Introduction
Tissue and organ fibrosis are the major causes of
disability and various life threatening diseases (1). Fibrotic diseases are typically
characterized by a progressive, harmful cycle of abnormally high
myofibroblast accumulation (2).
Myofibroblasts express high levels of α-smooth muscle actin (α-SMA)
under profibrogenic factor stimulation; however, their precise
origin remains to be fully elucidated (1). It was widely reported that
myofibroblasts may be derived from resident fibroblasts,
epithelial-mesenchymal transition and fibrocytes in
fibroproliferative disorders (3,4).
However, studies using genetic lineage tracing technology have
reported that mesenchymal stem cells (MSCs) and MSC-like cells may
be involved in myofibroblast generation during the development of
fibrosis (2,5,6).
Stem cell biology is becoming increasingly important
for use as novel therapies for several incurable chronic diseases,
including fibrotic disease; it has been reported that MSC
transplantation may attenuate organ fibrosis (7). However, a number of previous studies
have raised safety concerns regarding MSC transplantation, as
intrahepatic injection of human bone marrow (BM)-MSCs in mice has
been shown to contribute to myofibroblast formation, demonstrating
that MSCs can give rise to myofibroblasts in vivo following
injury (8,9). The information to date is
controversial as different studies have demonstrated either the
therapeutic or contributing effects of MSCs to organ fibrosis
(2). LeBleu et al (5) reported that up to 35% of renal
myofibroblasts are derived from BM-MSCs via the circulation. Tang
et al (10) revealed that
BM-MSCs are one of the major cell sources of myofibroblasts in the
fibrotic lung; ~40% of α-SMA-positive cells were derived from
BM-MSCs in the mouse lung following injury. Carlson et al
(11) reported that MSCs were a
source of scar forming myofibroblasts in heart fibrosis. Notably, a
previous report indicated that MSC-like cells have been implicated
in the myofibroblast formation of multiple organs during fibrosis
development, and up to 37–60% of the myofibroblasts in different
organs are derived from glioma-associated oncogene homolog
1+ MSC-like cells in mice (6). Preventing myofibroblast
differentiation has been demonstrated to attenuate organ fibrosis
(6,12). These results indicated that MSCs
and MSC-like cells may be the major cellular origins of organ
fibrosis, and demonstrated that these cells may be a relevant
therapeutic target to prevent solid organ dysfunction following
injury. Understanding the process and mechanism underlying
MSC-to-myofibroblast activation (fibrogenesis) is of particular
importance for the application of MSC therapies (13).
Transforming growth factor (TGF)-β1 is regarded as a
key regulator of myofibroblast differentiation in fibrosis
(14). TGF-β1 produces signals
through the TGF-β type I and type II receptors, and activates the
mothers against decapentaplegic (Smad) signaling pathway via the
phosphorylation of Smad2 and Smad3 (15). In addition, previous studies have
revealed that the TGF-β/Smad signaling pathway is closely
controlled by mitogen-activated protein kinase (MAPK) signaling
cascades, particularly extracellular signal-regulated kinase 1/2
(ERK1/2) (16). Previous studies
have also reported that TGF-β1 induced the transition of
fibroblasts from different sources to myofibroblasts in
vivo; however, its effect on the differentiation of MSCs into
myofibroblasts remains to be elucidated (16,17).
Tranilast [N-(3′,4′-dimethoxycinnamoyl)-anthranilic
acid] was originally developed as an antiallergic drug, used for
treating asthma, autoimmune diseases, and inhibiting angiogenesis
(18). Tranilast is also
considered to be an antiproliferative drug; previous studies have
investigated its applications against proliferative diseases,
particularly against hypertrophic scars and keloids (19,20).
Tranilast can reduce TGF-induced matrix production in different
types of cells, and also attenuates pathological fibrosis in the
kidneys and heart (21,22). Furthermore, in cultures of rat
cardiac fibroblasts, tranilast has been shown to attenuate
TGF-β1-stimulated fibrogenesis (23).
Previous studies have demonstrated that MSCs from
tracheal aspirates of premature infants and human adipose-derived
stem cells have the potential to differentiate into phenotypic
myofibroblasts under TGF-β1 stimulation (24,25).
Therefore, it was hypothesized that cultured rat MSCs may have the
potential to differentiate into myofibroblasts when induced by
TGF-β1, and tranilast may inhibit this process. The aim of the
present study was to evaluate the following hypotheses: i) TGF-β1
induces the differentiation of cultured rat MSCs into
myofibroblasts; ii) pretreating TGF-β1-induced cultured rat MSCs
with tranilast prevents myofibroblast differentiation and thus,
decreases collagen production; iii) tranilast inhibits
TGF-β1-mediated differentiation by inhibiting the Smad3 and ERK1/2
signaling pathways; and iv) tranilast inhibits cell proliferation
in TGF-β1-treated MSCs.
Materials and methods
Cell isolation and culture
A total of 10 Sprague-Dawley (SD) rats (specific
pathogen free, male, 3–4 weeks old, 60–100 g) were purchased from a
professional breeder (Hunan SJA Laboratory Animal Co., Ltd.,
Changsha, China). Animals were housed at 22±2°C and 50±10%
humidity, in a quiet environment with a 12/12 h light/dark cycle,
and were fed standard feed and water ad libitum. MSCs were
obtained from the femurs and tibias of the SD rats using an
established protocol (26). The
MSCs were cultured in low-glucose Dulbecco's modified Eagle's
medium (DMEM; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) with 10% fetal bovine serum (FBS; Biological Industries,
Kibbutz Beit-Haemek, Israel) and grown in plastic dishes to
confluence. The animal experiments were approved by the Animal
Ethics Committee and the Medical Ethics Committee of Hunan Normal
University (Changsha, China). The adherent cells were expanded as
monolayer cultures in a 5% CO2/95% O2 air
atmosphere at 37°C and the medium was replaced every 3 days. These
primary cells were referred to as passage 0. The confluent cells
were split with 0.25% trypsin and 0.01% EDTA at the ratio 1:2 or
1:3 every passage. When the MSCs reached 50–60% confluence, they
were cultured in DMEM containing 0.5% FBS at 37°C for 24 h, and the
majority of cells were in the quiescent state. Following this, the
cells were treated with or without tranilast (0.1, 1, and 10 µM;
Santa Cruz Biotechnology, Inc., Dallas, TX, USA) for 30 min at 37°C
and then exposed to TGF-β1 (10 ng/ml PeproTech, Inc., Rocky Hilly,
NJ, USA). MSCs cultured in 10% FBS + DMEM alone at 37°C were used
as a control.
For the differentiation assays, MSCs expanded for
passages 2–5 were cultured in differentiation media for 2–4 weeks
and images from at least six randomly chosen microscopic fields
were captured using an UOP DSZ2000 microscope (magnification, ×200;
Chongqing UOP Photoelectric Technology Co., Ltd., Chongqing,
China). For adipogenic differentiation, MSCs were cultured in DMEM
with 10% FBS, 1 mM dexamethasone, 0.5 mM isobutyl-1-methylxanthine,
100 mM indomethacin and 10 µg/ml insulin. After 14 days, cells were
fixed with 4% formaldehyde at room temperature for 10 min and
stained with 0.3% Oil Red O (Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany) at room temperature for 10 min to visualize lipid
droplets. Osteogenic differentiation was induced by DMEM with 10%
FBS, 10 nM dexamethasone, 10 mM β-glycerophosphate and 0.2 mM
ascorbic acid. After 28 days, the mineralization of the
extracellular matrix was determined by 1% Alizarin Red (pH 4.2;
Sigma-Aldrich; Merck KGaA) staining at 37°C for 30 min.
Chondrogenic differentiation was induced in pellet culture for
14–21 days and performed using SD rat MSC chondrogenic
differentiation medium (Cyagen Biosciences, Santa Clara, CA, USA)
according to the manufacturer's instructions, and chondrogenesis
was evaluated by 1% Alcian blue staining for 30 min at room
temperature.
Flow cytometric analysis of MSC
surface markers
Cells were harvested using 0.25% trypsin solution,
and following two washes in PBS, cells were single-stained with
fluorescent-labeled antibodies against cluster of differentiation
(CD)29 (cat no. 561796; 1:20), CD90 (cat no. 561973; 1:20),
CD-11b/c (cat no. 554862; 1:20) and CD45 (cat no. 554878; 1:20; all
BD Biosciences, San Jose, CA, USA) for 20 min at room temperature
in the dark. Subsequently, cells were resuspended in 300 µl PBS and
immediately analyzed using a flow cytometer (FC500; Beckman
Coulter, Inc., Brea, CA, USA). Data analysis was performed using
FACSDiva version 6.1.3 (BD Biosciences).
Immunocytochemical assay
The quiescent cells grown on a glass slide were
divided into five groups: i) Control (culture in 10% FBS + DMEM);
ii) TGF-β1 (10 ng/ml); iii) TGF-β1 + tranilast (0.1 µM); iv) TGF-β1
+ tranilast (1 µM); and v) TGF-β1 + tranilast (10 µM). The MSCs
were first either treated with or without tranilast for 30 min and
then exposed to TGF-β1 for 24 h at 37°C, with cells grown in 10%
FBS + DMEM alone as a control. The cells were fixed with 4%
paraformaldehyde for 15 min and permeabilized using 0.5% Triton
X-100 for 5 min. The cells were then treated with the 2-step plus
Poly-Horseradish Peroxidase Anti-Mouse/Rabbit Immunoglobulin G
Detection System (OriGene Technologies, Inc., Beijing, China).
Endogenous peroxide activity was quenched with 3%
H2O2 for 10 min, and cells were washed with
PBS. The cells were then incubated with a monoclonal antibody
against α-SMA (1:500, cat no. ab124964; Abcam, Cambridge, UK) for
12 h at 4°C. Following the addition of the 2-step agent, the cells
were incubated with 3,3′-diaminobenzidine at room temperature for 1
min. The cell nuclei were stained with hematoxylin. Brown color
staining was considered to indicate a positive result. Images were
captured using a fluorescence microscope (Olympus Corporation,
Tokyo, Japan).
Western blot analysis
Whole cell lysates were prepared in cell lysis
buffer (Wuhan Boster Biological Technology, Ltd., Wuhan, China)
supplemented with protease inhibitors (Nanjing KeyGen Biotech Co.,
Ltd., Nanjing, China) for 30 min. The protein concentration of
lysates was measured using a BCA protein assay (Beyotime Institute
of Biotechnology, Shanghai, China). Equal quantities (25 µg) of
protein were subjected to 10% SDS PAGE under reducing conditions
and electrotransferred onto a nitrocellulose membrane. The
membranes were blocked with 5% fat-free dry milk in TBS for 1 h at
room temperature and incubated overnight at 4°C with the following
primary antibodies: Rabbit monoclonal antibody against α-SMA
(1:1,000), mouse monoclonal antibody against collagen type I
(1:1,000; cat no. sc-59772; Santa Cruz Biotechnology, Inc.), and
rabbit polyclonal antibodies against Smad3 (1:500; cat no. YT4336;
ImmunoWay Biotechnology Company, Plano, TX, USA), phosphorylated
(p-)Smad3 (1:500; cat no. YP0585; ImmunoWay Biotechnology Company),
ERK1/2 (1:500; cat no. YT1623; ImmunoWay Biotechnology Company),
p-ERK1/2 (1:500; cat no. YP0100; ImmunoWay Biotechnology Company)
and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 1:500; cat no.
YT5052; ImmunoWay Biotechnology Company). Following washing three
times with TBS-T, membranes were incubated for 2 h at 37°C with
horseradish peroxidase-conjugated goat anti-rabbit (1:1,000; cat
no. A0208, Beyotime Institute of Biotechnology) or goat anti-mouse
(1:1,000; cat no. A0216; Beyotime Institute of Biotechnology)
immunoglobulin G secondary antibodies. The membranes were washed
with PBS and were exposed to ECL reagent (Beyotime Institute of
Biotechnology). The protein bands were quantified using Quantity
One software (version 4.6.3; Bio-Rad Laboratories, Inc., Hercules,
CA, USA) and normalized against the loading control, GAPDH.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
The quiescent cells were divided into four groups:
i) Control (10% FBS + DMEM); ii) tranilast (10 µM); iii) TGF-β1 (10
ng/ml); and iv) TGF-β1 + tranilast (10 µM). The cells were treated
with or without tranilast for 30 min, followed by the addition of
TGF-β1 for 24 h. Total RNA was isolated from cell cultures using
TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. cDNA was synthesized
using RNA (2 µg) with the RevertAid™ First Strand cDNA
Synthesis kit (Thermo Fisher Scientific, Inc.). The specific primer
sequences (Invitrogen; Thermo Fisher Scientific, Inc.) used were as
follows: α-SMA forward, 5′-TCCAGAGTCCAGCACAATACCAG-3′ and reverse,
5′-AATGACCCAGATTATGTTTGAGACC-3′; collagen type I forward,
5′-TGTTCGTGGTTCTCAGGGTAG-3′ and reverse,
5′-TTGTCGTAGCAGGGTTCTTTC-3′; and actin forward,
5′-CATCCTGCGTCTGGACCTGG-3′ and reverse, 5′-TAATGTCACGCACGATTTCC-3′.
The qPCR was performed using SYBRGreen PCR mix (ABI; Thermo Fisher
Scientific, Inc.). The qPCR conditions were as follows: 95°C for 10
min, followed by 40 cycles of 95°C for 10 sec and 55–59°C
(depending on the specific annealing temperatures of the primer
used) for 50 sec. The data were analyzed using the
2−ΔΔCq method (27) and
normalized to actin.
Measurement of collagen contents
The collagen contents of the MSC cultures were
measured using a hydroxyproline assay kit (Nanjing Jiancheng
Bioengineering Institute, Nanjing, China). The cells were cultured
to a quiescent state on 6-well plates, and were then divided into
five groups and treated as described above. The cells were then
exposed to TGF-β1 (10 ng/ml) for 48 h, using cells grown in 10%
FBS-DMEM as the control. The protein contents of the cell lysates
were measured using Coomassie reagent (Thermo Fisher Scientific,
Inc.). Data were recorded as µg/mg protein, assuming that collagen
contained an average of 13.5% hydroxyproline.
Cell viability assay
Cell viability was determined using a Cell Counting
kit-8 assay (CCK-8; Beyotime Institute of Biotechnology) according
to the manufacturer's protocol. The MSCs from passage three
(5×103/100 µl) were seeded on 96-well plates and
cultured at 37°C in 5% CO2 for 24 h, following which the
cells were made quiescent for 24 h. The cells were treated as
described above and then incubated for 24 h. Each well received 10
µl CCK-8 solution for 3 h. The optical density was measured at 450
nm using a microplate reader, with six samples for each group.
Statistical analysis
All experimental data are presented as the mean ±
standard deviation of at least three independent experiments and
were analyzed with SPSS 19.0 statistical software (IBM Corp.,
Armonk, NY, USA). Statistical analysis was performed using one-way
analysis of variance and Student-Newman-Keuls post hoc test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Identification of rat MSCs
Tri-lineage differentiation assays were performed;
the rat MSCs exhibited a fibroblast-like morphology, and possessed
osteogenic, adipogenic and chondrogenic differentiation capacity
in vitro. In addition, flow cytometric analysis demonstrated
that the majority of rat MSCs were positive for CD29 and CD90, but
negative for CD45 and CD-11b/c. These assays confirmed the rat MSC
differentiation potential as well as reliability of the isolation
and culture method (data not shown).
Tranilast attenuates the
TGF-β1-induced transformation of cultured rat MSCs to
myofibroblasts
Immunochemical staining with α-SMA, a phenotypic
marker of differentiated myofibroblasts, demonstrated that
treatment with TGF-β1 increased the intensity of α-SMA staining in
the MSCs; the control MSCs exhibited relatively few α-SMA-positive
cells (10±2%). By contrast, >70% of the TGF-β1-treated MSCs were
α-SMA-positive (Fig. 1A),
indicating that the majority of MSCs differentiated into
myofibroblasts following 24 h of treatment. Pretreatment with
tranilast markedly inhibited these changes in the TGF-β1-induced
MSCs; the number of α-SMA-positive cells was reduced in a
dose-dependent manner, with significant inhibitory effects
following treatment with 1 and 10 µM of tranilast (Fig. 1B).
The western blot analysis further confirmed the
immunocytochemistry results, revealing that treatment of the
cultured rat MSCs with TGF-β1 resulted in upregulation of the
expression of α-SMA by 2.6-fold, compared with that in the control,
and was significantly reduced by tranilast treatment (Fig. 1C and D). The mRNA levels of α-SMA
were also evaluated; the RT-qPCR analysis revealed that the levels
of α-SMA were significantly increased with TGF-β1 application, and
this upregulation was significantly inhibited by pretreatment with
tranilast (Fig. 1E). However,
treatment with tranilast alone had no significant effect on the
mRNA expression level of α-SMA.
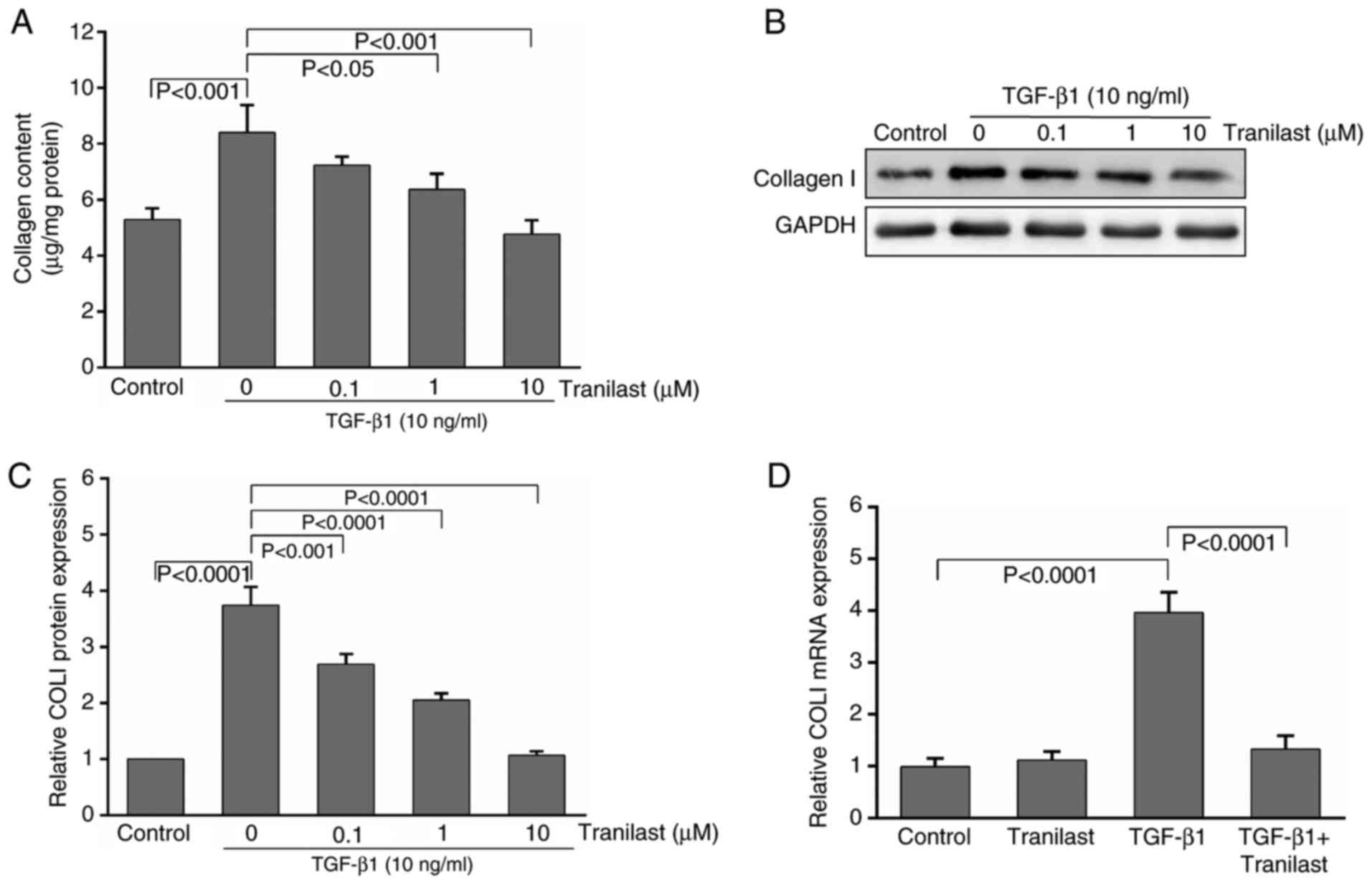
Tranilast decreases collagen
production in cultured rat MSCs induced by TGF-β1
To elucidate the effect of tranilast on
extracellular matrix (ECM) synthesis in the TGF-β1-induced MSCs,
the total collagen content was assessed using a hydroxyproline
assay. Treatment with TGF-β1 for 48 h increased the production of
collagen from control levels of 5.3±0.4 µg/mg protein to 8.4±1.1
µg/mg protein (P<0.01). Tranilast at 1 and 10 µM significantly
decreased TGF-β1-stimulated collagen production; this effect was
not evident at a lower concentration (0.1 µM, P>0.05; Fig. 2A).
The western blot analysis demonstrated that
treatment of the cultured rat MSCs with TGF-β1 resulted in
upregulation of the expression of collagen type I by 3.7-fold,
compared with that in the control, and the expression was
significantly decreased by pretreatment with tranilast (Fig. 2B and C).
Following induction by TGF-β1, the mRNA levels of
collagen type I were significantly increased by 3.9-fold in the
MSCs, compared with those in the control and tranilast-only treated
groups. By contrast, this effect was markedly inhibited by
pretreatment with 10 µM tranilast (Fig. 2D). Treatment with tranilast alone
had no significant effect on the mRNA levels of collagen type I
(P>0.05).
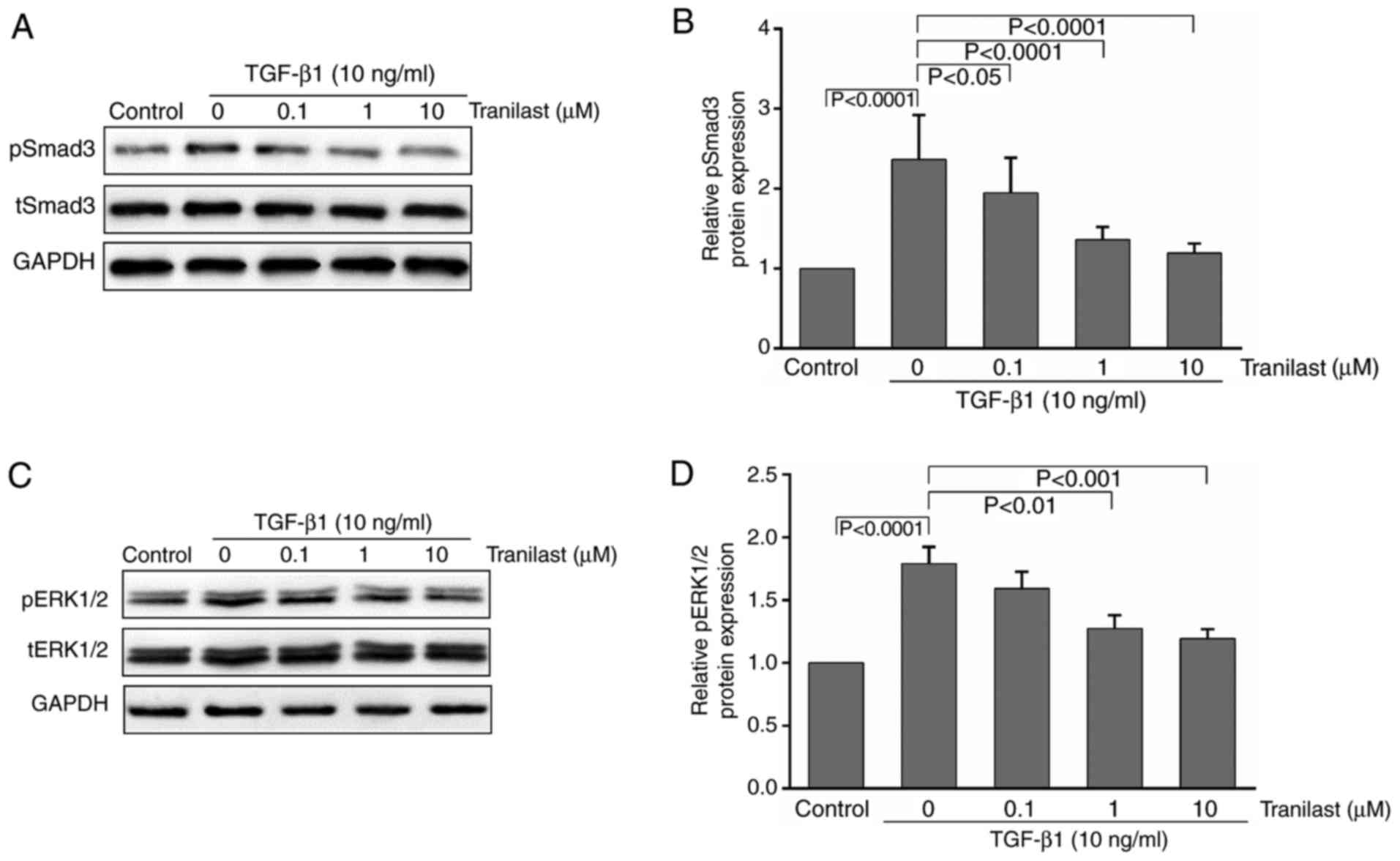
Tranilast inhibits the TGF-β1-induced
phosphorylation of Smad3 and ERK1/2 in cultured rat MSCs
To determine whether TGF-β1 stimulates the
transformation of cultured rat MSCs to myofibroblasts via the
activation of Smad3 and ERK1/2, and whether tranilast has any
effect on this process, the MSCs were pretreated with tranilast for
30 min and then incubated with TGF-β1 for 15–30 min. Western blot
analysis was performed to detect pSmad3/pERK1/2 and total
(t)-Smad3/tERK1/2. As presented in Fig. 3A-D, treatment with TGF-β1
significantly increased the phosphorylation of Smad3 and ERK1/2
compared with that in the control group; the phosphorylation was
significantly inhibited by pretreatment with tranilast.
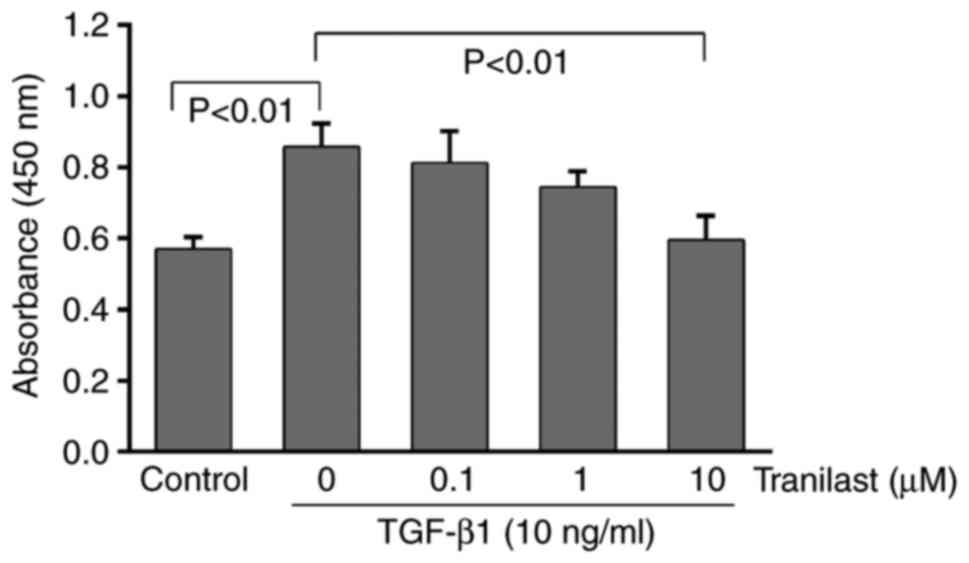
Tranilast inhibits the TGF-β1-induced
proliferation of cultured rat MSCs
As presented in Fig.
4, TGF-β1 administration significantly increased the viability
of the MSCs, as determined using the CCK-8 assay (P<0.01).
Following pretreatment with tranilast, the TGF-β1-induced
proliferation of the MSCs decreased in a dose-dependent manner and
was significantly inhibited at 10 µM (P<0.01). The lower
concentrations of tranilast (0.1 or 1 µM) did not have a
significant effect on the cell viability (P>0.05).
Discussion
The present study revealed that, as expected, TGF-β1
induced cultured rat MSC differentiation into myofibroblasts and
increased collagen production. Treating MSCs with tranilast
resulted in the inhibition of TGF-β1-induced myofibroblast
differentiation in vitro. It also decreased the expression
of α-SMA and collagen type I in a dose-dependent manner, and
suppressed the phosphorylation of Smad3 and ERK1/2 in the
TGF-β1-induced MSCs. It was also observed that the administration
of tranilast was able to inhibit the proliferation of
TGF-β1-induced cultured rat MSCs.
Although a number of studies have revealed the
beneficial effects of MSC therapy in treating several fibrotic
diseases, certain cultured MSCs may have the potential to become
myofibroblasts that substantially contribute to organ fibrosis
(2,5). Myofibroblasts are considered to be
important pathogenic cells in all fibrotic diseases and TGF-β1 may
be critical in the activation of fibrogenic myofibroblasts
(28). Cultured BM-MSCs can give
rise to myofibroblasts when transplanted into the murine liver
(8,29). In addition, BM-MSCs may contribute
to fibrosis by differentiating into tissue myofibroblasts in the
lungs (30). Differentiated
myofibroblasts are characterized by increased contractile capacity
and an elevated production of ECM; the most frequently used
molecular markers are α-SMA and collagen type І (3). Early reports indicated that α-SMA has
a low baseline expression, which is characterized by disorganized
intracellular α-SMA in cultured human cardiac fibroblasts and
neonatal lung MSCs (16,24). The results of the present study
showed that cultured rat MSCs became α-SMA-positive and contained
well-organized α-SMA filaments when stimulated by TGF-β1. To the
best of our knowledge, the results of the present study provide the
first evidence that co-culturing with exogenous TGF-β1 can induce
the transformation of cultured rat MSCs into myofibroblasts in
vitro.
Previous studies have revealed that tranilast has
therapeutic potential as an antifibrotic agent by inhibiting the
proliferation or differentiation of fibroblasts and leading to the
suppression of collagen synthesis (31,32).
The results of the present study revealed that tranilast prevented
the TGF-β1-induced myofibroblast differentiation of cultured rat
MSCs, and inhibited the expression of α-SMA and collagen type I at
the mRNA and protein levels. In addition, the mRNA expression
levels of α-SMA and collagen type I remained stable following
pretreatment with tranilast in the absence of exogenous TGF-β1. By
contrast, these expression levels were significantly increased by
TGF-β1, whereas pretreatment with tranilast markedly decreased this
process. These results suggested that the action of tranilast is
mainly associated with the TGF-β1 signaling pathway. The mechanism
underlying the action of tranilast remains to be fully elucidated,
however, its inhibitory effect on the activity of TGF-β has been
demonstrated in a range of cells (18).
The major mode of tranilast efficacy appears to be
via suppression of the expression and/or action of the TGF-β
signaling pathway (18). It is
hypothesized that TGF-β1 may be a key growth factor that mediates
organ fibrosis and operates via TGF-β receptors and Smad2/3/4
transcription factors (33).
Previous studies have revealed that Smad3, but not Smad2, was
essential in fibrosis and was observed to be required for
myofibroblast generation (34).
Smad3 has been implicated as a central mediator in the profibrotic
effects of TGF-β1 (35). It has
been demonstrated that the TGF-β1/Smad3 signaling pathway is
activated in myofibroblast differentiation, and inhibiting its
phosphorylation may attenuate myofibroblast differentiation and the
fibrogenic response (36). In
order to evaluate the possible signaling mechanism by which
tranilast suppresses the generation of myofibroblasts, the effects
of tranilast on the Smad3 and ERK1/2 signaling pathways in
TGF-β1-stimulated MSCs were investigated. The results of the
present study demonstrated that tranilast inhibited the
phosphorylation of Smad3 in the TGF-β-stimulated MSCs, suggesting
that it suppresses the transformation of MSCs to myofibroblasts and
collagen synthesis partly via the Smad signaling pathway. In
addition to the TGF-β/Smad signaling pathways, the ERK/MAPK
signaling pathways, particularly ERK1/2, mediate the biological
effects of TGF-β1 (16). In the
present study, tranilast suppressed pERK1/2 in the TGF-β-stimulated
MSCs, suggesting that its inhibitory effects on myofibroblast
differentiation may be partially mediated through inhibition of the
ERK/MAPK signaling pathway.
As demonstrated by the CCK-8 assays, the
proliferation of cultured rat MSCs stimulated with TGF-β1 increased
1.5-fold, and tranilast inhibited the increase in a dose-dependent
manner. These results indicated that one of the inhibitory
mechanisms of collagen synthesis by tranilast in cultured rat MSCs
may be involved in the inhibition of cell proliferation. The
antiproliferative effect of tranilast appears to be involved in the
inhibition of fibrosis. However, the detailed mechanisms through
which tranilast attenuates the TGF-β1-induced proliferation and
differentiation of cultured rat MSCs require further clarification.
In addition, further investigations are required to assess whether
tranilast has a cytotoxic effect.
In conclusion, to the best of our knowledge, the
present study demonstrated for the first time that in cultured rat
MSCs that tranilast inhibited TGF-β1-induced myofibroblast
differentiation and the Smad3/ERK1/2 signaling pathways. Tranilast
also inhibited the proliferation of MSCs stimulated by TGF-β1.
Tranilast has low adverse effects and is generally well tolerated
by patients (18). Therefore,
tranilast represents a potentially useful therapeutic agent in
fibrotic diseases. Furthermore, tranilast has potential activity as
an adjuvant treatment following the transplantation of MSCs for
antifibrotic activity. Therefore, the present study demonstrated
the role of tranilast in the differentiation of MSCs into
myofibroblasts, and may provide further support for future clinical
therapies.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural
Science Foundation of China (grant no. 81302883), the Natural
Science Foundation of Hunan Province (grant no. 14JJ4056) and the
Scientific Research Foundation of Hunan Educational Committee
(grant no. 12C0219).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
WT and BW conceived and designed the study. YZ, LT,
JZ and LX performed the experiments. WT and BW wrote the
manuscript. All authors read and approved the manuscript.
Ethics approval and consent to
participate
The animal experiments were approved by the Animal
Ethics Committee and the Medical Ethics Committee of Hunan Normal
University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wynn TA: Cellular and molecular mechanisms
of fibrosis. J Pathol. 214:199–210. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
El Agha E, Kramann R, Schneider RK, Li X,
Seeger W, Humphreys BD and Bellusci S: Mesenchymal stem cells in
fibrotic disease. Cell Stem Cell. 21:166–177. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hinz B: Formation and function of the
myofibroblast during tissue repair. J Invest Dermatol. 127:526–537.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kalluri R and Neilson EG:
Epithelial-mesenchymal transition and its implications for
fibrosis. J Clin Invest. 112:1776–1784. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
LeBleu VS, Taduri G, O'Connell J, Teng Y,
Cooke VG, Woda C, Sugimoto H and Kalluri R: Origin and function of
myofibroblasts in kidney fibrosis. Nat Med. 19:1047–1053. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kramann R, Schneider RK, DiRocco DP,
Machado F, Fleig S, Bondzie PA, Henderson JM, Ebert BL and
Humphreys BD: Perivascular Gli1+ progenitors are key contributors
to injury-induced organ fibrosis. Cell Stem Cell. 16:51–66. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bonfield TL and Caplan AI: Adult
mesenchymal stem cells: An innovative therapeutic for lung
diseases. Discov Med. 9:337–345. 2010.PubMed/NCBI
|
|
8
|
Baertschiger RM, Serre-Beinier V, Morel P,
Bosco D, Peyrou M, Clément S, Sgroi A, Kaelin A, Buhler LH and
Gonelle-Gispert C: Correction: Fibrogenic potential of human
multipotent mesenchymal stromal cells in injured liver. PLoS One.
4:e66572009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Russo FP, Alison MR, Bigger BW, Amofah E,
Florou A, Amin F, Bou-Gharios G, Jeffery R, Iredale JP and Forbes
SJ: The bone marrow functionally contributes to liver fibrosis.
Gastroenterology. 130:1807–1821. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tang N, Zhao Y, Feng R, Liu Y, Wang S, Wei
W, Ding Q, An MS, Wen J and Li L: Lysophosphatidic acid accelerates
lung fibrosis by inducing differentiation of mesenchymal stem cells
into myofibroblasts. J Cell Mol Med. 18:156–169. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Carlson S, Trial J, Soeller C and Entman
ML: Cardiac mesenchymal stem cells contribute to scar formation
after myocardial infarction. Cardiovasc Res. 91:99–107. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yang S, Cui H, Xie N, Icyuz M, Banerjee S,
Antony VB, Abraham E, Thannickal VJ and Liu G: miR-145 regulates
myofibroblast differentiation and lung fibrosis. FASEB J.
27:2382–2391. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bianco P, Cao X, Frenette PS, Mao JJ,
Robey PG, Simmons PJ and Wang CY: The meaning, the sense and the
significance: Translating the science of mesenchymal stem cells
into medicine. Nat Med. 19:35–42. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xu J, Lamouille S and Derynck R:
TGF-beta-induced epithelial to mesenchymal transition. Cell Res.
19:156–172. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang W, Koka V and Lan HY: Transforming
growth factor-beta and Smad signalling in kidney diseases.
Nephrology (Carlton). 10:48–56. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Peng H, Carretero OA, Peterson EL and
Rhaleb NE: Ac-SDKP inhibits transforming growth
factor-beta1-induced differentiation of human cardiac fibroblasts
into myofibroblasts. Am J Physiol Heart Circ Physiol.
298:H1357–H1364. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sun Q, Wu Y, Zhao F and Wang J: Maresin 1
inhibits transforming growth factor-β1-induced proliferation,
migration and differentiation in human lung fibroblasts. Mol Med
Rep. 16:1523–1529. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Darakhshan S and Pour AB: Tranilast: A
review of its therapeutic applications. Pharmacol Res. 91:15–28.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yamada H, Tajima S, Nishikawa T, Murad S
and Pinnell SR: Tranilast, a selective inhibitor of collagen
synthesis in human skin fibroblasts. J Biochem. 116:892–897. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Isaji M, Nakajoh M and Naito J: Selective
inhibition of collagen accumulation by
N-(3,4-dimethoxycinnamoyl)anthranilic acid (N-5′) in granulation
tissue. Biochem Pharmacol. 36:469–474. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kim TI, Lee H, Hong HK, Kim KS, Choi SI,
Maeng YS and Kim EK: Inhibitory effect of tranilast on transforming
growth factor-beta-induced protein in granular corneal dystrophy
type 2 corneal fibroblasts. Cornea. 34:950–958. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nakatani Y, Nishida K, Sakabe M, Kataoka
N, Sakamoto T, Yamaguchi Y, Iwamoto J, Mizumaki K, Fujiki A and
Inoue H: Tranilast prevents atrial remodeling and development of
atrial fibrillation in a canine model of atrial tachycardia and
left ventricular dysfunction. J Am Coll Cardiol. 61:582–588. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
See F, Watanabe M, Kompa AR, Bing HW,
Boyle AJ, Kelly DJ, Gilbert RE and Krum H: Early and delayed
tranilast treatment reduces pathological fibrosis following
myocardial infarction. Heart Lung Circ. 22:122–132. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Popova AP, Bozyk PD, Goldsmith AM, Linn
MJ, Lei J, Bentley JK and Hershenson MB: Autocrine production of
TGF-beta1 promotes myofibroblastic differentiation of neonatal lung
mesenchymal stem cells. Am J Physiol Lung Cell Mol Physiol.
298:L735–L743. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kakudo N, Kushida S, Suzuki K, Ogura T,
Notodihardjo PV, Hara T and Kusumoto K: Effects of transforming
growth factor-beta1 on cell motility, collagen gel contraction,
myofibroblastic differentiation, and extracellular matrix
expression of human adipose-derived stem cell. Human Cell.
25:87–95. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Song K, Huang M, Shi Q, Du T and Cao Y:
Cultivation and identification of rat bone marrow-derived
mesenchymal stem cells. Mol Med Rep. 10:755–760. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Phan SH: The myofibroblast in pulmonary
fibrosis. Chest. 122 6 Suppl:286S–289S. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
di Bonzo LV, Ferrero I, Cravanzola C,
Mareschi K, Rustichell D, Novo E, Sanavio F, Cannito S, Zamara E,
Bertero M, et al: Human mesenchymal stem cells as a two-edged sword
in hepatic regenerative medicine: Engraftment and hepatocyte
differentiation versus profibrogenic potential. Gut. 57:223–231.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tang N, Zhao Y, Feng R, Liu Y, Wang S, Wei
W, Ding Q, An MS, Wen J and Li L: Lysophosphatidic acid accelerates
lung fibrosis by inducing differentiation of mesenchymal stem cells
into myofibroblasts. J Cell Mol Med. 18:156–169. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yasukawa T, Kimura H, Dong J, Tabata Y,
Miyamoto H, Honda Y and Ogura Y: Effect of tranilast on
proliferation, collagen gel contraction, and transforming growth
factor beta secretion of retinal pigment epithelial cells and
fibroblasts. Ophthalmic Res. 34:206–212. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hattori T and Wang PL: Inhibition by
tranilast of nifedipine-induced proliferation of cultured human
gingival fibroblasts. Eur J Pharmacol. 498:79–81. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang S, Meng XM, Ng YY, Ma FY, Zhou S,
Zhang Y, Yang C, Huang XR, Xiao J, Wang YY, et al: TGF-β/Smad3
signalling regulates the transition of bone marrow-derived
macrophages into myofibroblasts during tissue fibrosis. Oncotarget.
7:8809–8822. 2016.PubMed/NCBI
|
|
34
|
Bonniaud P, Kolb M, Galt T, Robertson J,
Robbins C, Stampfli M, Lavery C, Margetts PJ, Roberts AB and
Gauldie J: Smad3 null mice develop airspace enlargement and are
resistant to TGF-beta-mediated pulmonary fibrosis. J Immunol.
173:2099–2108. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sato M, Muragaki Y, Saika S, Roberts AB
and Ooshima A: Targeted disruption of TGF-β1/Smad3 signaling
protects against renal tubulointerstitial fibrosis induced by
unilateral ureteral obstruction. J Clin Invest. 112:1486–1494.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gu L, Zhu YJ, Yang X, Guo ZJ, Xu WB and
Tian XL: Effect of TGF-beta/Smad signaling pathway on lung
myofibroblast differentiation. Acta Pharmacol Sin. 28:382–391.
2007. View Article : Google Scholar : PubMed/NCBI
|