Introduction
Rheumatoid arthritis (RA) and osteoarthritis (OA)
are the two most frequent types of degenerative joint diseases and
exhibit similar etiology (1,2). RA
is a complex, chronic inflammatory and autoimmune arthritis that
typically causes pain, swelling, stiffness and loss of function in
the joints (1). It has been
estimated that RA affects 0.5–1% of the adult population worldwide
with 20–50 novel cases per 100,000 people occurring annually, which
most frequently occurs in women aged >40 years old (3,4). RA
has become one of the most common causes of reduced productivity
and disability in affected patients and may additionally pose a
substantial financial burden on the family of the patient as well
as society (5). RA manifests as
osteoporosis around the joint and joint space narrowing in the
knees of patients (6). The bone
anatomy degeneration and cystic degeneration of the bone joint
surface may additionally occur with bone defects (7). During RA, the intercondylar fossa is
enlarged and the tibial plateau sinks (8,9).
Patients with late-stage RA may suffer from articular surface
sclerosis, joint subluxation or joint stiffness (10). Furthermore, OA, the most prevalent
form of arthritis worldwide, is a multi-gene and multi-factorial
disease, and is characterized by cartilage degeneration and
subchondral bone alterations, involving synovial tissue and
articular cartilage (10–12). OA may reduce the quality of life
for patients and eventually lead to disability due to pain. The
joint most commonly affected by OA is the knee (8). Similar to RA, OA additionally has an
increased occurrence rate in older adults, particularly in
women.
In routine clinical practice, the diagnostic
criteria for RA and OA are outlined by the American College of
Rheumatology (Atlanta, USA). RA and OA exhibit overlapping
symptoms, making differential diagnosis particularly challenging.
In addition, differentiation between RA and OA is difficult in
late-stage cases, primarily because disease progression frequently
begins prior to the onset of symptoms. Therefore, accurate
diagnosis of RA and OA may significantly improve the clinical
outcomes and prognosis for affected patients. However, the
mechanisms underlying the initiation and progression of RA and OA
remain unclear. Previously, important genes and diagnostic markers
that interact with each other and with environmental and stochastic
factors have been identified in the two diseases (13). However, these markers may not
entirely elucidate the complex pathogenesis of RA and OA.
Therefore, the present study aimed to investigate
the developmental differences between RA and OA. An updated
comprehensive analysis was performed to identify the potential
novel biomarkers associated with synovial tissues obtained from
patients with RA and OA. In the present study, three multicenter
genome-wide transcriptomic datasets, including 33 patients with RA
and 26 patients with OA were retrieved and analyzed. The present
study aimed to investigate the different mechanisms underlying the
differential pathogenesis of RA and OA, and thus improve the
diagnosis and treatment strategies available for patients suffering
from the two diseases in clinical practice.
Materials and methods
Microarray dataset source
A systematic search of microarray datasets was
performed to examine differentially expressed genes (DEGs) between
RA and OA. The National Center for Biotechnology Information's Gene
Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/) was utilized to
retrieve appropriate microarray datasets. The key words
‘Osteoarthritis’ and ‘Rheumatoid Arthritis’ were used for the
screening. Datasets were included if they met the following
inclusion criteria: i) were based on gene expression profiling of
synovial membrane samples from the same platform. When the
microarray datasets are obtained from the same platform, their
homogeneity is usually good. Subsequent to screening OA-associated
microarray datasets, the GPL96 platform was used at the highest
frequency. Therefore, the OA-associated microarray datasets
obtained from the GPL96 platform were included in the present
study; ii) case (RA)-control (OA) studies; iii) patients with RA
were diagnosed and classified based on the American College of
Rheumatology criteria (14) and
patients with OA were classified according to the criteria of
Diagnostic and Therapeutic Criteria Committee of the American
Rheumatism Association (15); and
iv) the number of synovial tissue samples in each group of patients
with RA and OA was ≥6. Three gene expression datasets, GSE55235
(nRA=10 and nOA=10), GSE55584
(nRA=10 and nOA=6) and GSE55457
(nRA=13 and nOA=10) met the inclusion
criteria and were included in the present study (16).
A further dependent dataset, GSE36700 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE36700)
(17), including microarray data
from synovial biopsies of patients with RA (n=7) and OA (n=5), was
used to validate the results obtained from the GSE55235, GSE55584
and GSE55457 datasets. The GSE36700 dataset was created based on
the Affymetrix Human Genome U133 Plus 2.0 Array (Affymetrix UK
Ltd., High Wycombe, UK) and was submitted by Nzeusseu Toukap et
al (17) March 22, 2012 and
updated on Aug 09, 2018. Patients with RA were diagnosed and
classified based on the 1987 American College of Rheumatology
criteria (14) and patients with
OA were classified according to X-ray evidence of osteoarthritis
(15).
Data preprocessing and differential
expression analysis
The three databases were created based on the
Affymetrix Human Genome U133A Array (Affymetrix UK Ltd.). A robust
multi-array average algorithm using the Affy package (justMRA;
http://ugrad.stat.ubc.ca/R/library/affy/html/00Index.html)
was conducted for background adjustment, normalization and
summarization of the three datasets to minimize data inconsistency
and heterogeneity. The probe sets were converted into corresponding
gene annotation using R/Bioconductor package (version 3.22.4;
http://www.bioconductor.org/packages/release/BiocViews.html#___ChipManufacturer)
and the Affymetrix Human Genome U133 Plus 2.0 Array. The probes
with no gene annotation were excluded from the analysis. The
expression values of all probes for a given gene were calculated
from the average expression value. DEGs [P<0.05; false discovery
rate (FDR) <0.05] between RA and OA from the three datasets were
investigated using R software (v3.4.0; http://bioconductor.org/biocLite.R). FDR was applied
based on the Benjamini & Hochberg method (18) and two independent sample Student's
t-test was performed to select sets of DEGs. To reduce the false
positive rate, DEGs of the three datasets were identified, and
subsequently Venn diagrams (Venn 2.1; http://bioinfogp.cnb.csic.es/tools/venny/index.html)
were used to screen the overlapping DEGs of the three datasets to
improve the stability of the subsequent results.
Biological functions and pathway
enrichment analyses
To further elucidate the biological functions of
DEGs between RA and OA, the identified DEGs were subjected to Gene
Ontology (GO; http://www.geneontology.org/) term enrichment and
Kyoto Encyclopedia of Genes and Genomes (KEGG; http://www.kegg.jp/) pathway analyses using Database
for Annotation, Visualization and Integrated Discovery (DAVID;
http://david.ncifcrf.gov/),
respectively. Significantly enriched pathways and GO terms (P≤0.05;
number of enrichment genes ≥2) were identified using Cytoscape
3.5.1 software (http://www.softpedia.com/get/Science-CAD/Cytoscape.shtml)
and GeneClip 2.0 (http://gsds.cbi.pku.edu.cn/). The identified genes
were classified into three functional categories, including the
Biological Process (BP), Molecular Function (MF) and Cellular
Component (CC). Fisher's exact tests (two-sided) or χ2
tests were performed to categorize the pathway and GO terms. The
FDR (Benjamini & Hochberg method) (18) was applied to obtain the corrected
P-values. Significantly enriched pathways and GO terms (P£0.05;
number of enrichment genes ≥2) were identified using Cytoscape
3.5.1 software.
Analysis of pathway networks
The establishment of a pathway network of DEGs
between RA and OA may help to identify important pathways
associated with the development of RA and OA. Furthermore, pathway
network analysis may reveal the possible interactions and crosstalk
among these pathways. Therefore, pathway networks were constructed
based on the identified DEGs using the ClueGO plugin on the
Cytoscape platform (http://www.softpedia.com/get/Science-CAD/Cytoscape.shtml).
Protein-protein interaction (PPI)
network analysis of DEGs
The Search Tool for the Retrieval of Interacting
Genes/Proteins (STRING) database (http://string-db.org/) was used to analyze the PPI of
DEGs, and Cytoscape software was used to construct the PPI network.
FDR<0.05 was considered as the cut-off criterion. A network was
constructed consisting of nodes and lines in which each node
represents a protein and the lines represent direct interactions
between proteins. The PPI network was constructed based on human
data alone. The number of nodes that may interact with a given node
was expressed as the degree of the node. The greater the degree
values of the included genes, the greater the degree in the whole
network.
Identification of candidate genes
between RA and OA
Identification of genes that may affect the
development of RA and OA within the genome may provide a
comprehensive understanding of the differences between the
pathogenesis of RA and OA. Candidate genes that may affect the
development of RA and OA based on the DEGs were predicted using
Molecular Complex Detection Algorithm (MCODE) in Cytoscape
(19). Furthermore, MCODE cluster
analysis was performed using Cytocluster 3.5.1 software (20) (Degree cutoff=2; Node score
cutoff=0.2; K-core=2; Max Depth=100) to identify the most
significant MCODE clusters, according to clustering scores. The
GeneMANIA Cytoscape plugin was used to identify and prioritize
novel candidate genes involved in RA and OA. The establishment of
entire PPI networks of DEGs between RA and OA were identified based
on the biological network using the GeneMANIA plugin (21). The PPI networks were composed of
genes included in the list of 80 DEGs and predicted candidate genes
that may affect the development of RA and OA. Following the
selection of Homo sapiens as the organism, common DEGs were
entered into the GeneMANIA search bar, and the PPI network was
constructed. Following this, the whole PPI network was filtered
using a degree-filtering approach to include the most critical
biomarkers in the occurrence of the two diseases using Cytoscape
3.5.1 software.
Statistical analysis
The raw expression data of patients with RA and OA
were obtained from GEO datasets and logarithmically transformed.
The means of two continuous, normally distributed variables were
compared by independent sample Student's t-tests. Mann-Whitney U
tests were used to compare the means of two groups of variables not
normally distributed. Receiver Operating Characteristic (ROC)
analysis was performed to identify a more accurate cut-off point in
the gene expression level, which may aid the classification of RA
and OA. ROC curves were generated by plotting the range of
sensitivity (true positive fractions) and specificity (false
positive fractions) pairs for each subject's error rate, with case
status (RA vs. OA) representing the classifier variable. Youden's
index was used for capturing the performance of a dichotomous
diagnostic test. Youden's index=sensitivity + specificity −1
(22).
The area under the ROC curve (AUC), which provides
an estimate of the accuracy of the diagnostic test for the
discrimination between patients with RA and patients with OA, was
used to assess the performance of the test. Binary logistic
regression using backward stepwise selection mode was performed to
screen out potential biomarkers that were positively correlated
with RA diagnosis when identified biomarkers were detected
together. Following this, ROC analysis was performed to determine
the performance of the established logistic regression models. All
statistical analyses in the present study (except for the screening
of DEGs) were performed using SPSS version 24.0 for Windows (IBM
Corp., Armonk, NY, USA) and GraphPad Prism 7.0 (GraphPad Software,
Inc., La Jolla, CA, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
Identification of DEGs between
patients with RA and OA
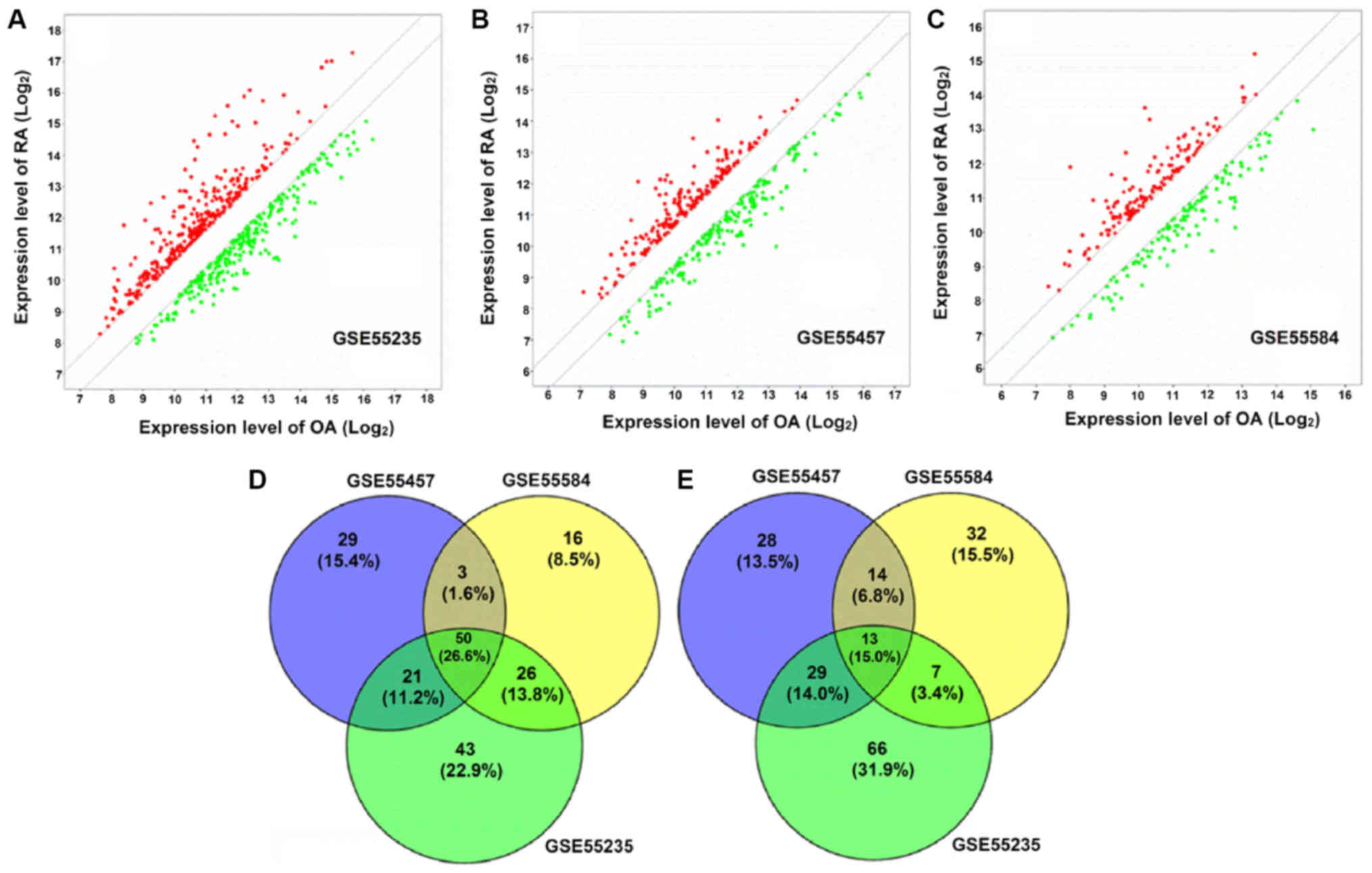
DEGs were identified by the t-test statistical
algorithm. Based on the cutoff criteria, 140, 103 and 95 genes were
identified in GSE55235, GSE55457 and GSE55584 datasets,
respectively, which were upregulated in patients with RA (Fig. 1A-C). In addition, 133, 102 and 84
genes were identified in GSE55235, GSE55457 and GSE55584 datasets,
respectively, which were downregulated in patients with RA
(Fig. 1A-C). Notably, 50
upregulated (Fig. 1D) and 31
downregulated DEGs (Fig. 1E) in
patients with RA were identified as being overlapped between the
three datasets. One DEG without a symbol was excluded from the
upregulated DEGs, therefore 80 DEGs in total, containing 49
upregulated and 31 downregulated, were included in the final
analysis. The list of 80 DEG symbols is available upon request.
Biological functions and KEGG
pathways
Cytoscape 3.5.1 and GeneClip 2.0 were used for
biological function and pathway enrichment analyses. The results of
these analyses identified 80 overlapped DEGs, which are presented
in Table I. The results
demonstrated that 80 overlapped DEGs were significantly enriched in
immune, inflammation, apoptosis and antioxidant stress-associated
functions and pathways.
 | Table I.Top five biological functions and top
ten KEGG pathways of the overlapped differentially expressed
genes. |
Table I.
Top five biological functions and top
ten KEGG pathways of the overlapped differentially expressed
genes.
| A, Biological
process |
|---|
|
|---|
| ID | GO term
description | Count | FDR |
|---|
| GO:0007166 | Cell surface
receptor signaling pathway | 38 | 1.10×10-13 |
| GO:0051239 | Regulation of
multicellular organismal process | 40 | 1.10×10-13 |
| GO:0030155 | Regulation of cell
adhesion | 23 | 1.23×10-13 |
| GO:2000026 | Regulation of
multicellular organismal development | 32 | 1.41×10-12 |
| GO:0070887 | Cellular response
to chemical stimulus | 37 | 6.78×10-12 |
|
| B, Molecular
function |
|
| ID | GO term
description | Count | FDR |
|
| GO:0005515 | Protein
binding | 51 | 3.83×10-11 |
| GO:0005102 | Receptor
binding | 25 | 1.05×10-09 |
| GO:0005003 | Ephrin receptor
activity | 6 | 4.91×10-08 |
| GO:0008201 | Heparin
binding | 10 | 5.03×10-07 |
| GO:0042802 | Identical protein
binding | 20 | 5.17×10-07 |
|
| C, Cellular
component |
|
| ID | GO term
description | Count | FDR |
|
| GO:0005615 | Extracellular
space | 28 | 4.35×10-11 |
| GO:0009986 | Cell surface | 19 | 2.35×10-08 |
| GO:0005576 | Extracellular
region | 42 | 8.80×10-07 |
| GO:0009897 | External side of
plasma membrane | 11 | 8.80×10-07 |
| GO:0098552 | Side of
membrane | 13 | 2.42×10-06 |
|
| D, KEGG
pathway |
|
| ID | GO term
description | Count | FDR |
|
| 4,060 | Cytokine-cytokine
receptor interaction | 16 | 3.66×10-12 |
| 4,360 | Axon guidance | 9 | 3.42×10-07 |
| 4,062 | Chemokine signaling
pathway | 10 | 3.69×10-07 |
| 5,340 | Primary
immunodeficiency | 6 | 4.88×10-07 |
| 4,151 | PI3K/AKT signaling
pathway | 10 | 8.41×10-05 |
| 4,630 | JAK-STAT signaling
pathway | 7 | 1.87×10-04 |
| 5,162 | Measles | 6 | 7.14×10-04 |
| 4,640 | Hematopoietic cell
lineage | 5 | 1.05×10-03 |
| 4,064 | NF-κB signaling
pathway | 5 | 1.11×10-03 |
| 4,660 | T cell receptor
signaling pathway | 5 | 1.52×10-03 |
| 5,142 | American
trypanosomiasis | 5 | 1.52×10-03 |
PPI network analysis
A PPI network was constructed based on the
biological interactions of the 80 identified DEGs to further
elucidate their associations at the protein level. As presented in
Fig. 2A, 32 nodes were screened
out, including 29 upregulated genes and three downregulated genes
(heparin-binding epidermal growth factor-like growth factor, ephrin
type-A receptor 3 and clusterin) in patients with RA. The PPI
network included three primary sub-clusters: i) A sub-cluster
including C-X-C motif chemokine receptor 4 (CXCR4)
and signal transducer and activator of transcription 1
(STAT1), and at its core was predominantly associated with
chemokines and immune functions (Fig.
2A; top circle); ii) a sub-cluster including LCK
proto-oncogene, Src family tyrosine kinase (LCK), and at
its core was predominantly correlated with the regulation of
developmental events, notably in the nervous system (Fig. 2A; middle circle); iii) a
sub-cluster including interleukin (IL)2 receptor, γ chain
(IL2RG) and CD3d molecule (CD3D), and at its
core was primarily associated with immunodeficiency (Fig. 2A; bottom circle).
Candidate genes and core network
To identify the potential pathological molecular
network of the 80 identified DEGs, the specific network among them
based on the human interactome network using MCODE algorithm was
extracted using the default settings on GeneMANIA. This approach
included maximal members of candidate genes with the minimal
interaction associations. The network, capturing the 80 DEGs as its
seeds, contained 323 nodes and 602 edges, including 30 genes of the
80 DEGs and 293 candidate genes that may affect RA and OA
progression. To identify the most important core network, networks
were filtered according to their degree using the degree-filtering
approach. Finally, a core network including six genes (CD3D,
CXCR4, IL2RG, IL7R, LCK and STAT1) was identified and
presented in Fig. 2B, which
suggested that these six genes may represent important biomarkers
associated with RA and OA development and diagnosis.
Gene-pathway network
To further understand how important genes in the
core PPI network affect RA development, a gene-pathway network was
constructed using ClueGO (Fig.
2C). From the gene-pathway network, five genes (CD3D, IL2RG,
IL7R, LCK and STAT1) were included in the PPI network.
Notably, the results demonstrated that CD3D, IL2RG, IL7R,
LCK and STAT1 interacted with the primary
immunodeficiency pathway, either directly or indirectly
(P<0.001), which suggested that upregulated genes may activate
the primary immunodeficiency pathway and increase the risk of RA
development.
Evaluation of the core network for RA
identification
ROC curves were constructed to calculate the AUCs of
CD3D (0.8357), CXCR4 (0.7855), IL2RG (0.8368),
IL7R (0.9161), LCK (0.8683) and STAT1 (0.9138;
all P<0.0001). Taking the maximum value of the Youden's index,
the Log2 expression value of CD3D (8.65),
CXCR4 (10.86), IL2RG (9.27), IL7R (8.11),
LCK (6.58) and STAT1 (7.36) were determined (Fig. 3A-F and Table II). For RA identification, at the
ROC-derived optimum cut-offs, the highest sensitivity exhibited by
STAT1 was 90.91%. The specificities exhibited by CXCR4,
IL2RG and IL7R were equal and reached a maximum value of
92.31%.
 | Figure 3.Receiver operating characteristic
curves of the six genes in the core network to distinguish
rheumatoid arthritis from osteoarthritis using data from the
GSE55235, GSE55457 and GSE55584 datasets. Receiver operating
characteristic curves of (A) CD3D, (B) CXCR4, (C) IL2RG, (D) IL7R,
(E) LCK, (F) STAT1 and (G) IL7R+STAT1 are presented. CD3D, T-cell
surface glycoprotein CD3 δ chain; CXCR4, C-X-C motif chemokine
receptor 4; IL2RG, interleukin 2 receptor γ; IL7R, interleukin 7
receptor; LCK, LCK proto-oncogene, Src family tyrosine kinase;
STAT1, signal transducer and activator of transcription 1. |
| Table II.Optimal cut-off points and associated
diagnostic values of six genes in the core network as determined by
receiver operator characteristic analysis. |
Table II.
Optimal cut-off points and associated
diagnostic values of six genes in the core network as determined by
receiver operator characteristic analysis.
| Genes | Cut-off value,
Log2 | Sensitivity, % | Specificity, % | AUC | AUC 95% CI |
|---|
| CD3D | 8.65 | 66.67 | 84.62 | 0.8357 | 0.7329–0.9384 |
| CXCR4 | 10.86 | 57.58 | 92.31 | 0.7855 | 0.6696–0.9015 |
| IL2RG | 9.27 | 63.64 | 92.31 | 0.8368 | 0.7381–0.9356 |
| IL7R | 8.11 | 81.82 | 92.31 | 0.9161 | 0.8466–0.9856 |
| LCK | 6.58 | 81.82 | 84.62 | 0.8683 | 0.7718–0.9648 |
| STAT1 | 7.36 | 90.91 | 69.23 | 0.9138 | 0.8404–0.9871 |
Subsequent binary logistic regression analysis
demonstrated that IL7R [odds ratio (OR) RA vs.
OA=4.551; OR 95% confidence interval (CI): 1.517–13.657;
P=0.007] and STAT1 (ORRA vs. OA=2.923; OR 95% CI:
1.091–7.829; P=0.033) were inputted into the regression, which
suggested that IL7R and STAT1 may be detected
together (Fig. 3G and Table III). Finally, ROC analyses
suggested that the detection of IL7R + STAT1 together
exhibited a higher diagnostic performance compared with the
detection of either IL7R or STAT1 alone (AUC=0.9464; 95% CI:
0.8962–0.9966), with a sensitivity of 93.94% and a specificity of
80.77% (data not shown).
| Table III.Binary logistic regression results of
the core network for rheumatoid arthritis diagnosis. |
Table III.
Binary logistic regression results of
the core network for rheumatoid arthritis diagnosis.
| Genes | β | S.E. | Wald | OR | OR 95% CI | P-value |
|---|
| IL7R | 1.515 | 0.561 | 7.307 | 4.551 | 1.517–13.657 | 0.007 |
| STAT1 | 1.073 | 0.503 | 4.551 | 2.923 | 1.091–7.829 | 0.033 |
Validation using an additional,
dependent dataset
To investigate the reliability of the results of the
ROC analyses obtained from all the three datasets and to identify
if there was any possible overlapping between them, the same ROC
analysis, including data from the GSE36700 dataset was performed,
and the results are presented in Fig.
4. Notably, it was demonstrated that the six genes in the core
network exhibited good performance in distinguishing RA from OA. In
addition, the AUCs of genes identified in the GSE36700 dataset were
increased compared with the results obtained from the
aforementioned three datasets. In conclusion, the results suggested
that the results obtained from the GSE36700 dataset were closely
associated with those obtained from the GSE55235, GSE55584 and
GSE55457 datasets, which further confirmed the reliability of the
aforementioned results.
 | Figure 4.Receiver operating characteristic
curves of the six genes in the core network to investigate the
differentiation between rheumatoid arthritis and osteoarthritis
using data from the GSE36700 dataset. Receiver operating
characteristic curves of (A) CD3D, (B) CXCR4, (C) IL2RG, (D) IL7R,
(E) LCK and (F) STAT1 are presented. CD3D, T-cell surface
glycoprotein CD3 δ chain; CXCR4, C-X-C motif chemokine receptor 4;
IL2RG, interleukin 2 receptor γ; IL7R, interleukin 7 receptor; LCK,
LCK proto-oncogene, Src family tyrosine kinase; STAT1, signal
transducer and activator of transcription 1. |
Discussion
RA and OA are the most common forms of degenerative
joint diseases. They are the leading cause of chronic disability
and may exhibit common clinical etiology (23,24).
However, there remains a paucity of studies investigating the
sensitivity and specificity of detection indicators for
identification of the two diseases, particularly for patients with
advanced-stage RA or OA. Recently, epigenetic dysregulation of
cartilage genes has been demonstrated to have an important role in
RA and OA development (24).
Despite advances in the field, biomarkers associated with the
pathogenesis and progression of RA and OA are not well
characterized. Therefore, investigation of the gene signatures
associated with disease development in RA and OA may elucidate the
molecular mechanisms underlying pathogenesis and identify potential
therapeutic strategies for the development of a biomarker of
differential diagnosis.
In previous years, bioinformatics has had an
increasingly important role in examining the pathogenesis of
multifactorial disorders (25). In
the present study, a comprehensive and systematic bioinformatics
analysis of three gene expression profile datasets identified 80
significant DEGs, including 49 upregulated and 31 downregulated
genes that may be associated with the development of RA and OA.
These results suggested that alterations in gene expression
profiles in synovial tissue may affect the development of RA and
OA. Therefore, detailed analysis of the biological functions of the
DEGs may be utilized to further understand the pathogenesis of the
two diseases and may additionally reveal biomarkers for more
accurate identification of RA and OA.
A previous study demonstrated that RA development
may depend on a common alteration in the expression pattern of
specific key genes (26), which
was consistent with the results of the present study. Numerous
previous studies have identified specific genes associated with RA
development. For example, Ma et al (27) identified numerous genes (including
adiponectin, C1Q and collagen domain containing,
3′-phosphoadenosine 5′-phosphosulfate synthase 1, DNA
methyltransferase 1 and TIMP metallopeptidase inhibitor
1) involved in immune responses and inflammatory responses.
Microarray analysis has additionally identified disease spectrum
features in rheumatology and identified additional genes that may
be associated with RA (28,29).
Biswas et al (30)
identified a number of different biomarkers, genes and pathways,
the majority of which have not been revealed in other studies.
Differential diagnoses of RA and OA remain clinically challenging
due to substantial etiological similarities (16). Microarray experiments performed by
Wang et al (31) identified
an overview of differences in OA gene expression compared with
healthy patients and identified 85 DEGs. In conclusion, these
studies suggested that RA and OA have complex pathogenic
mechanisms, and future studies should perform comprehensive and
systematic analyses to further elucidate these mechanisms.
Biological function and KEGG pathway enrichment
analyses identified that 80 overlapped DEGs were significantly
enriched in immune, inflammation, apoptosis and antioxidant
stress-associated functions and pathways, including
‘cytokine-cytokine receptor interaction’, ‘axon guidance’,
‘chemokine signaling pathway’ and ‘primary immunodeficiency’. A
constructed PPI network additionally demonstrated that RA
progression was associated with immunodeficiency. RA has been well
established to represent a progressive, chronic, inflammatory and
destructive joint disease (2).
These results were based on three high throughput microarray
datasets with multi-center design and containing large sample
numbers of synovial tissue, which may provide further evidence for
future research. In the present study, the PPI network studies
demonstrated that CXCR4, LCK, IL2RG and CD3D may
represent potential biomarkers associated with immunodeficiency in
RA. To confirm this inference, a more complete and specific
biological network based on GeneMANIA was determined, from which a
core network of 293 candidate genes that may affect RA and OA
development was obtained. Furthermore, the fact that the core
network was closely aligned with the constructed PPI network
further suggested that the six genes in the core network are
involved in the occurrence and development of RA and OA.
To further investigate how these genes exhibit their
biological function and affect the occurrence of RA, a gene-pathway
interaction network was constructed. The results demonstrated that
in the gene-pathway interaction network, five genes in the core
network (CD3D, IL2RG, IL7R, LCK and STAT1) were
included and notably, these genes were demonstrated to interact
with the primary immunodeficiency pathway either directly (CD3D,
IL2RG, IL7R and LCK) or indirectly (STAT1).
Therefore, the results suggested that altered expression levels of
CD3D, IL2RG, IL7R, LCK and STAT1 may activate the
primary immunodeficiency pathway and subsequently lead to primary
immune system dysfunction and the development of RA.
Primary immunodeficiencies are a heterogeneous group
of disorders that cause increased susceptibility to infection,
autoimmune disease and malignancy (32). From the primary immunodeficiency
pathway, IL7R, LCK and Janus kinase (JAK)3/STAT1
primarily affect T-cell differentiation and antibody production
(33), which may represent the
basis of RA development. Investigation of the diagnostic capacity
to distinguish RA from OA suggested that the genes in the core
network may be detected alone to predict and diagnose RA occurrence
with high sensitivity and specificity; however, the combined
detection of important indicators may improve the effectiveness of
this diagnostic strategy. Therefore, binary logistic regression
analysis was used to screen for IL7R and STAT1
simultaneously to improve RA diagnosis.
A previous study identified that STAT1 is important
in RA occurrence and is upregulated in patients with RA (34), which corroborates the results of
the present study. STAT1 has been widely regarded to represent an
important transcription factor involved in joint inflammation and
destruction (33,35). STAT1 may be activated by numerous
cytokines that are expressed in RA synovium, including interferon
(IFN)γ, type I IFNs, IL6, IL10 and IL27, which induce inflammation
via direct or indirect activation of mitogen-activated protein
kinase, JAK-STAT and nuclear factor-κB signaling pathways (36). The functional defects in important
proteins (including IL7R and IL7) associated with the IL7 signaling
pathway may be involved in the pathogenesis of severe combined
immunodeficiency (SCID) (36).
IL7R was identified as a novel molecule with a potential role in RA
in the present study. IL7R has been identified to have a critical
role in V(D)J recombination during lymphocyte development, and thus
mutations in this gene may increase the risk of SCID (37).
The identification of these two key biomarkers and a
key pathway associated with immunodeficiency in the development of
RA and reveals novel therapeutic targets for anti-immunotherapy for
patients with RA.
In conclusion, the present study demonstrated that
STAT1 and the primary immunodeficiency pathway may be precisely
utilized to differentiate RA from OA. In addition, the present
study additionally identified a previously unreported novel
biomarker (IL7R), which may serve as potential candidate biomarker
to differentiate RA from OA at the time of diagnosis. Therefore,
the present study demonstrated potential implications for future
clinical management of patients with RA and OA.
Acknowledgements
The authors would like to thank Dr Zhenzhong Li from
Beijing Compass Biotechnology Co., Ltd. (Beijing, China) for his
help with part of the data analysis.
Funding
The present study was supported by grants from the
National Natural Science Foundation of China (grant nos. 81773372
and 81573104).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the National Center for Biotechnology
Information's Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo/).
Authors' contributions
YX and RZ designed the study. JW, LH, XY, AY, JZ,
BL, DZ and ZL acquired the data. RZ, YX and JW analyzed and
interpreted the data, and drafted the manuscript. All authors
critically revised the manuscript, and read and approved the final
version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
García-Bermúdez M, López-Mejías R,
González-Juanatey C, Castañeda S, Miranda-Filloy JA, Blanco R,
Fernández-Gutiérrez B, Balsa A, González-Alvaro I, Gómez-Vaquero C,
et al: Lack of association between TLR4 rs4986790 polymorphism and
risk of cardiovascular disease in patients with rheumatoid
arthritis. DNA Cell Biol. 31:1214–1220. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Song YJ, Li G, He JH, Guo Y and Yang L:
Bioinformatics-based identification of MicroRNA-regulated and
rheumatoid arthritis-associated genes. PLoS One. 10:e01375512015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liu G, Jiang Y, Chen X, Zhang R, Ma G,
Feng R, Zhang L, Liao M, Miao Y, Chen Z, et al: Measles contributes
to rheumatoid arthritis: Evidence from pathway and network analyses
of genome-wide association studies. PLoS One. 8:e759512013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ballard DH, Aporntewan C, Lee JY, Lee JS,
Wu Z and Zhao H: A pathway analysis applied to genetic analysis
workshop 16 genome-wide rheumatoid arthritis data. BMC Proc. 3
(Suppl 7):S912009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang H, Guo J, Jiang J, Wu W, Chang X,
Zhou H, Li Z and Zhao J: New genes associated with rheumatoid
arthritis identified by gene expression profiling. Int J
Immunogenet. 44:107–113. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mc Ardle A, Flatley B, Pennington SR and
FitzGerald O: Early biomarkers of joint damage in rheumatoid and
psoriatic arthritis. Arthritis Res Ther. 17:1412015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Carlson A, Bothner B and June R: Toward OA
biomarkers: Metabolomic profiles of synovial fluid from OA, RA, and
healthy patients. Osteoarthritis Cartilage. 25 Suppl 1:S94–S95.
2017. View Article : Google Scholar
|
|
8
|
Atif U, Philip A, Aponte J, Woldu EM,
Brady S, Kraus VB, Jordan JM, Doherty M, Wilson AG, Moskowitz RW,
et al: Absence of association of asporin polymorphisms and
osteoarthritis susceptibility in US Caucasians. Osteoarthritis
Cartilage. 16:1174–1177. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bijsterbosch J, Kloppenburg M, Reijnierse
M, Rosendaal FR, Huizinga TW, Slagboom PE and Meulenbelt I:
Association study of candidate genes for the progression of hand
osteoarthritis. Osteoarthritis Cartilage. 21:565–569. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fang H, Zhang F, Li F, Shi H, Ma L, Du M,
You Y, Qiu R, Nie H, Shen L, et al: Mitochondrial DNA haplogroups
modify the risk of osteoarthritis by altering mitochondrial
function and intracellular mitochondrial signals. Biochim Biophys
Acta. 1862:829–836. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang M, Mu H, Lv H, Duan L, Shang Z, Li
J, Jiang Y and Zhang R: Integrative analysis of genome-wide
association studies and gene expression analysis identifies
pathways associated with rheumatoid arthritis. Oncotarget.
7:8580–8589. 2016.PubMed/NCBI
|
|
12
|
González-Huerta NC, Borgonio-Cuadra VM,
Zenteno JC, Cortés-González S, Duarte-Salazar C and Miranda-Duarte
A: D14 repeat polymorphism of the asporin gene is associated with
primary osteoarthritis of the knee in a Mexican Mestizo population.
Int J Rheum Dis. 20:1935–1941. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Young SP, Kapoor SR, Viant MR, Byrne JJ,
Filer A, Buckley CD, Kitas GD and Raza K: The impact of
inflammation on metabolomic profiles in patients with arthritis.
Arthritis Rheum. 65:2015–2023. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Arnett FC, Edworthy SM, Bloch DA, McShane
DJ, Fries JF, Cooper NS, Healey LA, Kaplan SR, Liang MH, Luthra HS,
et al: The American rheumatism association 1987 revised criteria
for the classification of rheumatoid arthritis. Arthritis Rheum.
31:315–324. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Altman R, Asch E, Bloch D, Bole G,
Borenstein D, Brandt K, Christy W, Cooke TD, Greenwald R, Hochberg
M, et al: Development of criteria for the classification and
reporting of osteoarthritis. Classification of osteoarthritis of
the knee. Diagnostic and therapeutic criteria committee of the
American rheumatism association. Arthritis Rheum. 29:1039–1049.
1986. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Woetzel D, Huber R, Kupfer P, Pohlers D,
Pfaff M, Driesch D, Häupl T, Koczan D, Stiehl P, Guthke R and Kinne
RW: Identification of rheumatoid arthritis and osteoarthritis
patients by transcriptome-based rule set generation. Arthritis Res
Ther. 16:R842014. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nzeusseu Toukap A, Galant C, Theate I,
Maudoux AL, Lories RJ, Houssiau FA and Lauwerys BR: Identification
of distinct gene expression profiles in the synovium of patients
with systemic lupus erythematosus. Arthritis Rheum. 56:1579–1588.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Green GH and Diggle PJ: On the operational
characteristics of the Benjamini and Hochberg false discovery rate
procedure. Stat Appl Genet Mol Biol. 6:Article272007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bader GD and Hogue CW: An automated method
for finding molecular complexes in large protein interaction
networks. BMC Bioinformatics. 4:22003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li M, Li D, Tang Y, Wu F and Wang J:
CytoCluster: A cytoscape plugin for cluster analysis and
visualization of biological networks. Int J Mol Sci. 18(pii):
E18802017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Montojo J, Zuberi K, Rodriguez H, Kazi F,
Wright G, Donaldson SL, Morris Q and Bader GD: GeneMANIA Cytoscape
plugin: Fast gene function predictions on the desktop.
Bioinformatics. 26:2927–2928. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Youden WJ: Index for rating diagnostic
tests. Cancer. 3:32–35. 1950. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bay-Jensen AC, Bihlet A, Byrjalsen I,
Andersen J, He Y, Siebuhr A, Thudium C, Guehring H, Michaelis M,
Ladel C, et al: Elevated levels of CRPM, an inflammatory biomarker
correlating with disease activity in RA, are prognostic of
radiographic knee OA. Osteoarthritis Cartilage. 25 Suppl 1:S322017.
View Article : Google Scholar
|
|
24
|
Castrejon I, Chua JR, Malfait AM, Block JA
and Pincus T: Disease burden in rheumatoid arthritis (RA) patients
who have secondary osteoarthritis (OA) is lower than in primary OA
but higher than in RA with no secondary OA. Osteoarthritis
Cartilage. 25:S218–S219. 2017. View Article : Google Scholar
|
|
25
|
Can T: Introduction to bioinformatics.
Methods Mol Biol. 1107:51–71. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Huber R, Hummert C, Gausmann U, Pohlers D,
Koczan D, Guthke R and Kinne RW: Identification of intra-group,
inter-individual, and gene-specific variances in mRNA expression
profiles in the rheumatoid arthritis synovial membrane. Arthritis
Res Ther. 10:R982008. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ma C, Lv Q, Teng S, Yu Y, Niu K and Yi C:
Identifying key genes in rheumatoid arthritis by weighted gene
co-expression network analysis. Int J Rheum Dis. 20:971–979. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li G, Han N, Li Z and Lu Q: Identification
of transcription regulatory relationships in rheumatoid arthritis
and osteoarthritis. Clin Rheumatol. 32:609–615. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yi CQ, Ma CH, Xie ZP, Cao Y, Zhang GQ,
Zhou XK and Liu ZQ: Comparative genome-wide gene expression
analysis of rheumatoid arthritis and osteoarthritis. Genet Mol Res.
12:3136–3145. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Biswas S, Manikandan J and Pushparaj PN:
Decoding the differential biomarkers of Rheumatoid arthritis and
Osteoarthritis: A functional genomics paradigm to design disease
specific therapeutics. Bioinformation. 6:153–157. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang X, Ning Y and Guo X: Integrative
meta-analysis of differentially expressed genes in osteoarthritis
using microarray technology. Mol Med Rep. 12:3439–3445. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nayan S, Alizadehfar R and Desrosiers M:
Humoral primary immunodeficiencies in chronic rhinosinusitis. Curr
Allergy Asthma Rep. 15:462015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Paciolla M, Pescatore A, Conte MI,
Esposito E, Incoronato M, Lioi MB, Fusco F and Ursini MV: Rare
mendelian primary immunodeficiency diseases associated with
impaired NF-κB signaling. Genes Immun. 16:239–246. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jiang LJ, Zhang NN, Ding F, Li XY, Chen L,
Zhang HX, Zhang W, Chen SJ, Wang ZG, Li JM, et al: RA-inducible
gene-I induction augments STAT1 activation to inhibit leukemia cell
proliferation. Proc Natl Acad Sci USA. 108:1897–1902. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lim JY, Im KI, Lee ES, Kim N, Nam YS, Jeon
YW and Cho SG: Enhanced immunoregulation of mesenchymal stem cells
by IL-10-producing type 1 regulatory T cells in collagen-induced
arthritis. Sci Rep. 6:268512016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yokota A, Narazaki M, Shima Y, Murata N,
Tanaka T, Suemura M, Yoshizaki K, Fujiwara H, Tsuyuguchi I and
Kishimoto T: Preferential and persistent activation of the STAT1
pathway in rheumatoid synovial fluid cells. J Rheumatol.
28:1952–1959. 2001.PubMed/NCBI
|
|
37
|
Pongratz G, Anthofer JM, Melzer M, Anders
S, Grässel S and Straub RH: IL-7 receptor α expressing B cells act
proinflammatory in collagen-induced arthritis and are inhibited by
sympathetic neurotransmitters. Ann Rheum Dis. 73:306–312. 2014.
View Article : Google Scholar : PubMed/NCBI
|