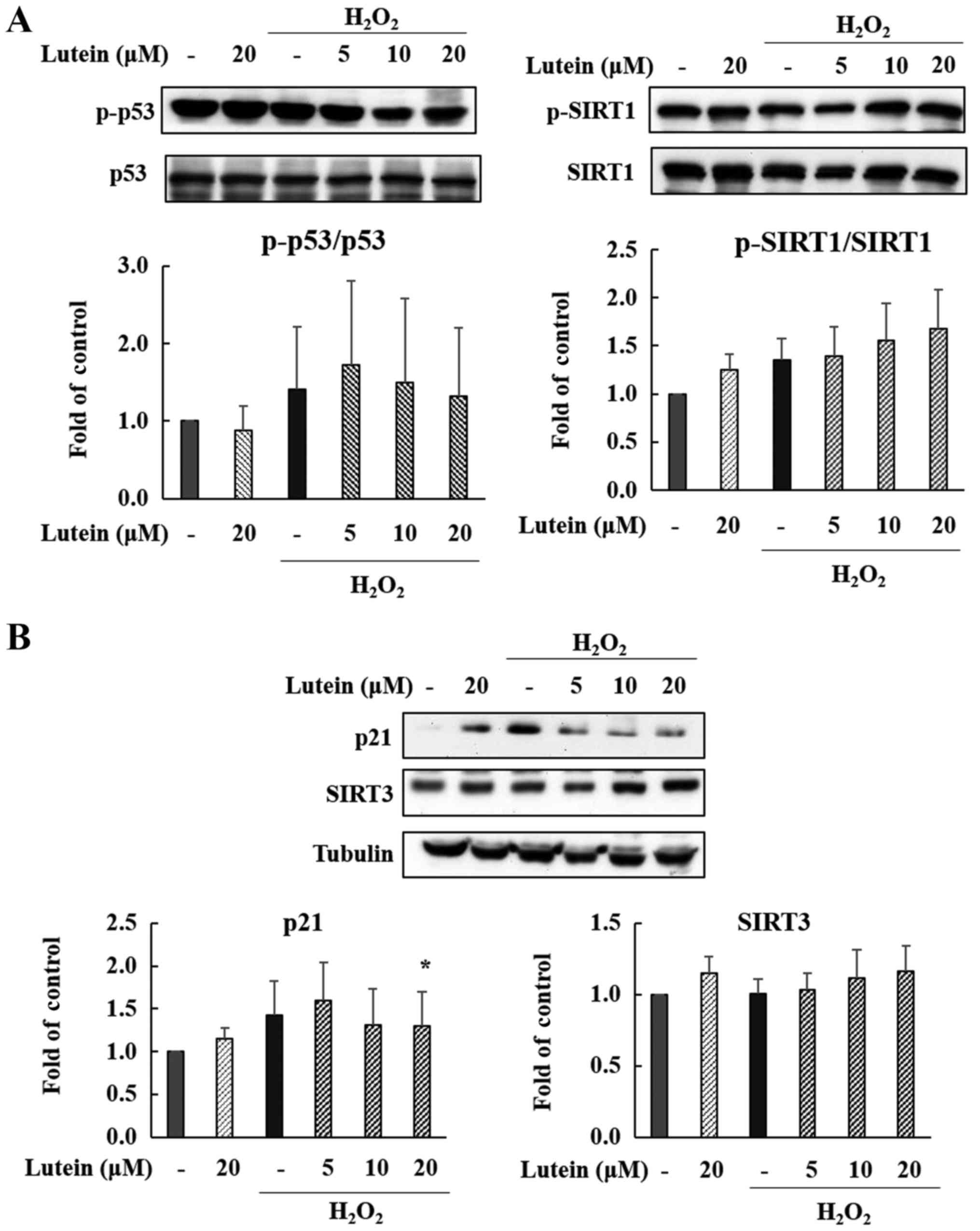
Expression of p53, p21 in
H2O2-treated ARPE-19 cells
We observed changes in the protein expression of p53
and p21 following lutein treatment in
H2O2-treated ARPE-19 cells. As shown in
Fig. 6A and B, the phosphorylation
of p53 protein was slightly elevated by H2O2
treatment, and this was reduced by 20 µM lutein treatment, but this
was not a statistically significant decrease. The expression of p21
protein was also increased after treatment with
H2O2, but this change was markedly decreased
by lutein treatment in a dose-dependent manner (Fig. 6B). Especially p21 showed a
statistically significant decrease at high concentrations. These
data suggest that the p53-p21 pathway is significantly activated by
H2O2, meaning that cell cycle changes and
cellular senescence may be induced via the p53-p21 pathway. Lutein
appears effective in controlling the p53-p21 pathway in these
H2O2-induced cells.
Expression of SIRT1 and SIRT3 in
H2O2-treated ARPE-19 cells
Sirtuins are typical aging-related proteins that
affect cellular processes. We confirmed that SIRT1 and SIRT3
expression is regulated by lutein exposure in
H2O2-treated ARPE-19 cells. SIRT3 was
increased by H2O2 and lutein treatment (Fig.
B), suggesting that lutein is associated with the inhibition of
senescence. Also, as shown in Fig.
6A, the phosphorylation of SIRT1 increased after treatment with
H2O2 and lutein in a dose-dependent manner.
Therefore, our data indicate that the protective effects of lutein
against H2O2-induced senescence are partially
controlled through the up-regulation of SIRT1 and SIRT3.
Discussion
The purpose of this study was to investigate the
effects of lutein on occurred cellular senescence and oxidative
stress induced by H2O2 treatment as well as
to determine which activation pathways lutein may act through.
Oxidative stress occurs when there is an imbalance between
antioxidants and free radicals, leading to an increase in free
radicals. ROS, a group of prevalent free radicals, are induced by
hypoxia, ER stress, metabolic defect, and oncogene activation.
Scavenging factors include Nrf2, glutathione, NADPH, and tumor
suppressor mechanisms. Excessive ROS lead to senescence processes
by promoting DNA damage and cellular oxidation, while also
contributing to the pathogenesis of various diseases. Oxidative
stress is an active field of research due to its involvement in
various diseases, including sepsis, mastitis, enteritis, pneumonia,
and respiratory and joint diseases (12).
Senescence-associated beta-galactosidase (SA-β-gal)
is a hydrolytic enzyme that catalyzes only in senescent cells.
Specifically, SA-β-galactosidase is overexpressed and accumulated
in senescent cells, therefore the most widely used biomarkers of
senescence and was the first marker for the detection of senescence
and senescent cells. Hydrogen peroxide (H2O2)
is a typical oxidizing agent and simplest peroxide, which causes
ROS, which can cause disease. H2O2 induces
oxidative stress by increasing lysosome contents and ROS
generation. Premature cellular senescence is confirmed by the
detection of increasing number of SA-β-gal positive cells. The
present study demonstrated that H2O2
treatment induced oxidative stress in ARPE-19 cells, leading to
cellular senescence. Lutein was applied at a various concentrations
but had no effect on cell viability at any of the treatment
concentrations or times. Lutein treatment markedly decreased
lysosome contents and ROS generation in a dose-dependent manner and
SA-β-gal positive cells were also considerably decreased. Our
results demonstrate that lutein treatment suppresses ROS and
decreases SA-β-gal positive cells. In other words, lutein protects
ARPE-19 cells from senescence caused by
H2O2-induced oxidative stress. In this
H2O2-induced cellular senescence model,
lutein suppressed the expected increase of cellular ROS, lysosome
contents and SA-β-gal positive cells as part of its protective
effects on ARPE-19 cells.
Nrf2 binds to keap1 and is inactivated, when
oxidative stress such as ROS occurs, it is activated and
dissociates and moves into the nucleus. And induces the expression
of antioxidant enzymes such as HO-1, NQO1 and SOD. Heme oxygenase
is an enzyme that catalyzes the degradation of heme and produces
biliverdin, iron, and carbon monoxide (13). HO-1 (Heme oxygenase-1) and NQO1
(NAD(P)H dehydrogenase 1) are important antioxidant enzymes. HO-1
and its byproducts play major roles in the regulation of oxidative
stress and inflammation. In addition, it is known to mediate
cellular defense mechanisms against diverse stresses (14,15).
NQO1 is part of the defense systems cells employ against oxidative
stress. Therefore, HO-1 and NQO1 are major enzymes that play
important roles in the intracellular, antioxidant defense systems
of cells. The induction of HO-1 and NQO1 is considered to be
cell-protective. Our study clearly indicates that lutein induces
HO-1 and NQO1 activation in ARPE-19 cells.
The p53 and p21 proteins are important to signaling
pathways in the DNA damage-related senescence response and are also
known to regulate apoptosis, be involved in cell cycling, and are
well known tumor suppressors (16). The p21 also known as
cyclin-dependent kinase inhibitor 1, it is a leading target of p53
activity, also is associated with linking DNA damage to cell cycle
arrest. Upon DNA damage, the p53 protein is activated and in turn
induces expression of p21. Activation of p21 can cause cell cycle
arrest and block cell growth (17). Lutein restored cell cycling in
cells induced towards cellular senescence via down-regulation of
p53 and thus also p21. Cell cycle analysis also showed that
H2O2-induced cell cycle arrest was restored
by lutein treatment.
Sirtuins are known to affect cellular processes in
response to cellular senescence, transcription, inflammation and
apoptosis and various stresses (18). SIRT1 is a well-known senescence
regulator that facilitates DNA damage repair in mouse and, in
particular, is reported to play a critical role in senescence
processes of the murine retina. In addition, SIRT1 activation
decreases in the retina are followed by the accumulation of DNA
damage (2). Our results suggest
that lutein exerts effects on ARPE-19 cells though SIRT1
activation, protecting them from senescence by oxidative stress.
The activation of SIRT1 also leads to reductions in the levels of
p53 and p21. SIRT3 has been reported to support the maintenance of
suitable mitochondrial function by limiting oxidative stress and by
reducing ROS generation (19).
Lutein treatment also increased the activity of SIRT3, again
supporting the hypothesis that lutein protects ARPE-19 cells
against H2O2-induced cellular senescence. Our
results indicate that activation of SIRT1 and SIRT3 also contribute
to the anti-senescence functions of lutein. And, in turn, lutein
may promote the expression and activation of SIRT1 and SIRT3.
Summarize, we analyzed on the production of
intracellular ROS in order to confirm the effect of lutein on the
cellular redox status. As a result, ROS generation is increased by
H2O2, and lutein was decreased in a
dose-dependent manner. The antioxidative effect of lutein was
confirmed through this results. Also, in our western results,
lutein showed anti-oxidative effects by up-regulates antioxidant
enzymes HO-1 and NQO1. Also, we analyzed the contents of lysosome.
As a result, we found that lysosome contents increased after
H2O2 treatment and decreased after lutein
treatment. These results suggest that lutein is also involved in
cell metabolism. Furthermore, our results suggest that lutein
prevents H2O2-induced cellular senescence
through the scavenging of SA-β-gal positive cells. Also, lutein
restores cell cycling though p53-p21 pathway regulation,
up-regulates anti-senescence related protein SIRT1 and SIRT3. Thus,
Lutein inhibits oxidative stress induced by ROS and protects
ARPE9-19 cells from cellular senescence, especially by oxidative
stress. As oxidative stress induced cellular senescence is an
important factor to the pathogenesis of AMD, and we have confirmed
that lutein protects ARPE-19 cells from oxidative stress induced
cellular senescence. We suggest that lutein may potentially be a
new, additional therapeutic strategy for the treatment of
retinal-based diseases such as AMD.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Basic Science
Research Program through the National Research Foundation of Korea
(NRF), funded by the Ministry of Education (grant nos.
NRF-2018R1D1A1B07047497 and NRF-2018R1D1A3B07047983).
Availability of data and materials
All data generated or analyzed during this study are
included in the published article.
Authors' contributions
SYC conducted the experimental work. SYP and GP
designed and performed the experiments, and analyzed the data. SYC,
SYP and GP wrote the manuscript. SYP and GP obtained financial
support and supervised the study.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
WST-1
|
water-soluble tetrazolium salt-1
|
|
CM-H2DCFDA
|
chloromethyl derivative of
2′,7′-dichlorodi-hydrofluorescein diacetate
|
|
ROS
|
reactive oxygen species
|
|
SA-β-gal
|
senescence-associated
β-galatosidase
|
|
PI
|
Propidium iodide
|
|
HO-1
|
heme oxygenase-1
|
|
NQO1
|
NAD(P)H quinone dehydrogenase 1
|
|
Nrf2
|
nuclear factor erythroid 2-related
factor 2
|
|
SIRT1
|
sirtuin-1
|
|
SIRT3
|
sirtuin-3
|
References
|
1
|
Alves-Rodrigues A and Shao A: The science
behind lutein. Toxicol Lett. 150:57–83. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Campisi J and d'Adda di Fagagna F:
Cellular senescence: When bad things happen to good cells. Nat Rev
Mol Cell Biol. 8:729–740. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Celi P: Biomarkers of oxidative stress in
ruminant medicine. Immunopharmacol Immunotoxicol. 33:233–240. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen Y, Fu LL, Wen X, Wang XY, Liu J,
Cheng Y and Huang J: Sirtuin-3 (SIRT3), a therapeutic target with
oncogenic and tumor-suppressive function in cancer. Cell Death Dis.
5:e10472014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fang Y, Su T, Qiu X, Mao P, Xu Y, Hu Z,
Zhang Y, Zheng X, Xie P and Liu Q: Protective effect of
alpha-mangostin against oxidative stress induced-retinal cell
death. Sci Rep. 6:210182016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Itahana K, Dimri G and Campisi J:
Regulation of cellular senescence by p53. Eur J Biochem.
268:2784–2791. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Izumi-Nagai K, Nagai N, Ohgami K, Satofuka
S, Ozawa Y, Tsubota K, Umezawa K, Ohno S, Oike Y and Ishida S:
Macular pigment lutein is antiinflammatory in preventing choroidal
neovascularization. Arterioscler Thromb Vasc Biol. 27:2555–2562.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jeong G, Li B, Lee D, Kim KH, Lee IK, Lee
KR and Kim Y: Cytoprotective and anti-inflammatory effects of
spinasterol via the induction of heme oxygenase-1 in murine
hippocampal and microglial cell lines. Int Immunopharmacol.
10:1587–1594. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kamoshita M, Toda E, Osada H, Narimatsu T,
Kobayashi S, Tsubota K and Ozawa Y: Lutein acts via multiple
antioxidant pathways in the photo-stressed retina. Sci Rep.
6:302262016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kuilman T, Michaloglou C, Mooi WJ and
Peeper DS: The essence of senescence. Genes Dev. 24:2463–2479.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Marazita MC, Dugour A, Marquioni-Ramella
MD, Figueroa JM and Suburo AM: Oxidative stress-induced premature
senescence dysregulates VEGF and CFH expression in retinal pigment
epithelial cells: Implications for age-related macular
degeneration. Redox Biol. 7:78–87. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Murthy RK, Ravi K, Balaiya S, Brar VS and
Chalam KV: Lutein protects retinal pigment epithelium from
cytotoxic oxidative stress. Cutan Ocul Toxicol. 33:132–137. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kikuchi G, Yoshida T and Noguchi M: Heme
oxygenase and heme degradation. Biochem Biophys Res Commun.
338:558–567. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Probin V, Wang Y, Bai A and Zhou D:
Busulfan selectively induces cellular senescence but not apoptosis
in WI38 fibroblasts via a p53-independent but extracellular
signal-regulated kinase-p38 mitogen-activated protein
kinase-dependent mechanism. J Pharmacol Exp Ther. 319:551–560.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Saviranta NMM, Veeroos L, Granlund LJ,
Hassinen VH, Kaarniranta K and Karjalainen RO: Plant flavonol
quercetin and isoflavone biochanin A differentially induce
protection against oxidative stress and inflammation in ARPE-19
cells. Food Res Int. 44:109–113. 2011. View Article : Google Scholar
|
|
16
|
Supanji, Shimomachi M, Hasan MZ, Kawaichi
M and Oka C: HtrA1 is induced by oxidative stress and enhances cell
senescence through p38 MAPK pathway. Exp Eye Res. 112:79–92. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Son Y, Byun SJ and Pae HO: Involvement of
heme oxygenase-1 expression in neuroprotection by piceatannol, a
natural analog and a metabolite of resveratrol, against
glutamate-mediated oxidative injury in HT22 neuronal cells. Amino
Acids. 45:393–401. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Preyat N and Leo O: Sirtuin deacylases: A
molecular link between metabolism and immunity. J Leukoc Biol.
93:669–680. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhuge CC, Xu JY, Zhang J, Li W, Li P, Li
Z, Chen L, Liu X, Shang P, Xu H, et al: Fullerenol protects retinal
pigment epithelial cells from oxidative stress-induced premature
senescence via activating SIRT1. Invest Ophthalmol Vis Sci.
55:4628–4638. 2014. View Article : Google Scholar : PubMed/NCBI
|