Introduction
Bone defects are commonly caused by traumatic
events, chronic infection or bone tumor resection, which pose
critical clinical issues and rapidly increasing morbidity (1). The severity of bone defects is
associated with the specific skeletal segment involved and the
degree of bone loss, specifically 6 cm for the humerus, 5 cm for
the femur and tibia, and 3 cm for the forearm (2). It has been noted that osteogenesis is
a key factor in the process of bone formation (3,4). The
gold standard for bone repair and healing is autologous and
allogeneic bone grafting in patients with large bone defects or
fracture non-unions (5). However,
this treatment method is limited due to donor shortages, infectious
diseases, nerve injury, persistent pain and potential new fractures
(6). These limitations motivate
additional investigations to develop improved techniques or
treatment regimens for bone defects, and therapies based on bone
marrow mesenchymal stem cells (BMSCs) may lead to the improved
repair of bone defects (7,8). Seed cells are essential for the
treatment of bone defects; therefore, genetically modified BMSCs
are widely employed in tissue engineering to improve osteogenesis
and angiogenesis in bone regeneration (9). Multiple studies have documented the
regulatory role of microRNAs (miRNAs/miRs) in the osteogenic
differentiation of BMSCs, including miR-125b and miR-16 (10–12).
miRNAs refer to a group of conserved, short
non-coding RNAs of 20–22 nucleotides that are closely associated
with gene expression at post-transcriptional levels (13,14).
Additionally, miRNAs modulate numerous cellular activities by
binding to the 3′-untranslated region (3′-UTR) of their target
mRNAs (15,16). miRNAs are commonly aberrantly
expressed in pathological alterations, and lentiviral vectors are
frequently adopted to regulate the expression of miRNAs, which
often serve as potential therapeutic targets (17,18).
In terms of the osteogenesis of bone healing or repair, miRNAs have
been demonstrated to be regulators, and miR-26a has already been
demonstrated to be involved in osteogenic differentiation by
targeting SMAD family member 1, as miR-26a is upregulated during
osteoblast differentiation (19,20).
Cytokines associated with osteogenesis, such as runt-related
transcription factor 2 (Runx2) and osteocalcin (OC), are crucial
osteogenic genes in bone regeneration of cranial bone defects
(21). Accordingly, the present
study investigated whether using BMSCs that overexpressed miR-26a,
via lentivirus-mediated transfection, as seed cells may improve
bone regeneration during the repair process of bone defects in a
mouse model.
Materials and methods
Experimental animals and
treatment
Male C57BL/6J mice aged 4 weeks (n=10; weight,
16.68±0.35 g; used for the cell separation) and 8 weeks (n=32;
weight, 20.25±0.57 g; used for bone defect model experiments) were
purchased from Shanghai SLAC Laboratory Animal Co., Ltd. (Shanghai,
China). Mice were housed under a controlled temperature of 18–28°C,
a relative humidity of 40–70%, air cleanliness (100,000, ISO 8),
background noise ≤60 dB and kept on a 12 h light/dark cycle. Free
access to food and water and regular ventilation and disinfection
were guaranteed for mice. Animal use and experimental procedures
were performed in accordance with the Declaration of Helsinki
(22) and the present study was
approved by the Experimental Animal Ethics Committee of the
People's Hospital of Dongsheng (Ordos City, China). The 32 male
C57BL/6J 8-week-old mice were equally assigned to groups: Blank
group (n=8, mice implanted with β-tricalcium phosphate (β-TCP)
scaffolds alone in the defect area), BMSC group (n=8, mice
implanted with β-TCP scaffolds co-cultured with BMSCs in the defect
area), NC-BMSC group (n=8, mice implanted with β-TCP scaffolds
co-cultured with negative control (NC)-transfected BMSCs in the
defect area) and miR-26a-BMSC group (n=8, mice implanted with β-TCP
scaffolds co-cultured with miR-26a-transfected BMSCs in the defect
area).
BMSC isolation and culture
The thigh bones of both sides were separated from
the male C57BL/6J mice (n=10; 4-weeks old) under sterile
conditions. Following washing with PBS, the metaphyses of both
sides were resected and attached muscles and fibrous tissues were
also scraped off. A syringe pipette (1 ml) was used to pipette 5 ml
α-minimum essential medium containing 10% fetal bovine serum (FBS;
both Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA),
which was also supplemented with 100 U/ml penicillin and 100 U/ml
streptomycin, to rinse the thigh bones. The mixture was triturated
evenly to resuspend cells. Subsequently, cells were inoculated in
culture flasks at a density of 1×106 cells/ml and
incubated in a 5% CO2 incubator at 37°C. The medium was
renewed every three days. When adherent cells grew to 80–90%
confluency, the culture medium was discarded. Following washing
with PBS twice, cells were digested with 0.25% trypsin at room
temperature and subcultured to a ratio of 1:3.
Flow cytometric analysis of BMSCs
Third generation BMSCs were digested with 0.25%
trypsin and washed using PBS containing bovine serum albumin (BSA;
20%; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany). The cells were
subsequently resuspended in complete medium (Suzhou Cyagen
Biosciences Inc., Suzhou, China) to a single cell suspension at a
density of 5×106 cells/ml. Eppendorf tubes were used to
contain the single cell suspension and each tube was filled with
200 µl of the suspension (5×106 cells/ml). Rat
anti-mouse antibodies against CD29 [fluorescein isothiocyanate
(FITC)-conjugated; cat. no. 555005; 1:500), CD34 (FITC-conjugated;
cat. no. 560238; 1:500), CD105 (phycoerythrin-conjugated; cat. no.
562759; 1:500) and CD45 (phycoerythrin-conjugated; cat. no. 553081;
1:500) and fluorescein isothiocyanate-labeled mouse IgG (cat. no.
555748; 1:500; all BD Biosciences, Franklin Lakes, NJ, USA) for
flow cytometric detection were supplemented to the EP tubes,
incubated at 4°C for 60 min, washed with PBS containing 3% FBS 3
times and resuspended following centrifugation at 112 × g at 4°C
for 5 min and fixed with 200 µl polyoxymethylene (4%) at 4°C for 1
h. A flow cytometer and Cell-Quest software (version 5.1; BD
Biosciences) were used to analyze the cell phenotype.
Construction and determination of the
lentiviral vector pLVTHM-miR-26a
The sequence of the miR-26a precursor pre-miR-26a-1
(accession no. MI0000083), with a full length of 77 bp, was
obtained from the miRBase Sequence database (http://www.mirbase.org/), and flanking sequences (50
bp) were added both upstream and downstream of pre-miR-26a-1, which
produced precursor fragments, including hairpin pre-miR-26a-1,
which had a full length of 177 bp. The primer sequences were
designed and synthesized by Shanghai Gene Pharma Co., Ltd.
(Shanghai, China). Forward, 5′-CGACGCGTCGAGCCAAGAGCAGGAGGAC-3′ and
reverse, 5′-CCATCGATGGTGGTGTTGGTGCCTCTGG-3′. Genomic DNA was
extracted from the blood (65–100 µl) of 4 week old mice, obtained
from retro-orbital blood using a heparinized microcapillary tube.
The blood was expelled immediately into a 1.5 ml microfuge tube
containing 20 µl of 10 mM EDTA and mixed immediately to prevent
clot formation. Then, 200 µl Lysis Buffer was added to each tube
and agitated to suspend evenly. Centrifugation (16,000 × g at 4°C
for 25 sec) was used to pellet nuclei. The supernatant was removed
and discarded and the above steps repeated 3 times. The nuclear
pellet was resuspended in 100 µl PBND buffer (50 mM KCl; 10 mM
Tris-HCl, pH 8.3; 2.5 mM MgCl2·6H2O; 0.1
mg/ml gelatin; 0.45% v/v Nonidet P-40; 0.45% v/v Tween-20) with 60
µg/ml proteinase K and incubated overnight at 4°C. Samples were
heated to 97°C for 10 min to inactivate proteinase K. Then, 1–5 µl
of DNA solution was added for a 25 µl PCR reaction. Using the
genomic DNA as a template, the primer sequences were amplified by
polymerase chain reaction (PCR) to obtain fragments. The
thermocycling conditions were: 98°C for 3 min; 95°C for 30 sec,
60°C for 30 sec and 72°C for 1.5 min, for 35 cycles. Following
agarose gel electrophoresis, the amplified products and vector
pLVTHM (held in our laboratory) were digested with Mlu I and
Cla I, respectively. The enzyme digestion products were
incubated with ligase at 16°C overnight and 20 µl transformed into
100 µl Escherichia coli DH-5α-competent cells (Transgen,
Beijing, China), with empty vector (0.1 µg/µl) as a control.
Positive recombinant clones were selected to conduct dual-enzyme
(Mlu I and Cla I) digestion and were verified by PCR.
Correct plasmids were verified via DNA sequencing by Shanghai
GenePharma Co., Ltd. Lipofectamine® 2000 (Invitrogen;
Thermo Fisher Scientific, Inc.) was used to transfect the
successfully verified recombinant plasmids into 293T cells
(>1×106/ml, Shanghai Genechem Co., Ltd., Shanghai,
China) at the logarithmic growth phase. Following 72 h
transfection, the supernatant of 293T cells was collected to
prepare the virus suspension. Following this, resuspended 293T
cells (>1×106/ml) were supplemented with the virus
suspension and incubated at 37°C for 24 h to measure the lentiviral
titer.
Infection of BMSCs with the lentiviral
vector pLVTHM-miR-26a
BMSCs of mice were inoculated in sterile petri
dishes (6 cm). When cells grew to 30–45% confluency, supernatant of
the lentiviral vector pLVTHM-miR-26a (8 µg/ml) and empty vector
pLVTHM (8 µg/ml) were added. Reverse transcription-quantitative PCR
(RT-qPCR) was applied to detect the expression levels of miR-26a 48
h later. BMSCs successfully infected with the lentiviral vector
pLVTHM-miR-26a were termed the miR-26a-BMSC group. BMSCs infected
with the empty vector pLVTHM were termed the NC-BMSC group.
MTT assay to detect cell
viability
BMSCs were inoculated in a 96-well plate with
5×104 cells per well, and each group was repeated in six
wells. When cells reached 80% confluency, they were grouped as
follows: BMSC group, NC-BMSC group (BMSCs infected with the pLVTHM
empty vector) and miR-26a-BMSC group (BMSCs infected with
pLVTHM-miR-26a). MTT solution (20 µl; Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) was added, followed by incubation at 37°C in an
incubator for 4 h. MTT solution was removed. A total of 150 µl
dimethyl sulfoxide (Sigma-Aldrich; Merck KGaA) was added to each
well. Subsequently, the mixture was shaken for 10 min. A microplate
reader was employed to determine the optical density value at 1, 2,
3, 4 and 5 days at 490 nm.
Marker of proliferation Ki67 (Ki67)
staining to detect cell proliferation
BMSCs were digested with trypsin and were incubated
in a 48-well plate, on which cover slips were placed. The culture
medium was aspirated out 24 h later, followed by washing in water
three times. 1×105/ml cells were then fixed in 4%
paraformaldehyde at room temperature for 20 min, followed by
washing with 0.1% Triton X-100 in PBS at room temperature for 5 min
on the shaking table and two PBS (0.01 mol/l) washes (5 min per
wash). Subsequently, the cells were blocked by 10% normal goat
serum (Abcam, Cambridge, UK) at 37°C in an incubator for 1 h.
Primary rabbit antibody against Ki67 (1:100; cat. no. ab16667;
Abcam) was added, followed by incubation at 4°C overnight. On the
next day, cells were rinsed by 0.1% Triton X-100 in PBS for 5 min
and 0.01 mol/l PBS twice (5 min per wash). A FITC-conjugated goat
anti-rabbit secondary antibody (1:1,000; cat. no. ab6717; Abcam)
was added, followed by incubation at 37°C in an incubator for 2 h.
The cells were washed with 0.01 mol/l PBS three times (5 min per
wash) on a shaking table. DAPI was incubated with the cells at 37°C
in an incubator for 10 min, followed by washing with 0.01 mol/l PBS
3 times (5 min per wash). Cells were mounted in antifade mounting
medium (Beyotime Institute of Biotechnology, Shanghai, China) and
observed under a fluorescence inverted microscope (magnification,
×400; Shanghai Dianying Optical Instrument, Co., Ltd., Shanghai,
China). A total of 10 fields of view were randomly selected from
each group to count the positive cells, and the positive staining
rate (%)=(number of Ki67 staining positive cells/total number of
cells) ×100 was calculated.
Alkaline phosphatase (ALP) staining to
detect osteogenesis capacity
The BMSCs of mice were cultured for 14 days and the
cell density was adjusted to 1×105 cell/ml. The culture
medium was removed and the cells were washed by PBS three times.
Following trypsin digestion, cells were centrifuged at 160 × g at
4°C for 5 min and the supernatant removed. Subsequently, 1 ml
culture medium was added to suspend the cells and the suspended
cells were cultured in a 6-well plate, on which sterile cover slips
had been placed. The cells grew to adhere to the wall of wells
fully for 6 h at 37°C. The cover slips of each well were taken out.
The revised calcium-cobalt method was adopted to stain cells
(23). The cells were observed
under a light microscope (magnification, ×100; Shanghai Dianying
Optical Instrument, Co., Ltd.) with 10 fields of view randomly
selected, and the ALP staining positive cells and total cells were
counted. The positive staining rate was calculated: ALP positive
staining rate (%)=(number of ALP staining positive cells/total
number cells) ×100.
Fabrication of β-tricalcium phosphate
(β-TCP) scaffolds
Cylindrical β-TCP material (Shanghai Bio-lu
Biomaterials Co., Ltd., Shanghai, China) was used as composite
scaffolds (24), with a porosity
of 90.7±1.9%, pore diameter of 300–500 µm, diameter of 22 mm and
thickness of 3 mm. On a clean table, 24 sterile packaged β-TCP
scaffolds were immersed in Dulbecco's modified Eagle's medium
(Gibco; Thermo Fisher Scientific, Inc.) in a 5% CO2
incubator at 37°C. After 48 h, β-TCP scaffolds were taken out and
rinsed with Dulbecco's modified Eagle's medium containing 10% FBS
five times. Both uninfected and infected BMSCs were centrifuged at
168 × g for 3 min at room temperature, followed by the addition of
DMEM low-glucose medium (Gibco; Thermo Fisher Scientific, Inc.) to
prepare cell suspension (5×106 cells/ml). The dry and
sterile β-TCP scaffolds were dripped with 1 ml of the cell
suspension and shaken gently to allow the suspension to fully
infiltrate the pores. The scaffolds were then incubated at 37°C in
5% CO2 and saturated at humidity for 8 h. The solution
was renewed every other day for seven days.
Mouse model establishment of repair of
cranial bone defects (25)
Under sterile conditions, the skin on the cranium of
anesthetized mice was cut open in the midline to 5 cm. The
full-thickness of the skin was separated, and the periosteum was
stripped. A ring with a diameter of 22 mm was drilled into the
parietal region of the mouse cranium to form a round full-thickness
bone defect with a diameter of 5 mm. The bone pieces were stripped
carefully without injuring the cerebral dura mater. During this
process, Hanks' balanced salt solution (Covidien, Mansfield, MA,
USA) was used for rinsing and cooling. After bleeding was stopped
with a gelatin sponge, corresponding prepared implants were
implanted into the defect areas to fill the whole area.
Subsequently, an absorbable suture was employed to suture the
incision on the skin.
Micro-computed tomography (micro-CT)
to detect bone regeneration
Two months following surgery, mice from all groups
were euthanized. The whole cranium was separated and scanned using
micro-CT. The working voltage was 55 kV, the working current was
145 mA, the depth of scanning layer was 20 µm and the scanning time
was 3,400 msec. The bone regeneration conditions were analyzed and
the bone volume of newly formed bones was calculated. Subsequently,
the cranium specimen was separated into two parts along the
midline. One part was immediately frozen in liquid nitrogen and
stored in a refrigerator at −80°C to perform RT-qPCR and western
blotting. The other part of the cranium was fixed in a 10% formalin
solution (Wuhan Boster Biological Technology, Ltd., Wuhan, China)
at 4°C for 24 h. The fixed sample was decalcified with 10% EDTA
(pH=7.4) for 2 weeks, followed by graded dehydration and
paraffin-embedding on the following day. The cranium was sliced,
with a thickness of a ~6 µm, successively parallel to the long
axis, from the inside out.
Histological examination of bone
regeneration
Hematoxylin and eosin (H&E) staining was
performed at room temperature to observe bone regeneration
conditions of the newly formed bones. Slices were put into xylene
for 15 min, washed with 100% anhydrous ethanol for 3 min, 90%
ethanol for 3 min and 75% ethanol for 3 min, followed by gradient
ethanol dewaxing to water. Hematoxylin staining was performed for 5
min, followed by washing and soaking in PBS for 5 min, eosin
staining for 2 min, washing with water, gradient alcohol
dehydration, xylene treatment twice for 5 min each and neutral
resin blocked cover slips. Subsequently, the newly formed bone
tissues were observed under an optical microscope (magnification,
×400). Toluidine blue staining was employed at room temperature to
observe the degradation conditions of the implanted scaffolds in
the area of bone defects: Descending ethanol series followed by
dewaxing and 1% toluidine blue staining for 3 min, washed with
water, gradient alcohol dehydration, xylene treatment twice for 5
min each and neutral resin blocked cover slips. The staining
results were observed under an optical microscope (magnification,
×400).
RT-qPCR to detect mRNA expression of
miR-26a, Runx2 and OC
Frozen cranium tissues from 4 mice were placed into
a mortar. Following supplementation with a small amount of liquid
nitrogen, the cranium tissues were ground to powder, which was
transferred to Eppendorf tubes containing 1 ml TRIzol (Life
Technologies; Thermo Fisher Scientific, Inc.). Shaking was
performed to fully lyse the tissues. The mixture was centrifuged at
16,100 × g at 4°C for 15 min and the supernatant was collected.
Subsequently, 200 µl chloroform was added. The mixture was shaken
for 15 sec to make it turn milky white and was incubated at room
temperature for 5 min, followed by centrifugation at 16,100 × g at
4°C for 15 min. The aqueous phase (400 µl) was collected.
Subsequently, acid phenolics (400 µl) were extracted twice,
followed by centrifugation at 16,100 × g at 4°C for 15 min.
Isopropyl alcohol (400 µl) was added, followed by inversion 4–5
times for blending and allowed to stand for 8 min at room
temperature (400 µl). Centrifugation was performed at 16,100 × g at
4°C for 10 min. The deposit was rinsed with 1 ml 75% ethanol twice
and centrifuged at 6,288 × g rpm at 4°C for 5 min. The deposit was
inverted and dried for 10 min. RNase-free water (30 µl) was
supplemented to dissolve the deposit. Ultra-violet analysis and
formaldehyde gel electrophoresis were used to validate the high
quality of the extracted RNA. RNA (1 µg) was reverse transcribed to
obtain cDNA using Prime Script™ RT-PCR kit (Takara Biotechnology
Co., Ltd., Dalian, China) according to the manufacturer's
protocols, and the reverse transcription sample were put into the
reverse transcriptome. The reverse transcription reaction
conditions were 16°C for 30 min, 42°C for 42 min and 85°C for 5
min. The RNA samples were reversely transcribed into cDNA (10 µl)
according to the manufacturer's protocols. The obtained cDNA was
diluted in 65 µl of diethyl phosphorocyanidate. The PCR mixture
comprised the following elements: 5 µl of SsoFast EvaGreen Supermix
(1708882; Bio-Rad Laboratories, Inc., Hercules, CA, USA), 0.5 µl of
forward primer (10 µM), 0.5 µl of reverse primer (10 µM) and 4 µl
of cDNA. Primers for qPCR were designed and synthesized by
Invitrogen (Thermo Fisher Scientific, Inc.), with GAPDH and U6 as
the internal control (Table I).
The thermocycling conditions for PCR were as follows:
Predenaturation at 94°C for 5 min, followed by 40 cycles of
denaturation at 94°C for 40 sec, annealing for 40 sec at 60°C and
DNA strand extension for 1 min at 72°C, and final extension for 10
min at 72°C. The CFX96 qPCR machine (Bio-Rad, Inc., Hercules, CA,
USA) was used. The results were analyzed by OpticonMonitor version
3 software (Bio-Rad Laboratories, Inc.). The quantification value
was manually selected at the lowest point of all of the parallel
rising logarithmic amplification curves, and the cycle
quantification (Cq) value of each reaction tube was recorded. The
2−ΔΔCq method (26) was
used to compare the association of target gene expression in the
experimental group and that of the blank group: ΔΔCq=(Cqtarget
gene-Cqcontrol gene)experimental
group-(Cqtarget gene-Cqcontrol
gene)blank group.
 | Table I.Primer sequences for reverse
transcription-quantitative polymerase chain reaction. |
Table I.
Primer sequences for reverse
transcription-quantitative polymerase chain reaction.
| Gene | Primer
sequence |
|---|
| miR-26a | F:
5′-GGATCCGCAGAAACTCCAGAGA-3′ |
|
| R:
5′-TTGGAGGAAAGACGATTTCCGT-3′ |
| U6 | F:
5′-ATTGGAACGATACAGAGAAGATT-3′ |
|
| R:
5′-GGAACGCTTCACGAATTTG-3′ |
| Runx2 | F:
5′-TGAGCGACGTGAGCCCGGTA-3′ |
|
| R:
5′-CGTGTGGAAGACAGCGGCGT-3′ |
| OC | F:
5′-CCTGGCAGGTGCAAAGCCCA-3′ |
|
| R:
5′-TGCGCTTGTAGGCGTCCTGG-3′ |
| GAPDH | F:
5′-CAAGTTCAACGGCACAGTCA-3′ |
|
| R:
5′-CCCCATTTGATGTTAGCGGG-3′ |
Western blot analysis to detect
protein expression of Runx2 and OC
The cranium specimens of the remaining 4 mice were
taken out and washed with ice-cold PBS three times. The samples
were then treated with radioimmunoprecipitation assay lysis buffer
(Beyotime Institute of Biotechnology). Following thorough grinding,
the samples were subjected to an ice bath for 30 min to lyse the
tissues. Tissues were then centrifuged at 894 × g at 4°C for 10 min
and the supernatant collected. The protein concentration was
measured by a BCA protein assay kit (Wuhan Boster Biological
Technology, Ltd., Wuhan, China), according to the manufacturer's
protocol. The extracted proteins were boiled with loading buffer at
95°C for 10 min and each well was loaded with 30 µg samples.
Subsequently, 10% polyacrylamide gel electrophoresis was performed
to separate proteins with a voltage of 80–120 V. The separated
proteins were transferred onto polyvinylidene difluoride membranes
by wet transfer with a transmembrane voltage of 100 mV for 45–70
min. The membranes were blocked with 5% BSA at room temperature for
1 h. Primary antibodies against Runx2 and OC (1:1,000; cat. no.
ab23981 and ab93876, respectively) and primary antibody against
β-actin (1:3,000; cat. no. ab8227; both Abcam) were added at 4°C
overnight. Following washing with Tris-buffered saline with 20%
Tween-20 (TBST) three times (5 min per wash), anti-rabbit secondary
antibody (1:2,000; cat. no. ab205718; Abcam) was added and
incubated at room temperature for 1 h. Chemiluminescence reagents
(BeyoECL PLUS kit; cat. no. P0018; Beyotime Institute of
Biotechnology) were added as a chromogen after the membranes were
washed three times (5 min per wash). β-actin was set as the
internal control. Images of the gels were captured in a Bio-Rad Gel
Doc EZ Imager (Bio-Rad Laboratories, Inc.). The target protein
bands were analyzed using ImageJ software (version 1.8.0; National
Institutes of Health, Bethesda, MD, USA). The experiment was
repeated three times and the mean value was calculated.
Statistical analysis
SPSS 21.0 software (IBM Corp., Armonk, NY, USA) was
used for data analysis. Data are presented as the mean ± standard
deviation. Comparisons among multiple groups were conducted by one-
or two-way analysis of variance followed by Tukey's post-hoc test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Analysis of isolated BMSCs by
morphology and immunophenotype
An inverted microscope was used to observe the
morphological changes of mouse BMSCs. Primary cultured BMSCs had a
long fusiform or oval shape that resembled fibroblasts. During
subculturing, BMSCs began to adhere to the wall at approximately 3
h; to proliferate at approximately 3 days, with accumulated cell
numbers, and to grow logarithmically for ~6–15 days, with
increasing growth rates and a monolayer growth adherent to the wall
(Fig. 1A and B).
The surface markers of separated BMSCs were detected
by flow cytometry, and the results indicated that cultured BMSCs of
the third generation highly expressed surface markers such as CD29
(94.2±2.1%) and CD105 (95.8±2.5%), but did not express surface
markers such as CD34 (2.5±0.7%) and CD45 (1.8±0.4%; Fig. 1C). The flow cytometric detection
findings were consistent with the immunophenotype of BMSCs,
confirming that mouse BMSCs were successfully separated from mouse
tissues.
DNA fragments of the full-length hairpin pre-miR-26a
and its flanking sequence were amplified by PCR and a specific band
of ~194 bp was identified by electrophoresis. Verified by
dual-enzyme (Mlu I and Cla I) digestion, recombinant
plasmids produced fragments of approximately 11 kb and 194 bp,
consistent with the predicted results. In addition, the sequencing
results were the same as the precursor miRNA sequences in the
miRBase database (data not shown). Following a graded dilution, the
packaged lentiviral particles were transfected into 293T cells and
the expression of green fluorescent protein (GFP) was observed
under a light microscope and a fluorescence inverted microscope
(Fig. 1D and E). The number of
GFP-positive 293T cells in each well was quantified, and the
determined lentiviral titer was 5×105 TU/ml.
Cell viability and expression levels
of miR-26a in BMSCs prior to and following lentiviral
infection
An MTT assay was conducted to identify the viability
of BMSCs in different groups (Fig.
2A). No significant differences were observed in BMSC viability
between uninfected BMSCs and BMSCs infected with the pLVTHM empty
vector (P>0.05). However, marked increases in the viability of
BMSCs infected with pLVTHM-miR-26a were observed from day 3
compared with uninfected BMSCs (P<0.05). The cells of all groups
grew continuously and the growth of these cells was not inhibited
by cytotoxicity. To validate successful lentiviral infection,
RT-qPCR was conducted to detect miR-26a expression prior to and
following lentiviral infection. Compared with uninfected BMSCs,
BMSCs infected with pLVTHM-miR-26a exhibited significantly
increased miR-26a expression (P<0.01), and BMSCs infected with
empty vector pLVTHM were not significantly different compared with
uninfected BMSCs (P>0.05), indicating that BMSCs were
successfully infected with the lentiviral vector pLVTHM-miR-26a
(Fig. 2B).
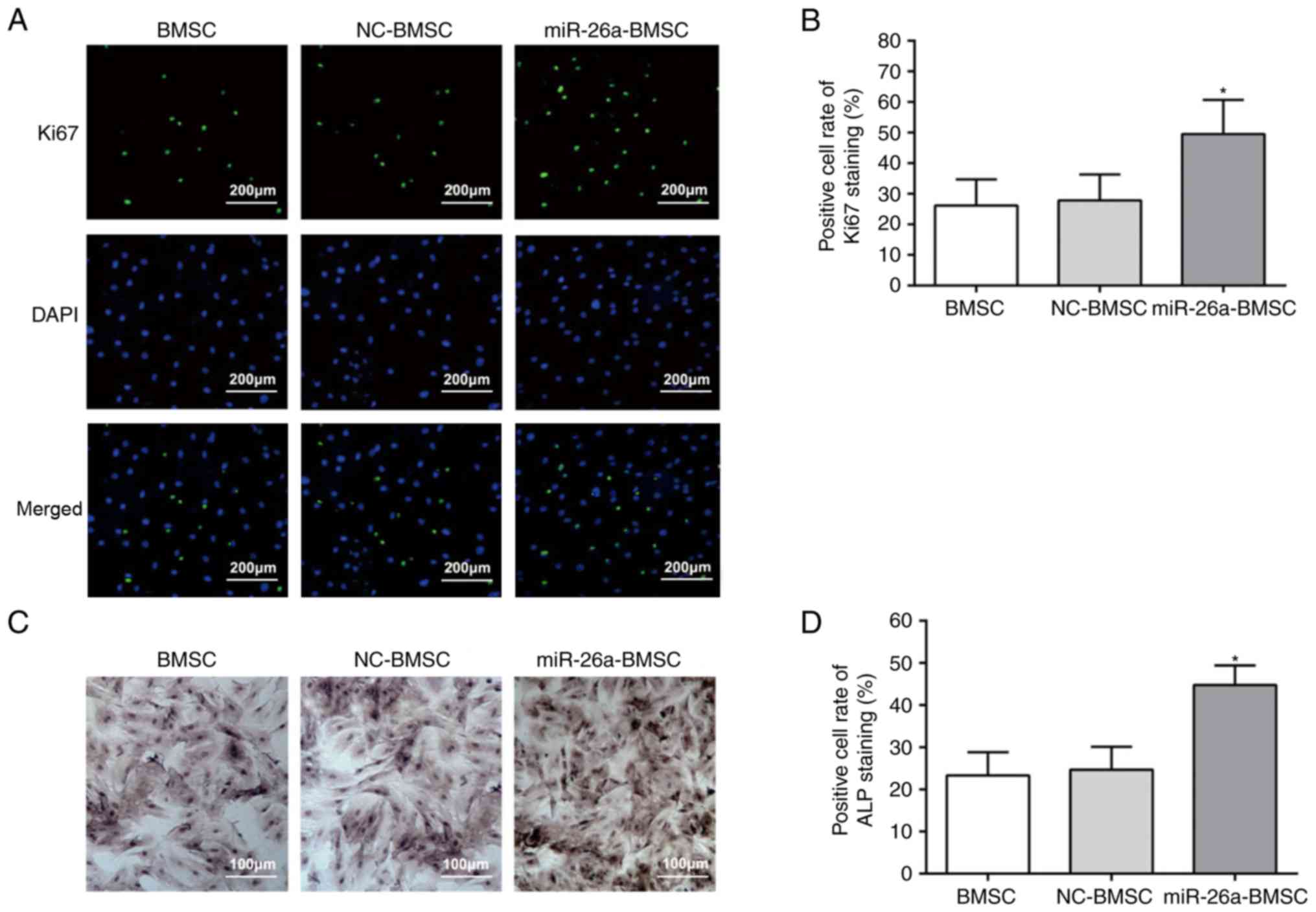
Effect of miR-26a on cell
proliferation and osteogenic capacity of BMSCs
Ki67 is a marker for cell proliferation, and a
higher positive rate of Ki67 reflects more active cell
proliferation (27). The results
of Ki67 and ALP staining are demonstrated in Fig. 3. The BMSCs of all groups had
Ki67-positive cells (Fig. 3A) and
the positive rate of the miR-26a-BMSC group was markedly higher
compared with the uninfected BMSC and BMSC-NC groups (Fig. 3B). No difference was identified
between the uninfected BMSC and BMSC-NC groups (Fig. 3B). Furthermore, the results of ALP
staining (Fig. 3C) indicated that,
following culture for 14 days, all groups exhibited varying
positive reactions, and brown or black granules and blocks appeared
in the cytoplasm. Compared with the uninfected BMSC group, the
miR-26a-BMSC group demonstrated an increased ALP positive rate
(P<0.05), while the uninfected BMSC group and the NC-BMSC group
were not significantly different (Fig.
3D). These findings indicated that miR-26a may facilitate the
osteogenic capacity of BMSCs.
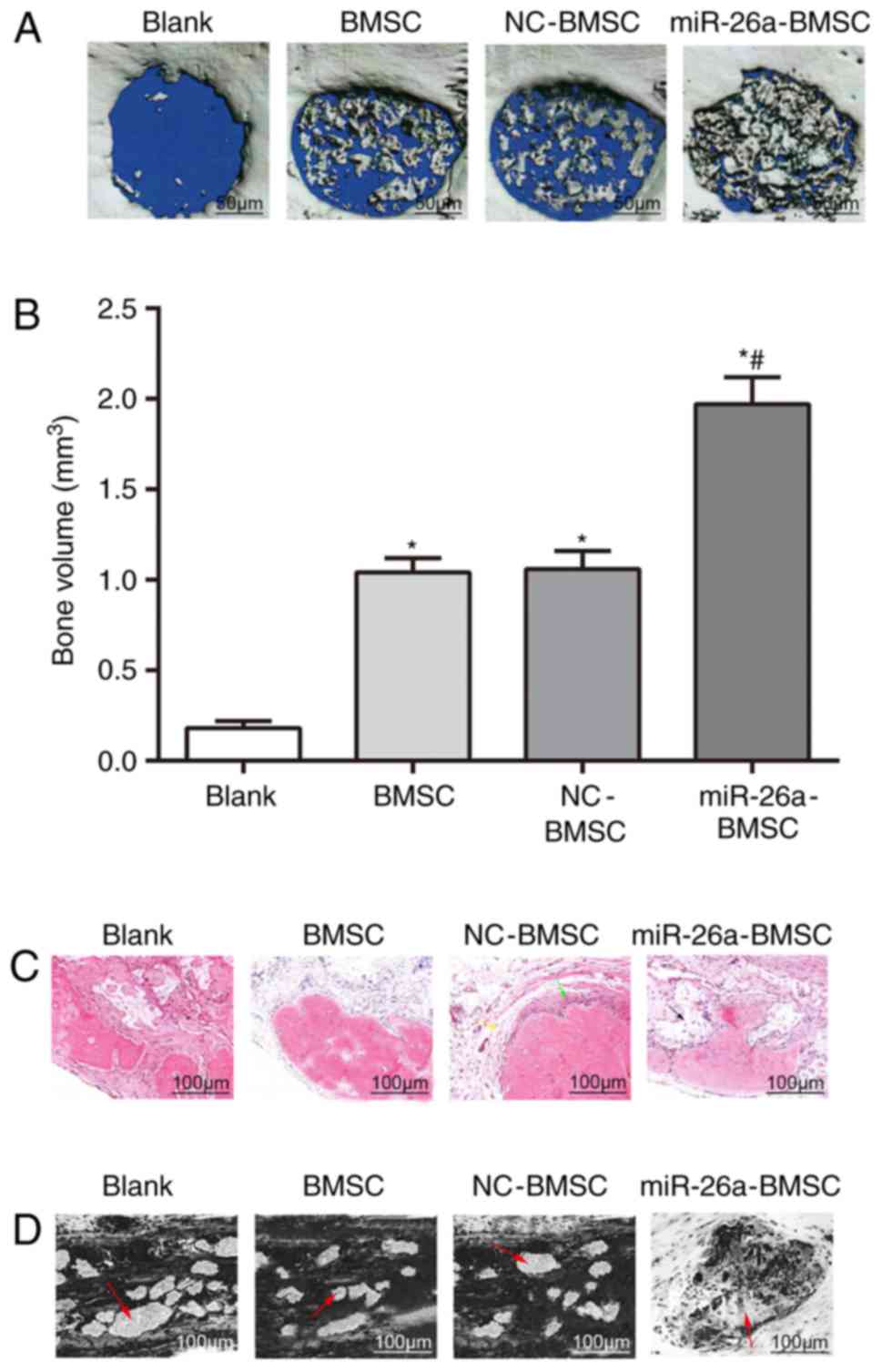
Bone regeneration in the defect areas
of mice from different groups
The conditions of newly formed bones in the defect
area were observed using micro-CT. The results (Fig. 4A) indicated that mice in the blank
group implanted with β-TCP scaffolds alone in the defect area did
not demonstrate newly formed bones. Mice in the BMSC group,
implanted with β-TCP scaffolds co-cultured with uninfected BMSCs in
the defect area, and mice in the NC-BMSC group, implanted with
β-TCP scaffolds co-cultured with NC-BMSCs in the defect area,
exhibited a few scattered newly formed bones. Mice in the
miR-26a-BMSC group, implanted with β-TCP scaffolds co-cultured with
miR-26a-BMSCs in the defect area, presented a marked increase in
newly formed bones compared with the BMSC and NC-BMSC groups, which
almost filled the whole defect area (Fig. 4A).
Bone volume of newly formed bones in
the defect areas of mice from different groups
Quantitative analysis was performed on the bone
volume of newly formed bones in the defect areas (Fig. 4B). Compared with the blank group,
the bone volume of newly formed bones in the BMSC and BMSC-NC
groups was significantly increased (P<0.05), indicating that
BMSCs promoted bone regeneration in bone defect repair.
Furthermore, compared with the BMSC group, the bone volume of newly
formed bones in the miR-26a-BMSC group was further increased
(P<0.05), indicating that lentivirus-mediated miR-26a
overexpression in BMSCs enhanced bone regeneration in bone defect
repair in mice.
Bone regeneration and degradation of
implanted scaffolds in the defect areas of mice from different
groups
To further verify the effects of miR-26a on bone
regeneration in the repair of cranial bone defects, H&E
staining was conducted to observe the bone regeneration conditions
in the defect areas. In the defect areas of mice from the 4 groups,
mice in the miR-26a-BMSC group demonstrated regeneration of bone
tissues. Even staining of newly formed bone tissues was observed.
Spindle-shaped osteoblasts were observed in the margin of newly
formed bones. Erythrocytes were observed inside and surrounding
newly formed bones and scattered in newly formed blood vessels.
Mice in the BMSC and BMSC-NC groups demonstrated minimal
regeneration of bone tissues in addition to some bone cells, and
mice in the blank group did not demonstrate regeneration of bone
tissues (Fig. 4C).
Toluidine blue staining was performed to observe the
degradation conditions of implanted scaffolds in the bone defect
areas of the cranium (Fig. 4D). In
the blank group, scaffolds that were not degraded and enveloped by
fibrosis were observed in the defect area. In the BMSC and BMSC-NC
groups, the scaffolds were gradually degraded and scaffolds that
were not degraded were scattered. In the miR-26a-BMSC group, the
majority of scaffolds were degraded and replaced by newly formed
bone tissues.
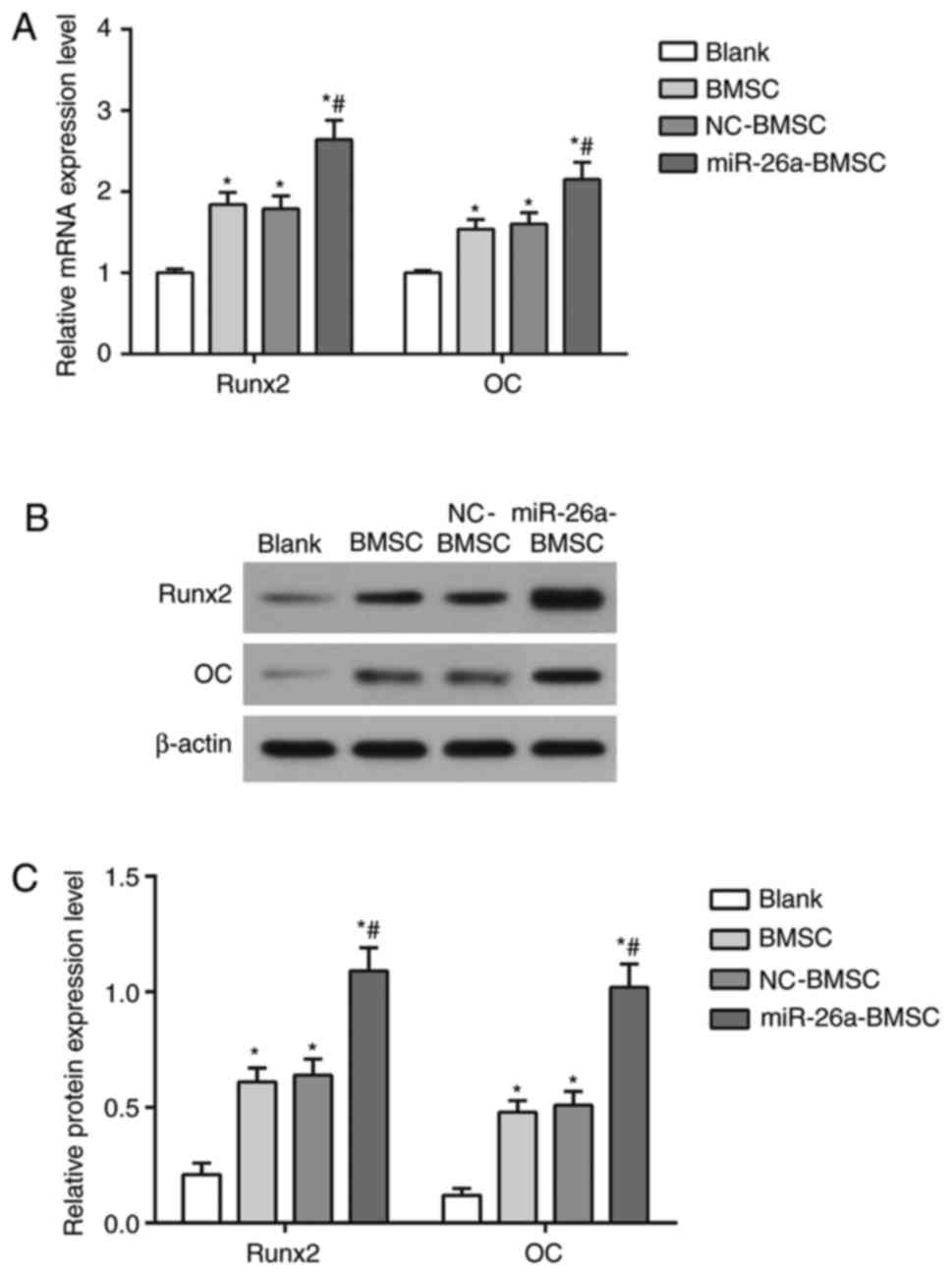
Expression levels of
osteogenesis-associated cytokines in the defect areas of mice from
different groups
RT-qPCR and western blot analysis were performed to
detect the mRNA and protein expression of the
osteogenesis-associated cytokines Runx2 and OC (Fig. 5). Compared with the blank group,
the mRNA and protein expression of Runx2 and OC in the BMSC and
BMSC-NC groups were elevated (P<0.05). The mRNA (Fig. 5A) and protein (Fig. 5B and C) expression of Runx2 and OC
in the miR-26a-BMSC group was also increased compared with the
blank group (P<0.05), and also significantly higher compared
with the BMSC group (P<0.05), indicating that
lentiviral-mediated miR-26a overexpression in BMSCs may enhance
bone regeneration in bone defect repair.
Discussion
Repair of bone defects presents a serious clinical
challenge as it is difficult to restore bone function and
regenerate bone loss (28). To
address this issue, the present study proposed miR-26a as a
potential therapeutic target, and lentivirus-mediated miR-26a
overexpression was used to genetically modify BMSCs in the repair
of cranial bone defects in mice. Delivery of BMSCs modified by
miR-26a to β-TCP scaffold-filled defect areas markedly enhanced
bone regeneration and novel bone formation, and also intensified
the BMSC proliferation capacity.
Initially, the present study successfully
constructed the lentiviral vector pLVTHM-miR-26a, which upregulated
the expression levels of miR-26a in BMSCs from mice. BMSCs
overexpressing miR-26a markedly increased bone regeneration and the
volume of newly formed bones, and also promoted BMSC proliferation.
Concerning the association between miRNAs and bone diseases,
Seeliger et al (29)
highlighted that five miRNAs, which included miR-21, miR-23a,
miR-24, miR-25 and miR-100, were elevated in the bone tissue and
serum of patients suffering from osteoporosis. Despite the
documented effects of miRNAs on osteoclastogenesis,
osteoblastogenesis and osteogenesis, their clinical value remains
poorly defined (30,31). The present study went further by
combining miR-26a with BMSCs and β-TCP scaffolds. The results of
in vitro and in vivo experiments in one study have
demonstrated that transfection of miR-26a significantly accelerated
the osteogenic differentiation of adipose-derived stem cells in
vitro and enhanced new bone formation following miR-26a
transfection in vivo (32).
Furthermore, the repair response to critical calvarial bone defects
was demonstrated to be strengthened through positive modulation of
miR-26a in angiogenic-osteogenic coupling (33). The underlying mechanism by which
miR-26a positively mediates angiogenic-osteogenic coupling may be
due to the fact that its high expression in newly formed bones
increases vascular endothelial growth factor (VEGF) secretion.
Bone, which is a highly vascularized tissue, relies on coordinated
angiogenic-osteogenic coupling to regenerate (34). miR-26a has been reported to be
implicated in VEGF-mediated angiogenesis through the regulation of
endothelial nitric oxide synthase activity, which is modulated by
its effect on NUS1 dehydrodolichyl diphosphate synthase subunit
(NgBR) expression by directly targeting the NgBR 3′-UTR (35). miRNAs are crucial regulators of the
differentiation of BMSCs. For example, upregulated miR-16
expression has been reported to promote BMSC arrest in the G1 phase
and enhance the differentiation of BMSCs of a cardiac niche towards
the myogenic phenotype (10,11).
In the present study, β-TCP scaffolds loaded with
miR-26a-modified and GFP-labeled BMSCs were implanted into defect
areas in mouse models of cranial bone defects. Subsequently, the
regeneration and volume of newly formed bones were demonstrated to
be markedly increased compared with the blank, uninfected BMSC and
BMSC-NC groups. Similar findings were identified in a previous
study, which demonstrated that β-TCP scaffolds seeded with
osteogenically induced BMSCs significantly repaired critically
sized mandibular defects in canine models as osteoclast-like cells
may originate in precursors of mononuclear myeloid cells and lead
to angiogenesis or migration from the microenvironment to scaffolds
(36). Multipotent and
undifferentiated BMSCs enable cells to transform into
differentiated types, producing similar phenotypic expression to
that of the resident cells of a particular tissue, such as bone
(37). Mesenchymal stem cells
(MSCs) and MSC-derived endothelial cells are reported to complement
one another and facilitate the vascularization of biomaterials and
the degree of bone regeneration (38). Furthermore, Dupont et al
(39) stated that porous scaffolds
augmented with stem cells accelerated the repair response to large
segmental bone defects. Notably, the microenvironment and
surrounding tissues facilitate MSC differentiation by secreting
growth factors, nutrients and extracellular matrices, and MSCs are
reprogrammed during gene expression (40).
To further confirm the initial findings of the
present study, the mRNA and protein expression of
osteogenesis-associated cytokines, including Runx2 and OC, in mice
treated with the various types of BMSCs and β-TCP scaffolds were
detected using RT-qPCR and western blot analysis. The results
demonstrated that mice implanted with β-TCP scaffolds co-cultured
with miR-26a-BMSCs in the defect area demonstrated significantly
elevated mRNA and protein expression of Runx2 and OC compared with
the blank group, and the levels were even higher compared with the
uninfected BMSC and BMSC-NC groups. Su et al (41) reported that overexpression of
miR-26a increased ALP activity and Runx2 mRNA expression levels in
BMSCs. Another study by Luzi et al (19) also demonstrated that upregulation
of miR-26a expression elevated the expression of OC in bone
diseases. It is widely accepted that the expression levels of Runx2
and OC may be detected to evaluate bone regeneration, and
osteogenesis influences BMSCs by enhancing the formation of blood
vessels, producing osteogenic cues and delivering osteoblastic
progenitors at the same time (7).
In conclusion, the present study provided promising
evidence that modified BMSCs by lentivirus-mediated miR-26a may
facilitate the repair of cranial bone defects in mouse models and
that β-TCP scaffolds seeded with BMSCs exhibit satisfactory
regeneration and formation of new bones. However, a limitation for
the present study is that quantitative examination in terms of
degradation rate was not performed, and only qualitative
degradation conditions of each group were observed based on the
toluidine blue staining. As the degradation of β-TCP matched the
regeneration of newly formed bone tissues, the degradation
conditions could be demonstrated by conditions of newly formed bone
tissues. Despite this limitation, the present study may provide a
potential therapeutic target for the treatment of bone defects.
Further investigations should focus on transferring the theoretical
hypothesis to clinical practice with quantitative examination of
the degradation rate.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The analyzed data sets generated during the study
are available from the corresponding author on reasonable
request.
Authors' contributions
ZL and BY designed the study. ZL, BY and HC wrote
the manuscript. HC and YH researched relevant references and
established the methodology. YW and ZZ acquired the data. MW and ZH
analyzed and interpreted the data.
Ethics approval and consent to
participate
The present study was approved by the Experimental
Animal Ethics Committee of the People's Hospital of Dongsheng.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kolambkar YM, Boerckel JD, Dupont KM,
Bajin M, Huebsch N, Mooney DJ, Hutmacher DW and Guldberg RE:
Spatiotemporal delivery of bone morphogenetic protein enhances
functional repair of segmental bone defects. Bone. 49:485–492.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Calori GM, Mazza E, Colombo M and
Ripamonti C: The use of bone-graft substitutes in large bone
defects: Any specific needs? Injury. 42 Suppl 2:S56–S63. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lv J, Xiu P, Tan J, Jia Z, Cai H and Liu
Z: Enhanced angiogenesis and osteogenesis in critical bone defects
by the controlled release of BMP-2 and VEGF: Implantation of
electron beam melting-fabricated porous Ti6Al4V scaffolds
incorporating growth factor-doped fibrin glue. Biomed Mater.
10:0350132015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kusumbe AP, Ramasamy SK and Adams RH:
Coupling of angiogenesis and osteogenesis by a specific vessel
subtype in bone. Nature. 507:323–328. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kolambkar YM, Dupont KM, Boerckel JD,
Huebsch N, Mooney DJ, Hutmacher DW and Guldberg RE: An
alginate-based hybrid system for growth factor delivery in the
functional repair of large bone defects. Biomaterials. 32:65–74.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li J, Hong J, Zheng Q, Guo X, Lan S, Cui
F, Pan H, Zou Z and Chen C: Repair of rat cranial bone defects with
nHAC/PLLA and BMP-2-related peptide or rhBMP-2. J Orthop Res.
29:1745–1752. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zou D, Zhang Z, Ye D, Tang A, Deng L, Han
W, Zhao J, Wang S, Zhang W, Zhu C, et al: Repair of critical-sized
rat calvarial defects using genetically engineered bone
marrow-derived mesenchymal stem cells overexpressing
hypoxia-inducible factor-1α. Stem Cells. 29:1380–1390.
2011.PubMed/NCBI
|
|
8
|
Zhang Y, Wang F, Chen J, Ning Z and Yang
L: Bone marrow-derived mesenchymal stem cells versus bone marrow
nucleated cells in the treatment of chondral defects. Int Orthop.
36:1079–1086. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lin CY, Chang YH, Lin KJ, Yen TC, Tai CL,
Chen CY, Lo WH, Hsiao IT and Hu YC: The healing of critical-sized
femoral segmental bone defects in rabbits using
baculovirus-engineered mesenchymal stem cells. Biomaterials.
31:3222–3230. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu JL, Jiang L, Lin QX, Deng CY, Mai LP,
Zhu JN, Li XH, Yu XY, Lin SG and Shan ZX: MicroRNA 16 enhances
differentiation of human bone marrow mesenchymal stem cells in a
cardiac niche toward myogenic phenotypes in vitro. Life Sci.
90:1020–1026. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen S, Yang L, Jie Q, Lin YS, Meng GL,
Fan JZ, Zhang JK, Fan J, Luo ZJ and Liu J: MicroRNA125b suppresses
the proliferation and osteogenic differentiation of human bone
marrowderived mesenchymal stem cells. Mol Med Rep. 9:1820–1826.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xu JF, Yang GH, Pan XH, Zhang SJ, Zhao C,
Qiu BS, Gu HF, Hong JF, Cao L, Chen Y, et al: Altered microRNA
expression profile in exosomes during osteogenic differentiation of
human bone marrow-derived mesenchymal stem cells. PLoS One.
9:e1146272014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang J, Han C and Wu T: MicroRNA-26a
promotes cholangiocarcinoma growth by activating β-catenin.
Gastroenterology. 143:246–256, e248. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mohamed JS, Lopez MA and Boriek AM:
Mechanical stretch up-regulates microRNA-26a and induces human
airway smooth muscle hypertrophy by suppressing glycogen synthase
kinase-3β. J Biol Chem. 285:29336–29347. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang X, Liang L, Zhang XF, Jia HL, Qin Y,
Zhu XC, Gao XM, Qiao P, Zheng Y, Sheng YY, et al: MicroRNA-26a
suppresses tumor growth and metastasis of human hepatocellular
carcinoma by targeting interleukin-6-Stat3 pathway. Hepatology.
58:158–170. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Leeper NJ, Raiesdana A, Kojima Y, Chun HJ,
Azuma J, Maegdefessel L, Kundu RK, Quertermous T, Tsao PS and Spin
JM: MicroRNA-26a is a novel regulator of vascular smooth muscle
cell function. J Cell Physiol. 226:1035–1043. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bahi A, Chandrasekar V and Dreyer JL:
Selective lentiviral-mediated suppression of microRNA124a in the
hippocampus evokes antidepressants-like effects in rats.
Psychoneuroendocrinology. 46:78–87. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sun BS, Dong QZ, Ye QH, Sun HJ, Jia HL,
Zhu XQ, Liu DY, Chen J, Xue Q, Zhou HJ, et al: Lentiviral-mediated
miRNA against osteopontin suppresses tumor growth and metastasis of
human hepatocellular carcinoma. Hepatology. 48:1834–1842. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Luzi E, Marini F, Tognarini I, Galli G,
Falchetti A and Brandi ML: The regulatory network menin-microRNA
26a as a possible target for RNA-based therapy of bone diseases.
Nucleic Acid Ther. 22:103–108. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Luzi E, Marini F, Sala SC, Tognarini I,
Galli G and Brandi ML: Osteogenic differentiation of human adipose
tissue-derived stem cells is modulated by the miR-26a targeting of
the SMAD1 transcription factor. J Bone Miner Res. 23:287–295. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ye JH, Xu YJ, Gao J, Yan SG, Zhao J, Tu Q,
Zhang J, Duan XJ, Sommer CA, Mostoslavsky G, et al: Critical-size
calvarial bone defects healing in a mouse model with silk scaffolds
and SATB2-modified iPSCs. Biomaterials. 32:5065–5076. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Williams JR: The declaration of Helsinki
and public health. Bull World Health Organ. 86:650–652. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mikheev AG: 2 modifications of the
calcium-cobalt method for cytochemical determination of alkaline
phosphatase in leukocytes of the blood and bone marrow. Lab Delo.
12:711–713. 1969.(In Russian). PubMed/NCBI
|
|
24
|
Li J, Jin L, Wang M, Zhu S and Xu S:
Repair of rat cranial bone defect by using bone morphogenetic
protein-2-related peptide combined with microspheres composed of
polylactic acid/polyglycolic acid copolymer and chitosan. Biomed
Mater. 10:0450042015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xu L, Lv K, Zhang W, Zhang X, Jiang X and
Zhang F: The healing of critical-size calvarial bone defects in rat
with rhPDGF-BB, BMSCs, and β-TCP scaffolds. J Mater Sci Mater Med.
23:1073–1084. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li TJ, Browne RM and Matthews JB:
Epithelial cell proliferation in odontogenic keratocysts: A
comparative immunocytochemical study of Ki67 in simple, recurrent
and basal cell naevus syndrome (BCNS)-associated lesions. J Oral
Pathol Med. 24:221–226. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Giannoudis PV, Faour O, Goff T, Kanakaris
N and Dimitriou R: Masquelet technique for the treatment of bone
defects: Tips-tricks and future directions. Injury. 42:591–598.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Seeliger C, Karpinski K, Haug AT, Vester
H, Schmitt A, Bauer JS and van Griensven M: Five freely circulating
miRNAs and bone tissue miRNAs are associated with osteoporotic
fractures. J Bone Miner Res. 29:1718–1728. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
van Wijnen AJ, van de Peppel J, van
Leeuwen JP, Lian JB, Stein GS, Westendorf JJ, Oursler MJ, Im HJ,
Taipaleenmäki H, Hesse E, et al: MicroRNA functions in osteogenesis
and dysfunctions in osteoporosis. Curr Osteoporosis Rep. 11:72–82.
2013. View Article : Google Scholar
|
|
31
|
Li KC, Chang YH, Yeh CL and Hu YC: Healing
of osteoporotic bone defects by baculovirus-engineered bone
marrow-derived MSCs expressing MicroRNA sponges. Biomaterials.
74:155–166. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang Z, Zhang D, Hu Z, Cheng J, Zhuo C,
Fang X and Xing Y: MicroRNA-26a-modified adipose-derived stem cells
incorporated with a porous hydroxyapatite scaffold improve the
repair of bone defects. Mol Med Rep. 12:3345–3350. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Paquet J, Moya A, Bensidhoum M and Petite
H: Engineered cell-free scaffold with two-stage delivery of
miRNA-26a for bone repair. Ann Transl Med. 4:2042016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li Y, Fan L, Liu S, Liu W, Zhang H, Zhou
T, Wu D, Yang P, Shen L, Chen J and Jin Y: The promotion of bone
regeneration through positive regulation of angiogenic-osteogenic
coupling using microRNA-26a. Biomaterials. 34:5048–5058. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jo HN, Kang H, Lee A, Choi J, Chang W, Lee
MS and Kim J: Endothelial miR-26a regulates VEGF-Nogo-B
receptor-mediated angiogenesis. BMB Rep. 50:384–389. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yuan J, Zhang WJ, Liu G, Wei M, Qi ZL, Liu
W, Cui L and Cao YL: Repair of canine mandibular bone defects with
bone marrow stromal cells and coral. Tissue Eng Part A.
16:1385–1394. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tian H, Bharadwaj S, Liu Y, Ma PX, Atala A
and Zhang Y: Differentiation of human bone marrow mesenchymal stem
cells into bladder cells: Potential for urological tissue
engineering. Tissue Eng Part A. 16:1769–1779. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhou J, Lin H, Fang T, Li X, Dai W, Uemura
T and Dong J: The repair of large segmental bone defects in the
rabbit with vascularized tissue engineered bone. Biomaterials.
31:1171–1179. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Dupont KM, Sharma K, Stevens HY, Boerckel
JD, Garcia AJ and Guldberg RE: Human stem cell delivery for
treatment of large segmental bone defects. Proc Natl Acad Sci USA.
107:3305–3310. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hasegawa N, Kawaguchi H, Hirachi A, Takeda
K, Mizuno N, Nishimura M, Koike C, Tsuji K, Iba H, Kato Y and
Kurihara H: Behavior of transplanted bone marrow-derived
mesenchymal stem cells in periodontal defects. J Periodontol.
77:1003–1007. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Su X, Liao L, Shuai Y, Jing H, Liu S, Zhou
H, Liu Y and Jin Y: MiR-26a functions oppositely in osteogenic
differentiation of BMSCs and ADSCs depending on distinct activation
and roles of Wnt and BMP signaling pathway. Cell Death Dis.
6:e18512015. View Article : Google Scholar : PubMed/NCBI
|