Introduction
Hypertrophic scars (HS) derive from the
overproliferation of fibroblasts following surgery, inflammation,
trauma or burns. They are often associated with functional
impairments, and severe physiological and psychological problems
(1). Despite numerous studies
(2,3) being conducted on the etiology and
pathogenesis of HS, the progression of fibrosis remains poorly
understood. Recently, increasing evidence has suggested that
noncoding RNAs (ncRNAs), including microRNAs (miRNAs/miRs) and long
noncoding RNAs (lncRNAs), serve important roles in the formation of
HS (4), and can regulate gene
expression at the levels of transcription, RNA processing and
translation (5).
MicroRNA is a class of small ncRNA, ranges from 18
to 25 nucleotides (nt). As we know it plays important roles in
fibrosis processes including transforming growth factor (TGF)-beta
signaling, ECM deposition, epithelial to mesenchymal transition
(EMT), fibroblast proliferation and differentiation (6). With the study of scarring, more and
more miRNAs and their function were found and identified. For
instance, by affecting the collagen I and III synthesis and
TGF-β1/α-SMA signalling, miR-200b could regulate the cell
proliferation of human hypertrophic scar fibroblasts and affected
hypertrophic scar (7).
Previous studies have shown that lncRNAs served a
crucial role in regulation of gene expression and disease processes
(8,9). It's reported that lncRNAs
participated in cell proliferation, differentiation, migration and
apoptosis (10). Notably, they can
regulate the stages of wound healing, including
re-epithelialization, angiogenesis, remodelling and scar formation
(11). lncRNA has become a new
research hotspot in wound healing.
In addition, a newly identified type of ncRNA,
circRNA, is molecularly stable and can function as a miRNA sponge,
thus affecting the functions of miRNAs (12). For example, circZNF91 contains 24
target sites for miR-23b-3p, which is known to play important roles
in keratinocyte differentiation (13). This means that circRNAs bind to
miRNAs and affect biological pathways of miRNAs and consequently
repress their function. Such as, circRNA_100782 can regulate BxPC3
cell proliferation by acting as miR-124 sponge through the
IL6-STAT3 pathway (14). Resistant
to exonuclease, circRNAs are stable molecules in cells which make
the possibility of circRNAs as biological markers in clinical
samples. To the best of the authors' knowledge, there is no report
about the functional roles of circRNA in HS.
LncRNAs and circRNAs can function as competing
endogenous RNAs (ceRNAs) and regulate each other by competing for
the same miRNA response elements (MREs) (15). ncRNAs have become a novel area of
research in wound healing; however, the potential roles of lncRNAs
and circRNAs as ceRNAs in human HS remain to be elucidated.
The present study aimed to investigate the
differentially expressed patterns of lncRNAs, circRNAs and mRNAs in
HS using high-throughput gene sequencing. Meanwhile, the functions
of the lncRNAs and circRNAs were predicted by investigating the
co-expression and ceRNA networks of these dysregulated RNAs, such
as AC048380.1 and LINC00299 were associated with metastasis-related
genes, including INHBA, SMAD7, COL1A1, TGF-β3, which might be
associated with HS.
Patients and methods
Patients and samples
The present study was approved by the Ethics
Committee of The First Affiliated Hospital of Nanchang University
(Nanchang, China). Written informed consent was obtained from all
subjects. A total of six pairs of HS and adjacent normal skin
tissues were collected from patients (4 men and 2 women; age range,
20–56 years old) who lived in the first affiliated hospital of
Nanchang University from April to September 2016. All 6 patients
underwent surgical treatment without preoperative drug therapy or
radiotherapy. Subsequently, the epidermis and subcutaneous tissue
were removed from the samples in a bacteria-free operating
environment within 15 min, and were stored in liquid nitrogen.
RNA isolation and quality control
Total RNAs were isolated from HS and normal skin
tissues using TRIzol® (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA), according to the
manufacturer's protocol. Subsequently, total RNA was cleaned using
the RNeasy Mini kit (Qiagen, Inc., Valencia, CA, USA), and RNA
quantity was analyzed using an Agilent 2100 Bioanalyzer (Agilent
Technologies, Inc., Santa Clara, CA, USA). RNA integrity and
chromosomal DNA contamination were assessed by denaturing agarose
gel electrophoresis (concentration of agarose gel is 1%, and 0.5
µg/ml ethidium bromide to help it visualized).
Library preparation for RNA sequencing
(RNA-seq)
Ribosomal RNA depletion was performed using a
5′-phosphate-dependent exonuclease (GeneRead rRNA Depletion kit
(Qiagen, Germany), which degraded transcripts with a 5′
monophosphate. Linear RNA depletion was performed using
Ribonuclease R. The dosage ratio of Ribonuclease R to RNA is 3U:1
µg and the digestive conditions were 37°C for 30 min. According to
experimental protocols, Rayscript cDNA Synthesis kit (GENEray,
GK8030) was used for the reverse transcription. RNA fragments were
used for sequential first-strand and second-strand cDNA synthesis,
and the cDNA templates were enriched. Libraries were constructed
using the TruSeq Stranded mRNA LT Sample Prep kit (Illumina, Inc.,
San Diego, CA, USA). Library quality was assessed using an Agilent
2100 Bioanalyzer (Agilent Technologies, Inc.) prior to sequencing.
Total concentrations were determined using a Qubit 12.0 Fluorometer
(Thermo Fisher Scientific, Inc.). Libraries were sequenced on the
Illumina NextSeq 500 platform (Illumina, Inc.) with 2×150 bp
paired-end reads. The libraries were constructed and sequenced by
Shanghai Personal Biotechnology Co., Ltd. (www.personalbio.cn/; Shanghai, China).
Data quality control
The quality of raw sequencing data was assessed by
using FASTQC (www.bioinformatics.babraham.ac.uk/projects/fastqc/).
All sequences were subjected to quality control to trim the adapter
contaminants and filter out low-quality reads. Cutadapt (version
1.2.1; pypi.org/project/cutadapt/1.2.1/#files), which can
perform various useful forms of trimming (16), was used for quality control of the
raw data.
Data arrangement and analysis
Raw reads were subjected to quality control and the
remaining ‘clean reads’ were mapped to the Ensembl database
(www.ensembl.org/) using Tophat version 2
(ccb.jhu.edu/software/tophat/index.shtml). The reads
per kilobase of transcript per million mapped reads was calculated
for each gene and used to analyze the fold change. The
differentially expressed genes (DEGs) were detected using DEGseq
Software (version 1.18.0) (17). A
fold-change >2, a P-value <0.05 and a false-discovery rate
(FDR) <0.05 were considered the criteria for differential
expression. The differentially expressed genes obtained from all
groups were analyzed by bidirectional clustering through Pheatmap
package. Euclidean method was used to calculate the distance, and
the Complete Linkage was used for clustering. The gene functional
annotation included: ENSEMBL ID, information on the chromosome
distribution in each locus, gene name [Human Genome Variation
Society Symbol, National Center for Biotechnology Information
(NCBI) Gene ID, UniProtKB ID], gene classification [Gene Ontology
(GO), Kyoto Encyclopedia of Genes and Genomes (KEGG) Orthology
(KO), enzyme classification]. The Gene Ontology Consortium
(www.geneontology.org/) was used for GO
functional enrichment analysis. It aided the description of the
biological function of the DEGs, including molecular functions,
cellular components and biological processes. The analysis first
mapped all DEGs to GO terms within the database and the gene
numbers were calculated for each term. Subsequently,
ultra-geometric tests were used to identify the GO terms markedly
enriched among the DEGs relative to the genomic background. KEGG
(www.kegg.jp/) includes KO and KEGG Pathway. KEGG
Pathway was used to conduct a pathway enrichment analysis of the
DEGs. It identified signal transduction pathways or metabolic
pathways associated with the DEGs, which were significantly
enriched. In the present study, significant pathways were
identified as those with P-values <0.05 and FDR <0.05. The
lower values indicated greater significance.
Validation with reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
The RNA-seq data were validated by RT-qPCR. RNA
extracted by using TRIzol™ Reagent (Beijing Solarbio Science &
Technology Co., Ltd., Beijing, China). cDNA was generated from RNA
using the Rayscript cDNA Synthesis kit (cat. no. GK8030; Shanghai
Generay Biotech Co., Ltd., Shanghai, China). cDNA was subjected to
RT-qPCR analysis using Power qPCR PreMix (cat. no. GK8020; Shanghai
Generay Biotech Co., Ltd.). The primers were designed using
NCBI-Primer BLAST (www.ncbi.nlm.nih.gov/tools/primer-blast/) and primers
were checked using OLIGO7.37 software in accordance with the
sequences of the corresponding human RNAs in GenBank (www.ncbi.nlm.nih.gov/genbank/). A list
of primers of the selected genes for RT-qPCR is provided in
Tables I–III. GAPDH and Human β-actin (H-actin)
were used as the control genes for normalization of cDNA loading
differences. Cycling conditions were: 95°Cx10 min→95°C ×15 sec,
total 45 cycles and it was repeated 3 times.
 | Table I.Primer sequences of long noncoding
RNAs used for reverse transcription-quantitative polymerase chain
reaction. |
Table I.
Primer sequences of long noncoding
RNAs used for reverse transcription-quantitative polymerase chain
reaction.
| Primer ID | Primer sequences
(5′-3′) | Annealing
temperature (°C) | Length (base
pairs) |
|---|
| ENSG00000258876
(TGFβ3-AS1) |
F:TTGAGTGAATGAATGGAGGA | 60 | 91 |
|
|
R:AAATGGAGGGAAGAGGACA |
|
|
| ENSG00000268262
(AC011445.1) |
F:CTTTGACGCCTTCCTGTCC | 60 | 111 |
|
|
R:AGCGTGAGGGTGTTTAGGTTC |
|
|
| ENSG00000223749
(STX17-AS1) |
F:AATAACCAGGGTCACAGAAAA | 60 | 147 |
|
|
R:AAAGCAGCATACAGTCATTCA |
|
|
| ENSG00000237548
(TTLL11-IT1) |
F:TACAAGTCCCCAGAAACCA | 60 | 196 |
|
|
R:AAAATCCACCCAGATAGCC |
|
|
| ENSG00000251085
(LINC01969) |
F:CAGACGGTCTCCTTAAAGATTCTCC | 60 | 89 |
|
|
R:GAACCCATCTTCACCCTTGCTAA |
|
|
| GAPDH |
F:GGACCTGACCTGCCGTCTAG | 60 | 100 |
|
|
R:GTAGCCCAGGATGCCCTTGA |
|
|
| Table III.Primer sequences of mRNA used for
reverse transcription-quantitative polymerase chain reaction. |
Table III.
Primer sequences of mRNA used for
reverse transcription-quantitative polymerase chain reaction.
| Primer ID | Primer
sequences | Annealing
temperature (°C) | Length (base
pairs) |
|---|
| COL1A1 |
F:AGACATCCCACCAATCACCT | 60 | 118 |
|
|
R:GTCATCGCACAACACCTTG |
|
|
| ASXL1 |
F:CCTACTGTCCTCCCAAACC | 60 | 118 |
|
|
R:CTCCTCATCATCACTTTCCC |
|
|
| HIPK3 |
F:GTTGTGATACGGTGGATGG | 60 | 135 |
|
|
R:CTGGCTTGGTTTCTGTGTC |
|
|
| CTGF |
F:TACCAATGACAACGCCTCCTG | 60 | 208 |
|
|
R:GCCGTCGGTACATACTCCACA |
|
|
| TGFb3 |
F:GCTACTATGCCAACTTCTGCTCA | 60 | 224 |
|
|
R:GCTACATTTACAAGACTTCACCACC |
|
|
| GAPDH |
F:GGACCTGACCTGCCGTCTAG | 60 | 100 |
|
|
R:GTAGCCCAGGATGCCCTTGA |
|
|
All reactions were analyzed using the
2−ΔΔCq method (18).
Student's t-test was applied to statistically analyze the data and
P<0.05 was considered to indicate a statistically significant
difference. The values are expressed as the mean ± standard
deviation. The data was analyzed by SPSS19 (IBM Corp., Armonk, NY,
USA).
Co-expression networks and ceRNA
network analysis
The expression levels of the lncRNAs/circRNAs and
mRNAs were analyzed to elucidate any meaningful association. The
sequences of lncRNAs/circRNAs and mRNAs were searched by Cytoscape
(www.cytoscape.org/) and the present
study aimed to discover their potential miRNA response elements.
The overlapping of the same miRNA-binding site on both
lncRNA/circRNA and mRNA may aid the prediction of the
lncRNA/circRNA-miRNA-mRNA interactions. The lncRNA-miRNA,
circRNA-miRNA, miRNA-mRNA interactions were predicted using miRanda
(www.microrna.org/), Targetscan (www.targetscan.org/) and psRobot (omicslab.genetics.ac.cn/psRobot/).
Results
Differential lncRNA, mRNA and circRNA
expression profiles, as determined by RNA-seq
High throughput sequencing is an efficient method
for studying the biological function of RNAs. Thousands of
transcripts were detected in HS and normal tissues by RNA-seq.
Among them, a total of 3,469 lncRNAs and 536 mRNAs were detected to
be significant differentially expressed (|fold change|>2.0,
P<0.05). Of these, 2,479 and 990 lncRNAs were upregulated and
downregulated, respectively. In addition, 345 and 191 mRNAs were
upregulated and downregulated in HS compared with in normal
tissues. Furthermore, the expression profiles included 3,171
circRNAs in HS and 4,519 in normal tissues respectively. A total of
11 circRNAs (fold change ≥2.0, P<0.05) were differentially
expressed between HS and normal tissues, 10 upregulated and 1
downregulated. In addition, a total of 644 lncRNAs exhibited a fold
change of ≥10, 493 of which were upregulated and 151 were
downregulated. AC022034.2 (fold change: 15,030,055) was the most
upregulated lncRNA. Meanwhile, the coding gene profile suggested
that 299 mRNAs had a fold change ≥10. Hierarchical clustering
demonstrated the distinguishable gene expression profiles of
lncRNA, circRNA and mRNA among the samples. These results suggested
that the expression profiles of lncRNAs, circRNAs and mRNAs in HS
tissues differed from those of normal skin tissues (Table IV; Fig. 1).
| Table IV.Significant differentially expressed
RNAs in hypertrophic scar tissues compared with the control group
(|fold change| >2.0, P<0.05). |
Table IV.
Significant differentially expressed
RNAs in hypertrophic scar tissues compared with the control group
(|fold change| >2.0, P<0.05).
| Category | Upregulated
genes | Downregulated
genes | Total
differentially expressed genes |
|---|
| mRNA | 345 | 191 | 536 |
| Long noncoding
RNA | 2,479 | 990 | 3,469 |
| Circular RNA | 10 | 1 | 11 |
All of these dysregulated RNAs were diffusely
distributed in all chromosomes, including sex chromosomes X and Y.
All forms of splicing transcripts were determined by StringTie
(ccb.jhu.edu/software/stringtie/index.shtml?t=manual#input),
mapped to the Ensembl database by gffcompare (ccb.jhu.edu/software/stringtie/gffcompare.shtml) and
novel transcripts were identified (Table V).
| Table V.Information regarding
transcripts. |
Table V.
Information regarding
transcripts.
| Group | Total
transcripts | Known
transcripts | Novel
transcripts |
|---|
| H | 119,518 | 26,160 | 93,358 |
| N | 190,765 | 29,093 | 161,672 |
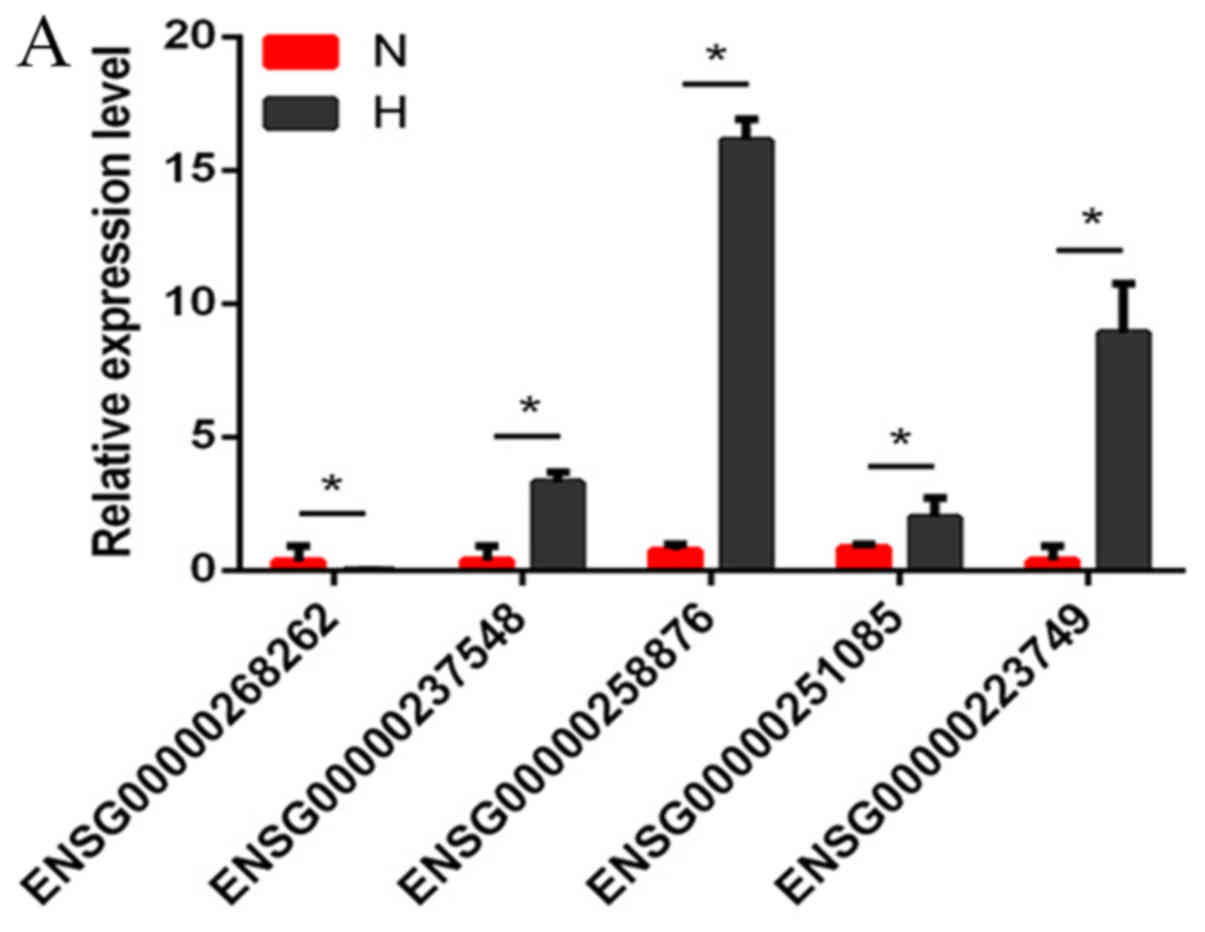
Validation with RT-qPCR
Several differentially expressed lncRNAs, circRNAs
and mRNAs were randomly selected and RT-qPCR was used to verify the
RNA-seq results. The results demonstrated that the fold changes
observed in the sequencing analysis were consistent with the
sequencing assay (Fig. 2A-C;
Tables VI–VIII).
| Table VI.Comparison of RT-qPCR data with
RNA-seq data for lncRNAs. |
Table VI.
Comparison of RT-qPCR data with
RNA-seq data for lncRNAs.
|
| ENSG0000 | ENSG0000 | ENSG0000 | ENSG0000 | ENSG0000 |
|---|
| lncRNA | 0251085 | 0223749 | 0258876 | 0237548 | 0268262 |
| RT-qPCR (H/N) | 2.20 |
8.14 | 15.42 |
6.78 | −6.84 |
| RNA-seq (H/N) | 5.48 | 192.80 | 21.11 | 54.27 | −13.05 |
| Table VIII.Comparison of RT-qPCR data with
RNA-seq data for mRNA. |
Table VIII.
Comparison of RT-qPCR data with
RNA-seq data for mRNA.
| mRNA | COL1A1 | ASXL1 | CTGF | TGFβ3 | HIPK3 |
|---|
| RT-qPCR (H/N) |
8.65 | 12.86 | 20.39 | 15.99 | 4.32 |
| RNA-seq (H/N) | 69.23 |
2.17 | 21.19 | 17.19 | 0.95 |
GO and KEGG pathway analyses
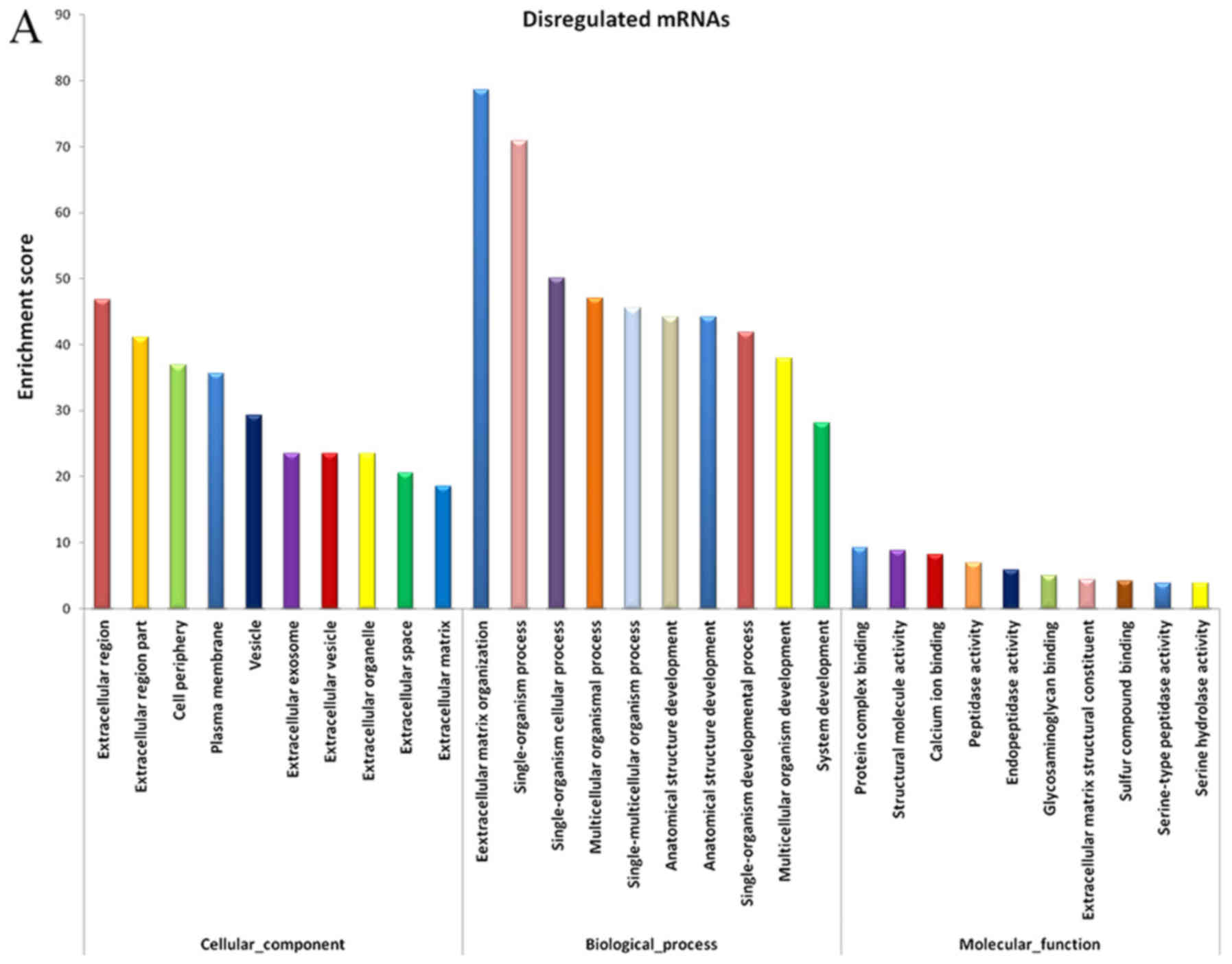
The evidently abnormal mRNAs were selected for
functional enrichment analysis. The results demonstrated that
certain mRNAs had important roles in various biological processes,
including extracellular matrix (ECM) organization, collagen
metabolic process, biological adhesion, negative regulation of
cellular response to growth factor stimulus and skin development
(Fig. 3A).
It is well known that lncRNAs influence DNA
sequences by regulating the neighboring and overlapping coding gene
expression levels (19).
Therefore, the functions of lncRNAs may be reflected in nearby
genes. GO enrichment analysis of the notably differentially
expressed mRNAs in the vicinity of the lncRNAs may help to reveal
the function of these lncRNAs. The data from the present study
suggested that the dysregulated mRNAs and lncRNAs were associated
with biological functions including regulation of collagen
metabolic process, regulation of transcription and DNA-template,
and ECM organization. The majority of these processes were
associated with ECM deposition, cell proliferation and gene
expression (Fig. 3B).
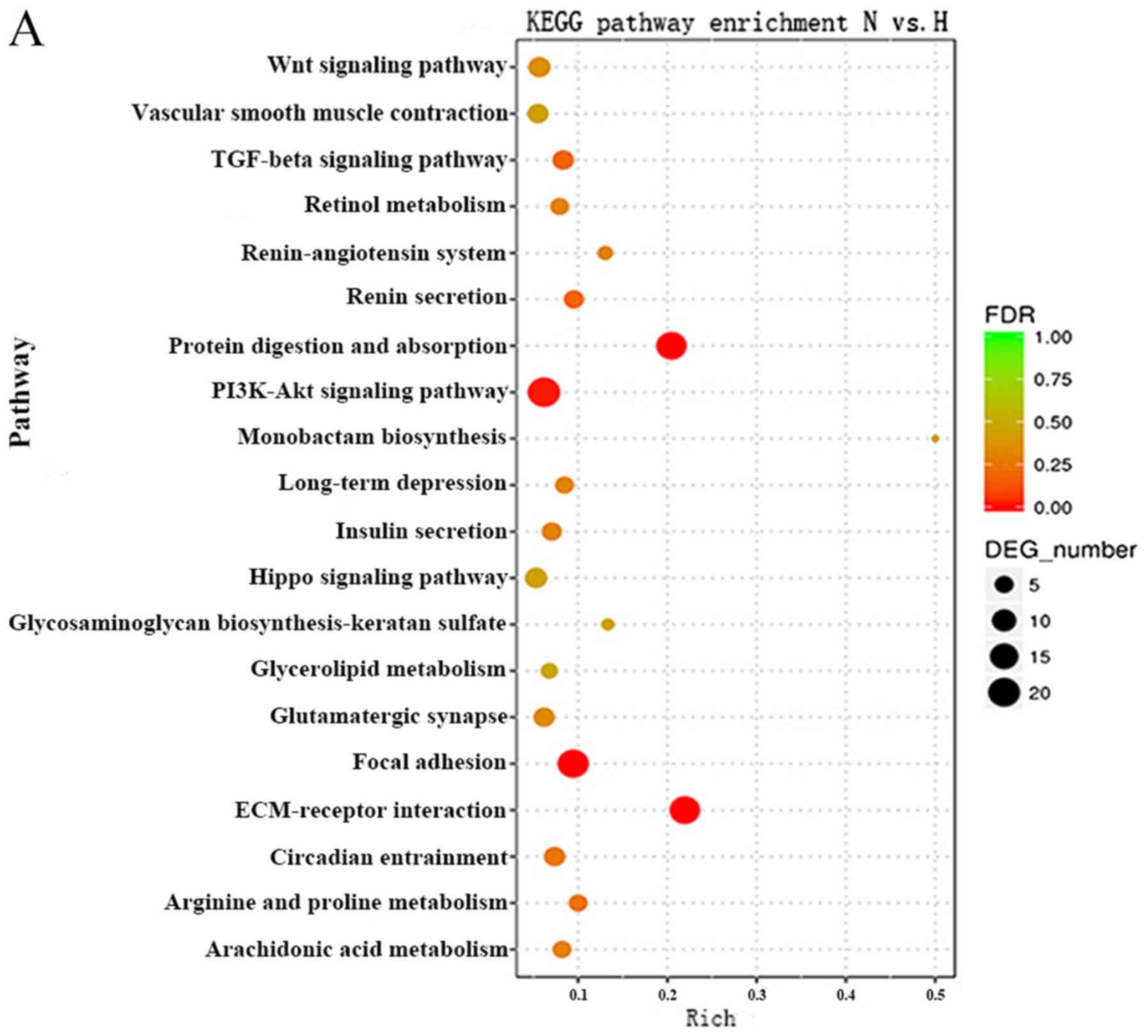
KEGG pathway analysis can reveal molecular
interactions and the pathways associated with genes. Accordingly,
the biological pathways associated with the significantly
differentially expressed RNAs were investigated. The P-value cutoff
was 0.05. The data demonstrated that the pathways associated with
the mRNAs were mainly involved in protein digestion and absorption,
the phosphoinositide 3-kinase (PI3K)-AKT serine/threonine kinase
(Akt) signaling pathway, focal adhesion and the TGFβ signaling
pathway (Fig. 4A). The Forkhead
box (Fox)O signaling pathway, prolactin signaling pathway and the
TGFβ signaling pathway were top pathways associated with the
lncRNAs (Fig. 4B). These results
suggested that these pathways may significantly contribute to the
pathophysiology and development of HS.
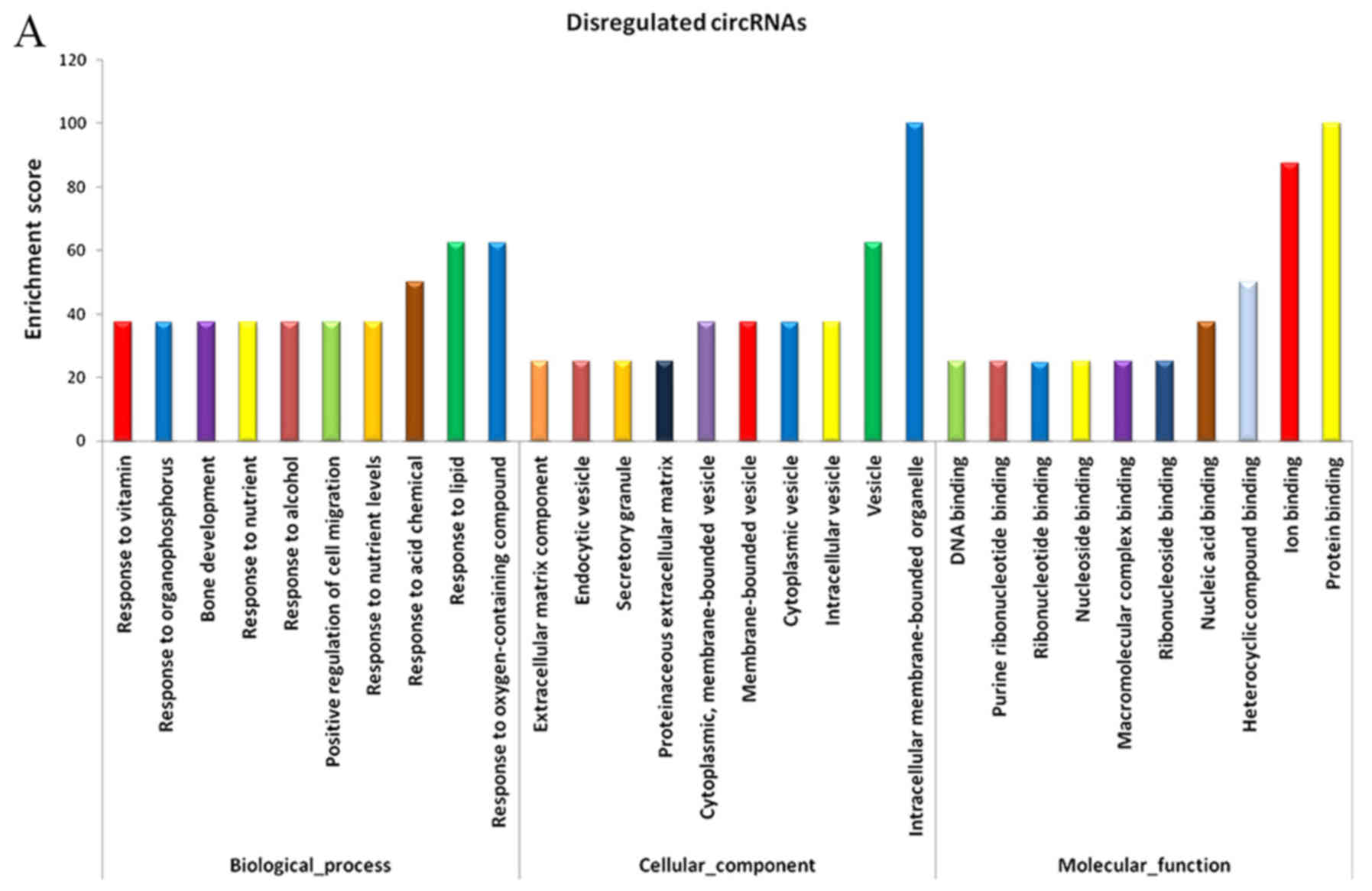
As miRNA sponges, circRNAs can regulate the function
of miRNAs. Differentially expressed circRNAs, and the parental gene
transcripts, were studied to determine their biological effects.
Compared with the adjacent normal skin tissues, GO analysis
revealed that the differentially overexpressed circRNAs in HS were
associated with ECM component, response to nutrient and bone
development, among others (Fig.
5A). Furthermore, KEGG analysis demonstrated that dysregulated
circRNAs were associated with the PI3K-Akt signaling pathway,
ECM-receptor interaction, and protein digestion and absorption
(Fig. 5B). Based on these data,
the biological functions of these circRNAs may contribute to the
development of HS.
Co-expression networks of lncRNA-mRNA
and circRNA-mRNA, and functional prediction
Thus far, the functions of lncRNAs and circRNAs have
yet to be annotated; The prediction of lncRNAs or circRNAs function
mainly depend on the annotation of the co-expressed mRNAs To obtain
the core lncRNAs and circRNAs associated with fibroblast
hyperplasia, significantly associated pairs of genes were selected
to build the coding-noncoding gene co-expression (CNC) network, in
order to identify the interactions and significance among the
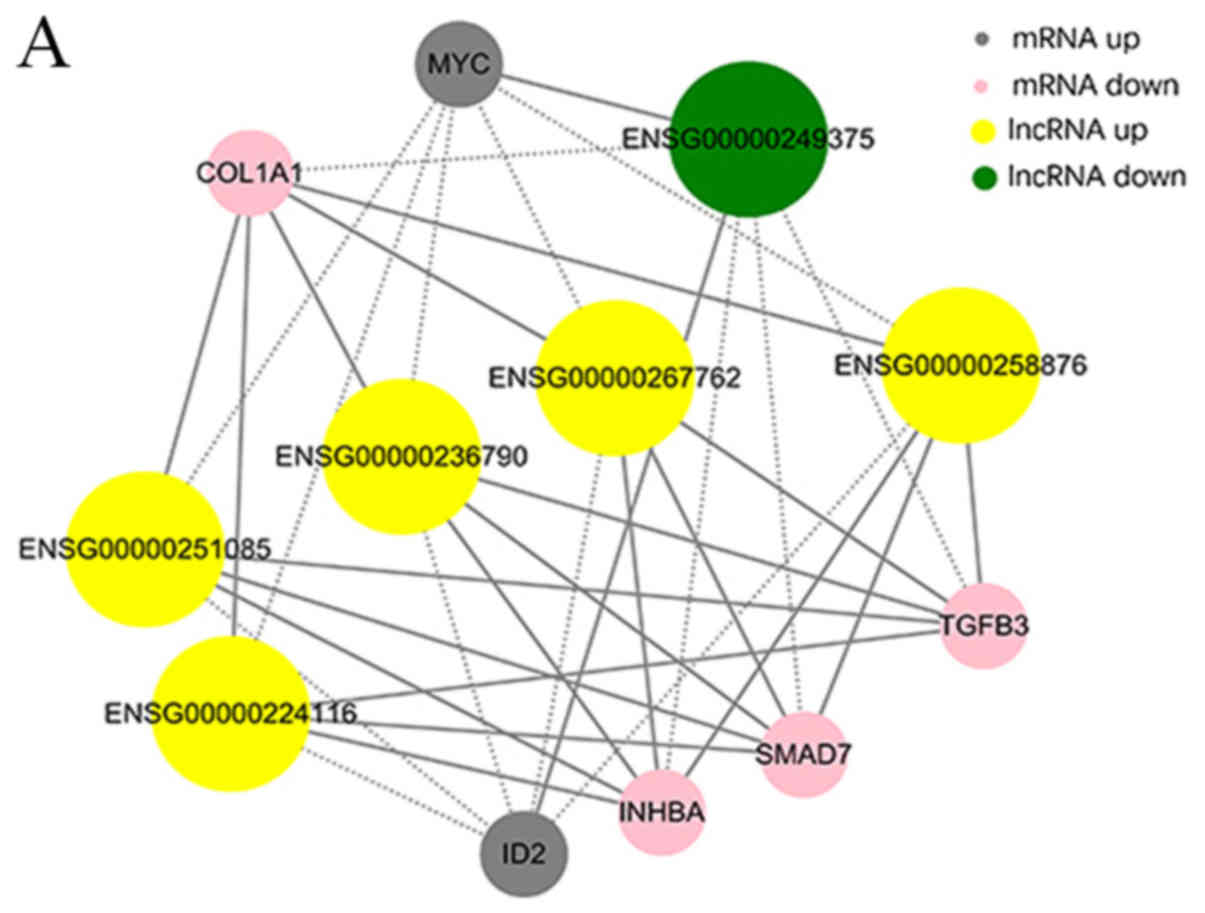
differentially expressed lncRNAs, circRNAs and mRNAs (Fig. 6A and B).
The co-expression networks were associated with
numerous biological processes, including cell proliferation,
adhesion, metastasis and differentiation. The network demonstrated
that lncRNAs AC048380.1 (ENSG00000267762) and LINC00299
(ENSG00000236790) were associated with metastasis-related genes,
including inhibin subunit βA (INHBA), SMAD family member 7 (SMAD7),
collagen type I A1 chain (COL1A1), TGFβ3 and MYC proto-oncogene,
bHLH transcription factor (MYC). Inhibitor of DNA binding 2 (ID2)
was associated with lncRNA cancer susceptibility 11 (CASC11,
ENSG00000249375), TGFβ3-antisense RNA 1 (AS1; ENSG00000258876),
INHBA-AS1 (ENSG00000224116), AC048380.1 (ENSG00000267762),
LINC00299 (ENSG00000236790) and LINC01969 (ENSG00000251085). In
addition, the circRNA-mRNA co-expression network suggested that
circ-Chr17:50187014_50195976_-, circ-Chr17:50189167_50194626_-,
circ-Chr17:50189167_ 50198002_- and circ-Chr17:50189858_50195330_-
were also associated with INHBA, SMAD7, COL1A1, TGFβ3 and MYC.
COL1A1 and TGFβ3 were associated with
circ-Chr9:125337017_125337591_+ and
circ-Chr12:120782654_120784593_-. The co-expression networks
demonstrated that one lncRNA, circRNA or mRNA may be associated
with one or more mRNA, lncRNA or circRNA. This finding may aid the
search for the regulation between lncRNAs, circRNAs and mRNAs
implicated in HS.
Construction of a ceRNA network
According to the ceRNA hypothesis, numerous lncRNAs,
circRNAs and mRNAs may function as ceRNAs, which compete for the
same MREs and regulate each other. Analysis of ceRNA interactions
aids the functional characterization of such noncoding transcripts.
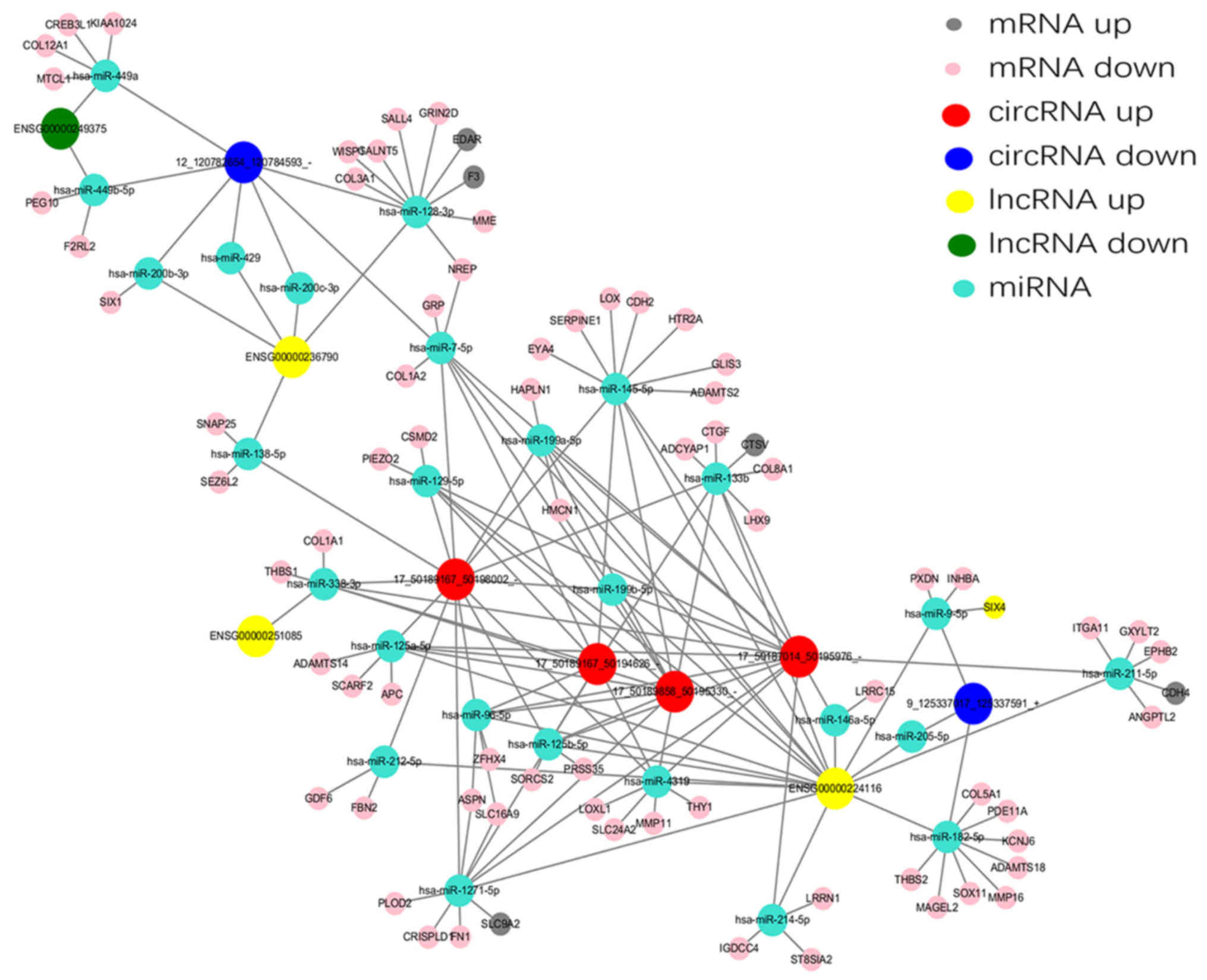
A ceRNA network associated with HS was established in the present
study using the high-throughput sequencing data (Fig. 7). Five differentially expressed
lncRNAs and six differentially expressed circRNAs, sharing a common
MRE site were selected. For example, INHBA-AS1 and
circ-Chr9:125337017_125337591_+ were ceRNAs of miR-182-5p targeting
potassium voltage-gated channel subfamily J member 6, ADAM
metallopeptidase with thrombospondin type 1 motif 18, SRY-box 11,
MAGE family member L2, matrix metallopeptidase 16, thrombospondin
(THBS)2, phosphodiesterase 11A and collagen type V A1 chain. In
addition, LINC01969 and circ-Chr17: 50187014_50195976_-,
circ-Chr17:50189167_50194626_-, circ-Chr17: 50189167_50198002_- and
circ-Chr17: 50189858_50195330_- were ceRNAs of miR-338-3p targeting
THBS1 and COL1A1. These RNA interactions may supply novel insights
into the mechanisms underlying HS.
Discussion
It has previously been reported that dysregulated
lncRNAs serve an important role in tumorigenesis and tumor
progression, and may be used as potential prognostic biomarkers
(20). Recently, circRNAs have
also been considered to possess this potential function in cancer
(21). HS are characterized by
excessive deposition of ECM molecules and the invasive growth of
fibroblasts. Although it remains a benign disease, the fibroblasts
express malignant features (22);
therefore, it has been speculated that ncRNAs may serve an
important role in HS. Existing studies have revealed that miRNAs
serve a key role in HS (7,23). Previous studies reported that
miR-21 promotes fibrosis via modulating the expression of TGFβ and
SMAD7 at the post-transcriptional level (24,25).
In addition, whether lncRNA8975-1 is overexpressed in HS tissues
and dermal fibroblasts has been investigated, and it has been
demonstrated that overexpression of lncRNA8975-1 inhibits cell
proliferation and reduces the protein expression levels of collagen
type I A2 chain, COL1A1, collagen type III A1 chain and α-smooth
muscle actin in HS fibroblasts (26). These data suggested that the
expression of ncRNAs undergoes significant alterations in HS, which
may be closely associated with the development of HS. However, to
the best of our knowledge, lncRNA and circRNA expression profiles,
and their associated co-expression and ceRNA networks, have not
been fully elucidated in HS.
In the present study, the expression profiles of
lncRNAs, circRNAs and mRNAs were evaluated in HS and normal skin
using RNA-seq, after which, CNC and ceRNA networks were
constructed. The RNA-seq analysis demonstrated that a total of
3,649 lncRNAs, 536 mRNAs and 11 circRNAs were differentially
expressed between the studied groups. Hierarchical clustering
demonstrated that these differentially expressed genes were
distinguishable. Functional annotation of the DEGs highly expressed
in HS was shown. A total of 77 mRNAs were associated with ECM
organization, 26 mRNAs were associated with collagen metabolic
process; 693 lncRNAs were associated with the regulation of gene
expression, and 591 lncRNAs were associated with nucleic
acid-templated transcription; 4 circRNAs were associated with
regulation of the RNA metabolic process and 2 circRNAs were
associated with ECM organization.
KEGG pathway analysis demonstrated that the greatest
number of genes was associated with pathways involved in the
accumulation of ECM components, including the TGFβ signaling
pathway, the PI3K-Akt signaling pathway and ECM receptor
interaction. These genes may serve important roles during cell
proliferation, differentiation, metastasis and apoptosis, all of
which are associated with the stages of wound repair, inflammation,
formation of new tissues, remodeling and formation of HS.
Previous studies have revealed that lncRNA H19 is
aberrantly regulated in a wide range of cancers (27,28).
The data from the present study demonstrated that H19 was markedly
higher compared with in normal skin. According to previous studies,
H19 mediates the expression of genes involved in invasion,
promotion of epithelial-mesenchymal transition (EMT) and Wnt
signaling (29), and promotes cell
migration by acting as a sponge for miR-138 and miR-200a (30). In addition, the PI3K-Akt pathway
has been reported to be of importance in EMT and wound healing
(31,32), serving important roles in
inflammation, angiogenesis, re-epithelialization and connective
tissue regeneration.
The data from the present study also detected
significant increases in INHBA-AS1, TGFβ3-AS1, LINC00299,
AC048380.1 and AC037198.1 expression, and decreases in CASC11,
AC012065.4 and AC016866.3 expression in HS tissues. These RNAs are
associated with the TGFβ signaling pathway. Numerous studies
(33,34) have focused on analysis of the
expression patterns of lncRNAs and their potential crosstalk with
adjacent protein-coding genes (PCGs). INHBA is a ligand belonging
to the TGFβ superfamily, which promotes cell proliferation and
metastasis (35,36). INHBA-AS1 has also been revealed to
be increased in gastric cancer and chronic kidney disease (37,38).
The data from the present study indicated that the expression of
INHBA-AS1 was increased in HS. As an antisense lncRNA, INHBA-AS1
may influence the expression of its nearby PCGs through various
mechanisms.
In the present study, the nearest PCGS was about 100
kb from LINC00299, and identified ID2 as a potential, corresponding
cis-regulated target gene. Introduction of the ID2 gene results in
Akt phosphorylation and negative regulation of cell
differentiation. In terms of cell proliferation, the ID2 protein
induces a malignant phenotype (39,40).
Studies showed that ID2 is a negative regulator of EMT.
Downregulated ID2 might associated with EMT such as cancer and
fibrosis (41,42). To further investigate the
mechanisms underlying LINC00299, a CNC co-expression network was
constructed and 292 PCGs were identified to be associated with it.
The majority of these genes were involved in cell proliferation,
differentiation and metastasis. These findings suggested that
LINC00299 may be an important regulator in HS via its co-expressed
genes. In addition, certain major mRNAs were associated with
different lncRNAs, including AC048380.1, LINC01969 and CASC11.
Therefore, it was hypothesized that these lncRNAs may be associated
with the pathophysiology of HS by regulating co-expressed
genes.
Functional pathway enrichment analysis was performed
on the co-expressed genes of lncRNAs. The results indicated that
these lncRNAs were involved in signaling pathways including FoxO,
TGFβ and Hippo, all of which serve pivotal roles in regulating cell
growth, proliferation, differentiation and apoptosis, and mediate
the regulatory effects of cells in response to ECM adhesion
(43–45). These results provided further
insight into the molecular mechanisms of action underlying these
lncRNAs.
circRNAs have been reported to be involved in
transcriptional and post-transcriptional gene expression
regulation. They are enriched with functional miRNA binding sites
and work as miRNA sponges. For example, circZNF91 contains 24
target sites for miR-23b-3p, which is known to serve important
roles in keratinocyte differentiation (13). Furthermore, circRNA_100782 can
regulate BxPC3 cell proliferation by acting as a miR-124 sponge via
the interleukin 6-signal transducer and activator of transcription
3 pathway (14). The present study
performed a GO analysis on the dysregulated circRNAs in HS to
analyze their functions. The results suggested that certain
biological processes and molecular functions may be involved in the
development of HS. KEGG pathway analysis for the differentially
expressed circRNAs identified six pathways including PI3K-Akt
signaling pathway, protein digestion and absorption, gap junction,
platelet activation, AMPK signaling pathway, which may serve
important roles during the wound repair stages, inflammation,
formation of new tissues and remodeling, and may be strongly
associated with the mechanisms underlying HS.
The concept of ceRNA indicates that RNA molecules
harboring MREs can communicate with each other by competing for the
same miRNA. Understanding this novel RNA crosstalk may provide a
novel perspective to account for the function of uncharacterized
ncRNAs involved in various types of disease (46). Prediction of ceRNA crosstalk is
dependent on the identification of MREs on the relevant transcripts
of interest. Previous studies (15,47)
have reported that ceRNAs are widely implicated in various
biological processes and associated with various diseases,
including cancer. However, to the best of our knowledge, there are
currently no studies regarding the functional roles of this novel
RNA crosstalk in HS. In the present study, a ceRNA network of HS
was constructed based on RNA-seq data. This network indicated the
lncRNA-miRNA-circRNA-mRNA associations in HS, and included four
lncRNAs, six circRNAs, 72 mRNAs and 24 miRNAs. It has previously
been reported that miR-145 serves an important role in the
differentiation and function of skin myofibroblasts (48), which may participate in the
pathophysiology of HS.
With the development of RNA-seq, it has been
revealed that ncRNAs serve a key role in the occurrence and
development of disease. To date, the functions of the majority of
lncRNAs and circRNAs are not well understood. The present study
demonstrated that numerous ncRNAs, including lncRNAs and circRNAs,
were markedly differentially expressed in HS compared with in
normal skin. Although the results of the present study require
further experimental verification, the profile of these
dysregulated lncRNAs and circRNAs may aid identification of
prospective clinical markers, and contribute to the understanding
of the pathophysiology and development of HS.
Acknowledgements
Authors would like to thank Dr Shangfeng Fu from the
First Affiliated Hospital of Nanchang University for cell culture
support, and Professor Jianhua Fu from the First Affiliated
Hospital of Nanchang University for providing samples of
hypertrophic scar and normal skin tissue for the present study.
Funding
The present study was supported by grants from the
National Natural Science Foundation of China (grant nos. 81060323
and 81460293).
Availability of data and materials
The datasets used during the current study are
available from the corresponding author on reasonable request.
Authors' contributions
ML and DL designed the experiments. ML, JW and HH
performed the experimental work. JW also participated in the data
analysis. ML wrote the manuscript with help from JW and DL. All
authors read and approved the final version of the manuscript.
Ethics approval and consent to
participate
The study procedures were approved by the Ethics
Committee of the First Affiliated Hospital Nanchang University.
Written informed consent was obtained from all subjects.
Patient consent for publication
Identifying information, including names, initials,
date of birth or hospital numbers, images or statements did not be
included in this manuscript. The patients consented for the
publication of the associated data and accompanying images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Zhu Z, Ding J, Shankowsky HA and Tredget
EE: The molecular mechanism of hypertrophic scar. J Cell Commun
Signal. 7:239–252. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Curran TA and Ghahary A: Evidence of a
role for fibrocyte and keratinocyte-like cells in the formation of
hypertrophic scars. J Burn Care Res. 34:227–231. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Butzelaar L, Soykan EA, Garre Galindo F,
Beelen RH, Ulrich MM, Niessen FB and van der Molen Mink AB: Going
into surgery: Risk factors for hypertrophic scarring. Wound Repair
Regen. 23:531–537. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen L and Li J, Li Q, Yan H, Zhou B, Gao
Y and Li J: Non-coding RNAs: The new insight on hypertrophic scar.
J Cell Biochem. 118:1965–1968. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Arnvig KB, Cortes T and Young DB:
Noncoding RNA in Mycobacteria. Microbiol Spectr. 2:2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fatica A and Bozzoni I: Long non-coding
RNAs: New players in cell differentiation and development. Nat Rev
Genet. 15:7–21. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li P, He QY and Luo CQ: Overexpression of
miR-200b inhibits the cell proliferation and promotes apoptosis of
human hypertrophic scar fibroblasts in vitro. J Dermatol.
41:903–911. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mehra M and Chauhan R: Long noncoding RNAs
as a key player in hepatocellular carcinoma. Biomark Cancer.
9:1179299X–17737301X. 2017. View Article : Google Scholar
|
|
9
|
Rühle F and Stoll M: Long non-coding RNA
databases in cardiovascular research. Genomics Proteomics
Bioinformatics. 14:191–199. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li X, Wu Z, Fu X and Han W: lncRNAs:
Insights into their function and mechanics in underlying disorders.
Mutat Res Rev Mutat Res. 762:1–21. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Herter EK and Xu Landén N: Non-coding
RNAs: New players in skin wound healing. Adv Wound Care (New
Rochelle). 6:93–107. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Haque S and Harries LW: Circular RNAs
(circRNAs) in health and disease. Genes (Basel). 8:E3532017.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kristensen LS, Okholm TLH, Venø MT and
Kjems J: Circular RNAs are abundantly expressed and upregulated
during human epidermal stem cell differentiation. RNA Biol.
15:280–291. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen G, Shi Y, Zhang Y and Sun J:
CircRNA_100782 regulates pancreatic carcinoma proliferation through
the IL6-STAT3 pathway. Onco Targets Ther. 10:5783–5794. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li S, Chen X, Liu X, Yu Y, Pan H, Haak R,
Schmidt J, Ziebolz D and Schmalz G: Complex integrated analysis of
lncRNAs-miRNAs-mRNAs in oral squamous cell carcinoma. Oral Oncol.
73:1–9. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Didion JP, Martin M and Collins FS:
Atropos: Specific, sensitive, and speedy trimming of sequencing
reads. Peer J. 5:e37202017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang L, Feng Z, Wang X and Zhang X:
DEGseq: An R package for identifying differentially expressed genes
from RNA-seq data. Bioinformatics. 26:136–138. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Adnan M, Morton G and Hadi S: Analysis of
rpoS and bolA gene expression under various stress-induced
environments in planktonic and biofilm phase using 2(−∆∆CT) method.
Mol Cell Biochem. 357:275–282. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ørom UA, Derrien T, Beringer M, Gumireddy
K, Gardini A, Bussotti G, Lai F, Zytnicki M, Notredame C, Huang Q,
et al: Long noncoding RNAs with enhancer-like function in human
cells. Cell. 143:46–58. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhong Y, Chen Z, Guo S, Liao X, Xie H,
Zheng Y, Cai B, Huang P, Liu Y, Zhou Q, et al: TUG1, SPRY4-IT1, and
HULC as valuable prognostic biomarkers of survival in cancer: A
PRISMA-compliant meta-analysis. Medicine (Baltimore). 96:e85832017.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Han YN, Xia SQ, Zhang YY, Zheng JH and Li
W: Circular RNAs: A novel type of biomarker and genetic tools in
cancer. Oncotarget. 8:64551–64563. 2017.PubMed/NCBI
|
|
22
|
Liu BH, Chen L, Li SR, Wang ZX and Cheng
WG: Smac/DIABLO regulates the apoptosis of hypertrophic scar
fibroblasts. Int J Mol Med. 32:615–622. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kwan P, Ding J and Tredget EE: MicroRNA
181b regulates decorin production by dermal fibroblasts and may be
a potential therapy for hypertrophic scar. PLoS One.
10:e01230542015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li G, Zhou R, Zhang Q, Jiang B, Wu Q and
Wang C: Fibroproliferative effect of microRNA-21 in hypertrophic
scar derived fibroblasts. Exp Cell Res. 345:93–99. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhou R, Zhang Q, Zhang Y, Fu S and Wang C:
Aberrant miR-21 and miR-200b expression and its pro-fibrotic
potential in hypertrophic scars. Exp Cell Res. 339:360–366. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li J, Chen L, Cao C, Yan H, Zhou B, Gao Y,
Li Q and Li J: The long non-coding RNA LncRNA8975-1 is upregulated
in hypertrophic scar fibroblasts and controls collagen expression.
Cell Physiol Biochem. 40:326–334. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Huang C, Cao L, Qiu L, Dai X, Ma L, Zhou
Y, Li H, Gao M, Li W, Zhang Q, et al: Upregulation of H19 promotes
invasion and induces epithelial-to-mesenchymal transition in
esophageal cancer. Oncol Lett. 10:291–296. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhou X, Yin C, Dang Y, Ye F and Zhang G:
Identification of the long non-coding RNA H19 in plasma as a novel
biomarker for diagnosis of gastric cancer. Sci Rep. 5:115162015.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Weidle UH, Birzele F, Kollmorgen G and
Ruger R: Long non-coding RNAs and their role in metastasis. Cancer
Genomics Proteomics. 14:143–160. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liang WC, Fu WM, Wong CW, Wang Y, Wang WM,
Hu GX, Zhang L, Xiao LJ, Wan DC, Zhang JF and Waye MM: The lncRNA
H19 promotes epithelial to mesenchymal transition by functioning as
miRNA sponges in colorectal cancer. Oncotarget. 6:22513–22525.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yeh YH, Wang SW, Yeh YC, Hsiao HF and Li
TK: Rhapontigenin inhibits TGF-β-mediated epithelialmesenchymal
transition via the PI3K/AKT/mTOR pathway and is not associated with
HIF-1alpha degradation. Oncol Rep. 35:2887–2895. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Baek SH, Ko JH, Lee JH, Kim C, Lee H, Nam
D, Lee J, Lee SG, Yang WM, Um JY, et al: Ginkgolic acid inhibits
invasion and migration and TGF-beta-induced EMT of lung cancer
cells through PI3K/Akt/mTOR inactivation. J Cell Physiol.
232:346–354. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu T, Zhang X, Yang YM, Du LT and Wang
CX: Increased expression of the long noncoding RNA CRNDE-h
indicates a poor prognosis in colorectal cancer, and is positively
correlated with IRX5 mRNA expression. Onco Targets Ther.
9:1437–1448. 2016.PubMed/NCBI
|
|
34
|
Ding LJ, Li Y, Wang SD, Wang XS, Fang F,
Wang WY, Lv P, Zhao DH, Wei F and Qi L: Long noncoding RNA
lncCAMTA1 promotes proliferation and cancer stem cell-like
properties of liver cancer by inhibiting CAMTA1. Int J Mol Sci.
17:E16172016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lee HY, Li CC, Huang CN, Li WM, Yeh HC, Ke
HL, Yang KF, Liang PI, Li CF and Wu WJ: INHBA overexpression
indicates poor prognosis in urothelial carcinoma of urinary bladder
and upper tract. J Surg Oncol. 111:414–422. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Katayama Y, Oshima T, Sakamaki K, Aoyama
T, Sato T, Masudo K, Shiozawa M, Yoshikawa T, Rino Y, Imada T and
Masuda M: Clinical significance of INHBA gene expression in
patients with gastric cancer who receive curative resection
followed by adjuvant S-1 chemotherapy. In Vivo. 31:565–571. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Smyth LJ, McKay GJ, Maxwell AP and
McKnight AJ: DNA hypermethylation and DNA hypomethylation is
present at different loci in chronic kidney disease. Epigenetics.
9:366–376. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ke D, Li H, Zhang Y, An Y, Fu H, Fang X
and Zheng X: The combination of circulating long noncoding RNAs
AK001058, INHBA-AS1, MIR4435-2HG, and CEBPA-AS1 fragments in plasma
serve as diagnostic markers for gastric cancer. Oncotarget.
8:21516–21525. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Talkowski ME, Maussion G, Crapper L,
Rosenfeld JA, Blumenthal I, Hanscom C, Chiang C, Lindgren A,
Pereira S, Ruderfer D, et al: Disruption of a large intergenic
noncoding RNA in subjects with neurodevelopmental disabilities. Am
J Hum Genet. 91:1128–1134. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kamata YU, Sumida T, Kobayashi Y, Ishikawa
A, Kumamaru W and Mori Y: Introduction of ID2 enhances invasiveness
in ID2-null oral squamous cell carcinoma cells via the SNAIL axis.
Cancer Genomics Proteomics. 13:493–497. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wen XF, Chen M, Wu Y, Chen MN, Glogowska
A, Klonisch T and Zhang GJ: Inhibitor of DNA binding 2 inhibits
epithelial-mesenchymal transition via up-regulation of Notch3 in
breast cancer. Transl Oncol. 11:1259–1270. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Gervasi M, Bianchi-Smiraglia A, Cummings
M, Zheng Q, Wang D, Liu S and Bakin AV: JunB contributes to Id2
repression and the epithelial-mesenchymal transition in response to
transforming growth factor-β. J Cell Biol. 196:589–603. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mahajan SG, Fender AC, Meyer-Kirchrath J,
Kurt M, Barth M, Sagban TA, Fischer JW, Schrör K, Hohlfeld T and
Rauch BH: A novel function of FoxO transcription factors in
thrombin-stimulated vascular smooth muscle cell proliferation.
Thromb Haemost. 108:148–158. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wennmann DO, Vollenbröker B, Eckart AK,
Bonse J, Erdmann F, Wolters DA, Schenk LK, Schulze U, Kremerskothen
J, Weide T and Pavenstädt H: The Hippo pathway is controlled by
Angiotensin II signaling and its reactivation induces apoptosis in
podocytes. Cell Death Dis. 5:e15192014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Li X, Zhang L, Yin X, Gao Z, Zhang H, Liu
X, Pan X, Li N and Yu Z: Retinoic acid remodels extracellular
matrix (ECM) of cultured human fetal palate mesenchymal cells
(hFPMCs) through down-regulation of TGF-β/Smad signaling. Toxicol
Lett. 225:208–215. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Tay Y, Rinn J and Pandolfi PP: The
multilayered complexity of ceRNA crosstalk and competition. Nature.
505:344–352. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Sui J, Li YH, Zhang YQ, Li CY, Shen X, Yao
WZ, Peng H, Hong WW, Yin LH, Pu YP and Liang GY: Integrated
analysis of long non-coding RNA associated ceRNA network reveals
potential lncRNA biomarkers in human lung adenocarcinoma. Int J
Oncol. 49:2023–2036. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Gras C, Ratuszny D, Hadamitzky C, Zhang H,
Blasczyk R and Figueiredo C: miR-145 contributes to hypertrophic
scarring of the skin by inducing myofibroblast activity. Mol Med.
21:296–304. 2015. View Article : Google Scholar : PubMed/NCBI
|