Introduction
Lung cancer remains a leading cause of
cancer-related mortality worldwide (1). Non-small cell lung cancer (NSCLC) is
a major type of lung cancer and is characterized by a poor
prognosis with relatively low 5-year survival rate (2,3).
Cisplatin-based chemotherapy is a standard procedure for the
treatment of patients with NSCLC (4). Many patients initially respond to
cisplatin-based chemotherapy, whereas certain patients with
intrinsic resistance do not initially respond to cisplatin and
others develop acquired resistance to cisplatin (5). As a result, the 5-year survival rate
for patients with NSCLC is only 17% (6).
MicroRNAs (miRNAs) are non-coding, short
single-chain nucleotide molecules (7). Through binding to the 3′-untranslated
region (UTR) of their target genes, miRNAs control a variety of
physiological processes, including cell proliferation, cell
migration and the cell cycle (8).
Dysregulation of miRNAs has been reported to contribute to the
development of multiple diseases such as cancer (9,10).
In addition, a number of miRNAs have been reported to be involved
in the development of chemoresistance (11,12).
miRNA (miR)-29 has been demonstrated to sensitize ovarian cancer
cells to cisplatin treatment, and represents a potential
therapeutic target (13). However,
whether and how miR-29 contributes to the development of cisplatin
resistance in NSCLC remains unknown.
REV3-like DNA-directed polymerase ζ catalytic
subunit (REV3L) encodes the catalytic subunit of DNA polymerase ζ,
which is responsible for translesional replication (14); this function makes REV3L a cancer
susceptibility candidate gene. A previous study reported that a
c.460T>C variant in the REV3L 3′UTR affected the binding ability
of miRNAs on REV3L mRNA and may contribute to lung cancer
initiation (15). Despite its role
in cancer initiation, overexpression of REV3L has been reported to
promote cell survival and the development of cisplatin resistance
in human fibroblasts (16).
In the present study, the expression of miR-29a was
demonstrated to determine the sensitivity of A549 and H1650 cells
to cisplatin. Furthermore, miR-29a expression was reduced in the
cisplatin resistant A549 cell line (A549rCDDP), and increased
miR-29a expression resensitized A549rCDDP cells to cisplatin. REV3L
was confirmed to be a target gene of miR-29a. Further
investigations revealed that the silencing of REV3L expression and
the overexpression of miR-29a in A549 cells that were treated with
a low concentration of cisplatin may significantly reduce cell
proliferation, inhibition and cell cycle arrest at the G2/M phase.
In addition, a decrease in miR-29a expression and an increase in
REV3L expression were observed in cisplatin-resistant A549rCDDP
cells. Gene expression analysis in tumor tissues from patients with
NSCLC revealed a negative correlation between miR-29a and REV3L
mRNA expression. In conclusion, results from the present study
indicated that miR-29a may enhance NSCLC cell sensitivity to
cisplatin treatment through the regulation of REV3L expression.
Materials and methods
Cell culture
The human NSCLC cell lines A549 and H1650, and 293
cells were purchased from American Type Culture Collection
(Manassas, VA, USA), The A549 cisplatin-resistant sub-line,
A549rCDDP, was obtained from The Cancer Hospital of Peking Union
Medical College, Chinese Academy of Medical Sciences (Beijing,
China). All cell lines were cultured in RPMI-1640 medium (Gibco;
Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10%
fetal bovine serum (HyClone; GE Healthcare Life Sciences, Logan,
UT, USA) in a humid incubator with 5% CO2. For A549rCDDP
cells, the complete culture medium was supplemented with 2 mg/l
cisplatin (Selleck Chemicals, Houston, TX, USA). For cisplatin
treatment conditions, the culture medium of A549, H1650 or
A549rCDDP cells was supplemented with cisplatin (2.5, 5, 10 and 20
μg/ml; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) for the
indicated times (24, 48 and 72 h).
Small interfering (si)RNA
transfection
Two REV3L siRNAs (#1, 5′-GAUCACAGGUUUGUGCCAG-3′; and
#2, 5′-AGACUGAGUGAGUCACCUG-3′) and a control siRNA were purchased
from Invitrogen (Thermo Fisher Scientific, Inc.). Cells
(2×105) were seeded in 6-well plates and cultured for 24
h; the siRNAs were individually mixed with
Lipofectamine® RNAiMAX (Invitrogen; Thermo Fisher
Scientific, Inc.) and added into the cell culture medium at a final
concentration of 0.01 µM and incubated for 72 h according to the
manufacturer's instructions. At 72 h after transfection, cells were
collected for the subsequent experiments.
miRNA transfection
Cells were transfected with 50 nmol/l miR-29a mimics
(5′-UAGCACCAUCUGAAAUCGGUUA-3′) or miR-NC mimics
(5′-UAACCACUUUCACAUGGUCCUA-3′), miR-29a inhibitor
(5′-UAACCGAUUUCAGAUGGUGCUA-3′) or miR-NC inhibitor
(5′-UAACCGAAUUCACAUGGUCCUA-3′) using
Lipofectamine® 2000 (Thermo Fisher Scientific,
Inc.). In brief, cells (2×105) were seeded in a
6-well plate and incubated to 60–70% confluence. At 24 h after
transfection, cells were collected for the subsequent
experiments.
Cell cycle assay
For cell cycle analysis, cells were stained with
propidium iodide (PI; Invitrogen; Thermo Fisher Scientific, Inc.).
Briefly, following different treatments (control siRNA + miR-NC
mimics + vehicle; control siRNA + miR-NC mimics + cisplatin; REV3L
siRNA1 + miR-NC mimics + cisplatin; REV3L siRNA2 + miR-NC mimics +
cisplatin; and control siRNA + miR-29a mimics + cisplatin) in five
groups, cells were collected, washed with PBS and fixed in 70%
ethanol at 4°C overnight. Annexin V (5 μl) and PI (2.5 μl) were
subsequently added to the cell suspension, and cell distribution
was analyzed by flow cytometry. The cell number at each phase was
analyzed using FloJo software (version 7.6.3; FlowJo LLC, Ashland,
OR, USA).
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
MiRNeasy Mini kit (Qiagen, Inc. Valencia, CA, USA)
was used to extract total RNA from cells, according to the
manufacturer's instructions. An M-MLV Reverse Transcriptase kit
(Thermo Fisher Scientific, Inc.) was used to synthesize cDNA. qPCR
was performed using SYBR Premix Ex Taq (Takara Bio, Inc.,
Otsu, Japan) on a CFX96 Real-Time PCR Detection System (Bio-Rad
Laboratories, Hercules, CA, USA). GAPDH and U6 were used as
internal controls for mRNA and miRNA, respectively. The primers
were as follows: miR-29a, 5′-TAGCACCATCTGAAATCG-3′ (forward) and
5′-CACACCAGCACTGACTA-3′ (reverse); GAPDH,
5′-TGAACTGAAAGCTCTCCACC-3′ (forward) and 5′-CTGATGTACCAGTTGGGGAA-3′
(reverse); U6, 5′-CTCGCTTCGGCAGCACA-3′ (forward),
5′-AACGCTTCACGAATTTGCGT-3′ (reverse); REV3L,
5′-GCTCCAGTATGTGTACCATCTTGT-3′ (forward) and
5′-ATGGATATCTCGAAGTAACACGTC-3′ (reverse). The 2−ΔΔCq
method was used to calculate relative gene expression (17).
Western blotting
Cell lysates (100 µl; 2×106 cells) were
prepared using radioimmunoprecipitation assay lysis buffer
(Beyotime Institute of Biotechnology, Haimen, China) containing 2
µl protease inhibitor (Sigma-Aldrich; Merck KGaA). Briefly, the
concentration of each protein sample was determined by
bicinchoninic acid assay kit (Beyotime Institute of Biotechnology),
and the total protein (20 μg/lane) extracted from each sample was
separated by SDS-PAGE on 8% gels and transferred to polyvinylidene
fluoride membranes (EMD Millipore, Billerica, MA, USA). The
membranes were blocked in 5% non-fat milk and incubated with
primary antibodies against REV3L (1:1,000; catalog no. GTX17515;
GeneTex, Inc., Irvine, CA, USA) and GAPDH (1:10,000; catalog no.
G8795; Sigma-Aldrich; Merck KGaA) at 4°C overnight, followed by
incubation with anti-rabbit peroxidase-conjugated secondary
antibody (1:80,000; catalog no. a0545; Sigma-Aldrich; Merck KGaA)
at room temperature for 1 h. Protein bands were visualized using
Enhanced Chemiluminescence detection reagents (Thermo Fisher
Scientific, Inc. USA). GAPDH served as a loading control.
Cell viability assay
Cell viability was determined by Cell Counting Kit-8
(Dojindo Molecular Technologies, Inc., Kumamoto, Japan). For the
detection of miR-29a on cisplatin induced cell viability, cells
were seeded in a 96-well plate and subsequently exposed to vehicle
(0.9% NaCl as control for ciaplatin) or cisplatin treatments (2.5,
5, 10 and 20 µg/ml) for 72 h.
For the determining the effect of miR-29a on
cisplatin induced changes of cell proliferation, cells were treated
with cisplatin (5 µg/ml) for 72 h. Subsequently, cells
(2×105) were seeded in a 6-well plate and transfected
with 50 nmol/l miR-29a mimics, miR-29a inhibitor or NC using
Lipofectamine® 2000 (Thermo Fisher Scientific, Inc.).
Subsequently, at 24 h after transfection, cells were collected for
the subsequent experiments.
To determine the effect of REV3L on cell viability,
REV3L siRNA (0.01 µM) or control siRNA (Thermo Fisher Scientific,
Inc.) was transfected into cells which were treated with cisplatin
(2 µg/ml) by Lipofectamine® RNAiMAX (Invitrogen; Thermo
Fisher Scientific, Inc.). At 72 h after transfection, cells were
collected for the subsequent experiments. Briefly, 10 µl CCK-8 was
added to the culture medium of each well and incubated for 2 h. The
absorbance was measured at 450 nm with a microplate reader (Bio-Rad
Laboratories, Inc.).
Subsequently, 10 µl CCK-8 was added to the culture
medium of each well and incubated for 2 h. The absorbance was
measured at 450 nm with a microplate reader (Bio-Rad
Laboratories).
Cell apoptosis assay
Cells were collected by trypsinization and cell
apoptosis was detected using an Annexin V-fluorescein
isothiocyanate/PI cell apoptosis kit (Invitrogen; Thermo Fisher
Scientific, Inc.), according to the manufacturer's instructions.
Briefly, cells were suspended in Annexin binding buffer, and PI and
Annexin V were added to the cell suspension. Cells were analyzed
with a BD FACSCalibur flow cytometer (BD Biosciences, Franklin
Lakes, NJ, USA).
Luciferase reporter assay
The wild-type (WT) REV3L 3′UTR sequence was
amplified from cDNA of 293 cells and inserted into pGL-3 (Promega
Corporation, Madison, WI, USA). REV3L 3′UTR-mutant (Mut) was
constructed using PrimeSTAR Mutagenesis Basal kit (Takara Bio,
Inc.). The 293 cells were co-transfected with pGL3-REV3L 3′UTR-WT
or pGL3-REV3L 3′UTR-Mut and miR-29a mimics or miR-negative control
(NC) mimics, and an internal control Renilla plasmid.
Luciferase activity and Renilla activity were measured at 24
h post-transfection using a Dual Luciferase Reporter Assay kit
(Promega Corporation), according to the manufacturer's
instructions.
Patients
NSCLC tissues and adjacent normal liver tissues
(located ≥2 cm from the tumor margins) were obtained from 30
patients (20 male and 10 female; 9 patients aged <60 years old
and 21 patients aged ≥60 years old) who received surgery at
Zhejiang Cancer Hospital (Zhejiang, China) and Shaoxing People's
Hospital (Shaoxing, China; Table
I). Patients that received chemotherapy or radiotherapy were
excluded from the study. Tissues were removed and stored at −80°C.
The present study was approved by the ethics committee of Zhejiang
Cancer Hospital, and written informed consent was obtained from
each patient prior to surgery and enrolment in the study.
 | Table I.Association between miR-29a or REV3L
and clinicopathological factors. |
Table I.
Association between miR-29a or REV3L
and clinicopathological factors.
|
|
| miR-29a
expression | REV3L
expression |
|---|
|
|
|
|
|
|---|
| Clinicopathological
parameters | n | Mean ± SD | P-value | Mean ± SD | P-value |
|---|
| Age |
|
| 0.686 |
| 0.435 |
|
<60 | 9 | 0.749±0.094 |
| 1.422±0.141 |
|
|
≥60 | 21 | 0.707±0.054 |
| 1.305±0.076 |
|
| Sex |
|
| 0.817 |
| 0.927 |
|
Male | 20 | 0.735±0.101 |
| 1.349±0.122 |
|
|
Female | 10 | 0.712±0.051 |
| 1.336±0.083 |
|
| TNM
classification |
|
| 0.007a |
| 0.003a |
|
I–II | 13 | 0.867±0.061 |
| 1.108±0.059 |
|
|
III–IV | 17 | 0.621±0.056 |
| 1.495±0.089 |
|
| Metastasis |
|
| 0.264 |
| 0.059 |
|
Yes | 18 | 0.676±0.063 |
| 1.443±0.085 |
|
| No | 12 | 0.784±0.067 |
| 1.185±0.099 |
|
Prediction of the target of miR-29a. TargetScan 7.1
was used to predict target sequences of miR-29a in the 3′UTR of
REV3L (www.targetscan.org/vert_71/).
Statistical analysis
All statistical analyses were carried out using
GraphPad Prism 5.0 software (GraphPad Software, Inc., La Jolla, CA,
USA). Data are presented as the mean ± standard deviation.
Statistically significant differences between two groups were
analyzed using Student's t-test. Differences between multiple
groups were analyzed with one-way ANOVA, followed by a Newman-Keuls
post-hoc test. Correlations were made using Pearson's correlation
coefficient. P<0.05 was considered to indicate a statistically
significant difference.
Results
miR-29a expression is associated with
cisplatin sensitivity in NSCLC cells
To explore whether miR-29a expression affected the
sensitivity of NSCLC cells to cisplatin, miR-29a mimics were used
to increase miR-29a expression in A549 cells (Fig. 1A). miR-29a overexpression reduced
the viability and increased cisplatin sensitivity of
cisplatin-sensitive A549 and H1650 cells, compared with the cells
transfected with miR-NC mimics (Fig.
1B and C, respectively). Conversely, cells transfected an
miR-29a inhibitor exhibited decreased miR-29a expression levels,
increased viability and a reduction in sensitivity of A549 and
H1650 cells to cisplatin exposure, compared with miR-NC-transfected
cells (Fig. 1D-F). These data
suggested that miR-29a expression may be involved in cisplatin
sensitivity of NSCLC cells.
Reduced miR-29a expression is
associated with development of cisplatin resistance of A549
cells
The contribution of miR-29a dysregulation to the
development of cisplatin resistance in NSCLC cells was
investigated. Compared with parental A549 cells, treatment of
cisplatin only slightly reduced cell viability of
cisplatin-resistant A549rCDDP cells (Fig. 2A), which indicated a relative
insensitivity of these cells to cisplatin. RT-qPCR confirmed a
significantly decreased expression level of miR-29a in A549rCDDP
cells compared with A549 cells (Fig.
2B). Transfection of miR-29a mimics enhanced the reduction in
viability induced by cisplatin treatment in A549rCDDP cells,
compared with miR-NC transfected cells (Fig. 2C). It was also revealed that
miR-29a overexpression significantly reduced cell proliferation in
A549rCDDP cells treated with 5 µg/ml cisplatin, compared with
miR-NC-transfected cell (Fig. 2D).
In addition, miR-29a overexpression significantly increased
cisplatin-induced apoptosis (Fig.
2E). These data suggested that reduced miR-29a expression may
be involved in the development of cisplatin resistance in NSCLC
cells.
miR-29a downregulates REV3L expression
in NSCLC cells
REV3L is involved in the DNA repair pathway and is
an indicator of chemotherapy sensitivity in several cancer types
(18). In A549 and H1650 cells,
overexpression of miR-29a significantly decreased REV3L mRNA
expression, compared with miR-NC-transfected cells (Fig. 3A). Western blot analysis further
demonstrated a notable reduction of REV3L protein expression in
miR-29a-transfected cells (Fig.
3B). In addition, higher REV3L mRNA and protein expression
levels were detected in A549rCDDP cells, compared with A549 cells
(Fig. 3C and D). A549rCDDP cells
transfected with miR-29a mimics exhibited a significant reduction
in REV3L mRNA and protein expression levels, compared with
miR-NC-transfected cells (Fig. 3E and
F). These results suggested a potential role for miR29a and
REV3L in the development of cisplatin resistance.
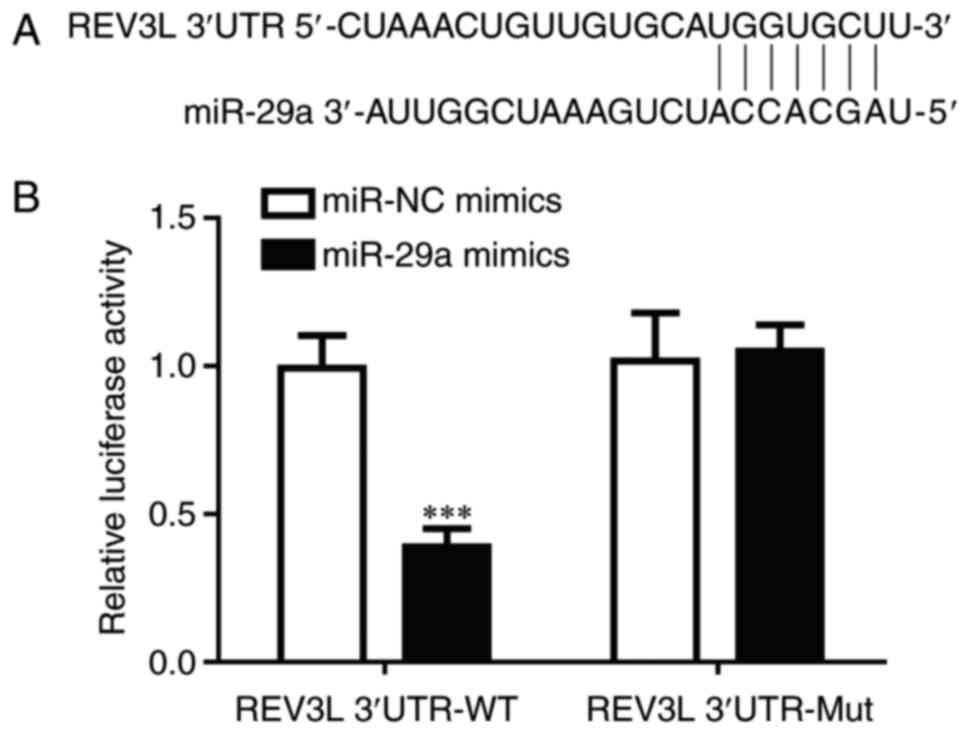
REV3L was a direct target of
miR-29a
TargetScan 7.1 was used to predict target sequences
of miR-29a in the 3′UTR of REV3L (Fig.
4A). To confirm the direct regulatory relationship between
miR-29a and REV3L, a dual luciferase assay was performed in 293
cells. Transfection of miR-29a mimics, but not miR-NC mimics,
significantly reduced luciferase activity in cells transfected with
REV3L 3′UTR-WT (Fig. 4B); no
significant differences in luciferase activity were identified in
cells co-transfected with REV3L 3′UTR-Mut. These data demonstrated
that miR-29a may inhibit REV3L expression by binding to its
3′UTR.
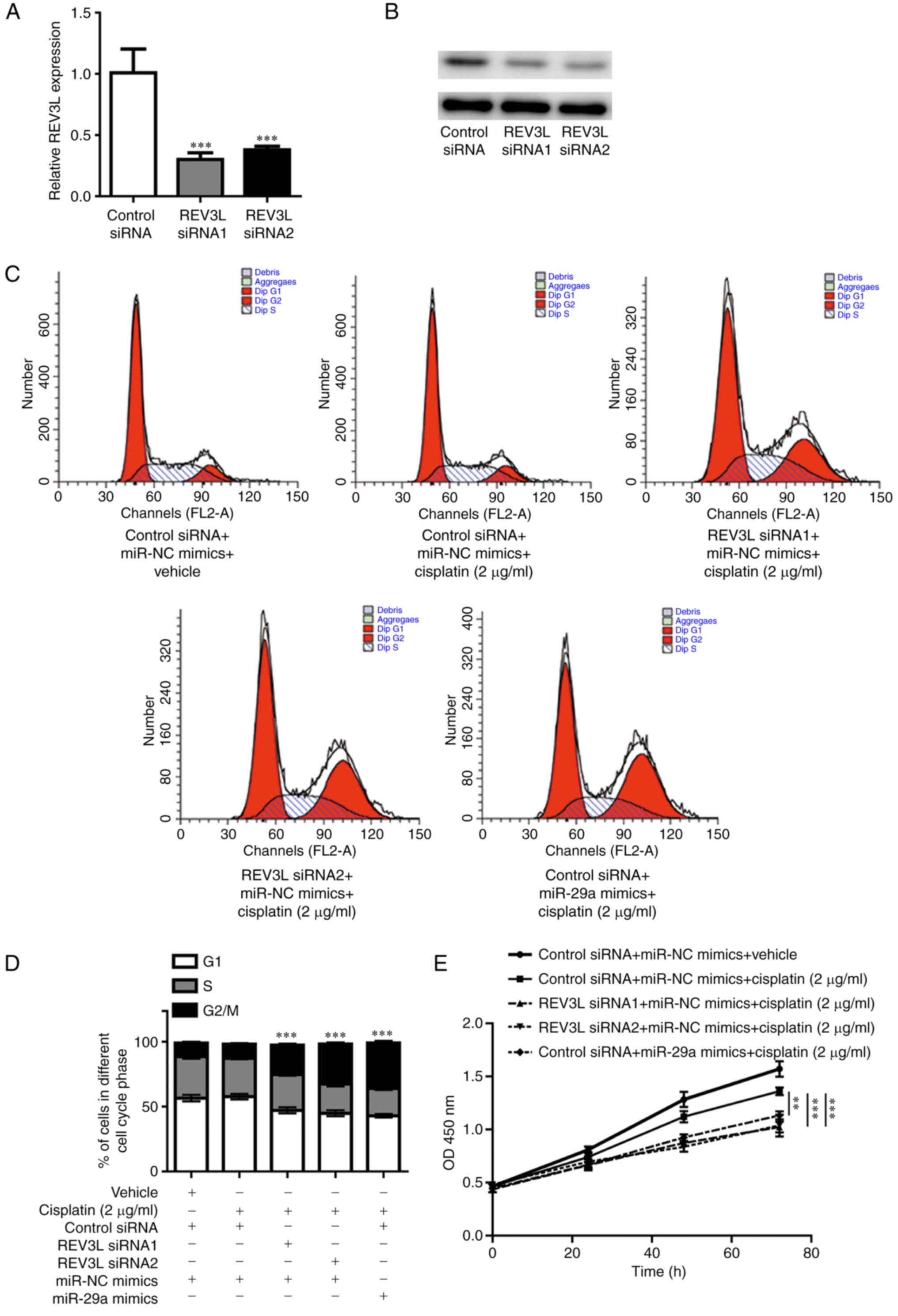
miR-29a regulates cisplatin
sensitivity of NSCLC cells through REV3L
REV3L is the catalytic subunit of DNA polymerase ζ,
which is involved in translesional DNA synthesis (14). Variations in REV3L have previously
been demonstrated to lead to altered cell cycle distribution and,
therefore, an altered sensitivity to chemotherapy (18). REV3L siRNA1 and REV3L siRNA2
significantly decreased REV3L mRNA (Fig. 5A) and protein level (Fig. 5B) in A549 cells. Although treatment
with a low concentration of cisplatin (2 µg/ml) alone (control
siRNA + miR-NC mimics + cisplatin) did not affect the cell cycle
compared with untreated control cells, a significant enrichment of
cells in the G2/M phase was observed in REV3L-siRNA-transfected
A549 cells treated with 2 µg/ml cisplatin (Fig. 5C and D). Similarly, miR-29a
overexpression also induced G2/M arrest in A549 cells exposed to
low-dose cisplatin (Fig. 5C and
D). In addition, knockdown of REV3L expression or miR-29a
overexpression significantly inhibited cell proliferation in the
presence of cisplatin (2 µg/ml) compared with cells treated with
cisplatin (2 µg/ml) only (Fig.
5E). These results further validated the potential role of
miR-29a in the regulation of cisplatin sensitivity of A549 cells;
this regulation may be achieved through the downregulation of REV3L
expression and increased cisplatin-induced G2/M arrest.
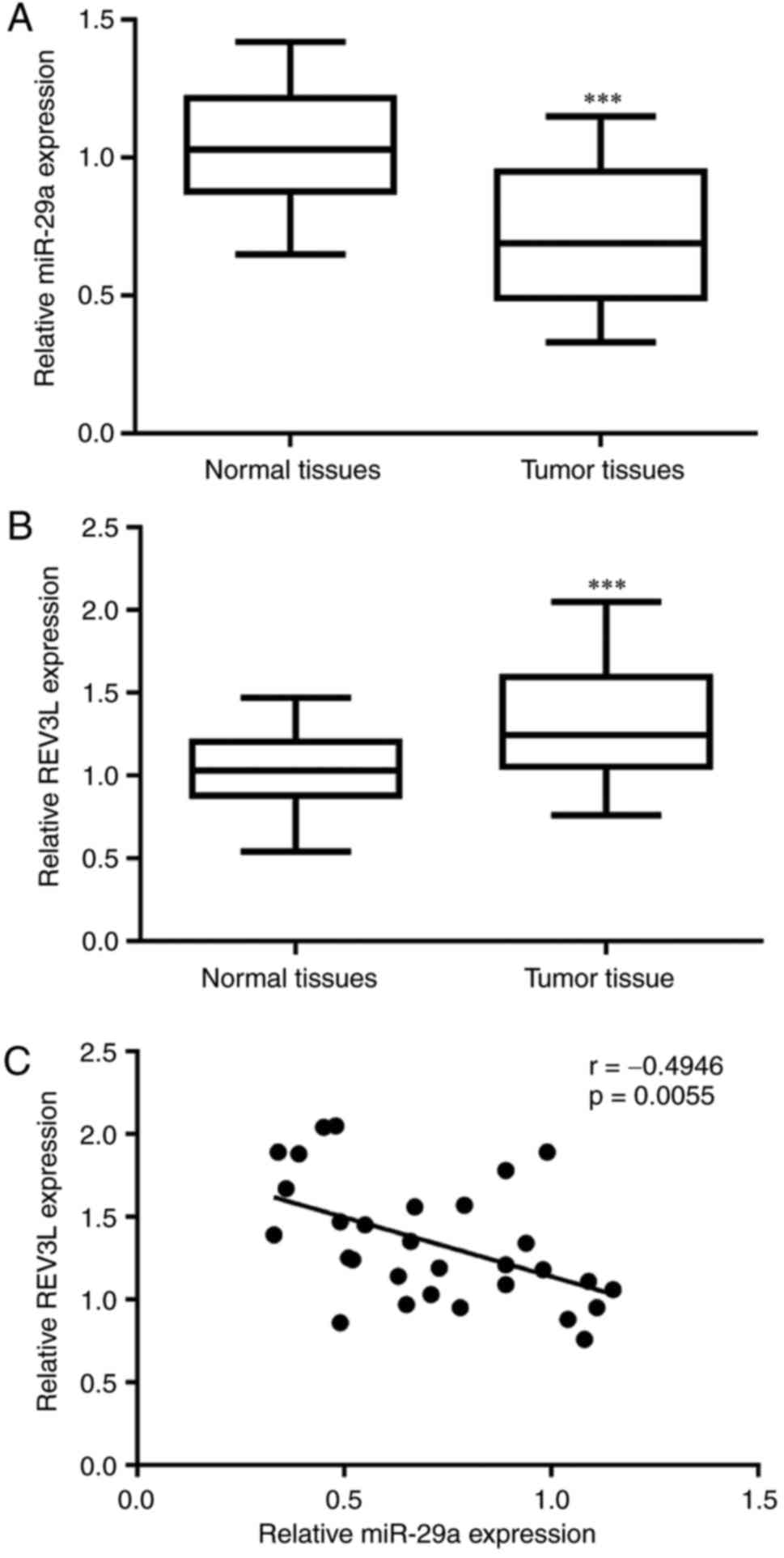
miR-29a expression is inversely
correlated with REV3L mRNA expression in tumor tissues from
patients with NSCLC
To investigate the function of miR-29a and REV3L in
patients with NSCLC, RT-qPCR was used to detect miR-29a and REV3L
mRNA expression levels in tumoral and adjacent normal tissues
(Fig. 6). A significant decrease
in miR-29a expression was observed in tumoral tissues compared with
adjacent normal tissues (Fig. 6A),
whereas REV3L mRNA expression was elevated in tumoral tissues
compared with normal tissues (Fig.
6B). Notably, correlation analysis indicated that miR-29a
expression was inversely correlated with REV3L mRNA expression in
tumor tissues from patients with NSCLC (Fig. 6C). Statistical analysis of the
associations between miR-29a expression, REV3L mRNA expression and
clinicopathological factors reveled that low expression of miR-29a
and high expression of REV3L were closely associated with advanced
TNM stage (Table I). No
significant associations were identified between miR-29a and REV3L
expression with age, sex or metastatic status of the patients with
NSCLC (Table I).
Discussion
Resistance towards cisplatin-based chemotherapy is a
major obstacle for the treatment of patients with NSCLC (19). Although many proteins and miRNAs
have been identified as drivers of chemoresistance (20–22),
further investigation is required to elucidate the complicated
underlying mechanisms. In the present study, miR-29a was revealed
to potentially regulate the sensitivity of NSCLC cells to cisplatin
treatment. miR-29a was also predicted and confirmed to directly
regulate REV3L expression, and therefore antagonize cisplatin
resistance in A549rCDDP cells.
Recently, a number of reports have demonstrated that
dysregulation of miRNA networks leads to the development of drug
resistance in a number of cancers (23,24).
In NSCLC, several miRNAs have been reported to contribute to the
development of cisplatin resistance (25,26).
miR-29a was demonstrated to act as a tumor suppressor in NSCLC by
regulating oncogene expression, such as LIM and SH3 protein 1, in
several in vitro studies (27,28).
Consistent with these findings, the present study observed a
decrease in miR-29a expression in NSCLC tumoral tissues.
Downregulation of miR-29a has also been reported to be involved in
the development of cisplatin resistance in ovarian cancer cells
in vitro and in vivo (13). The present study revealed that
altered miR-29a expression affected the sensitivity of NSCLC cells
to cisplatin treatment. In A549 and H1650 cells, downregulation of
miR-29a partially reversed cisplatin-induced cell growth arrest,
whereas upregulation of miR-29a sensitized cells to cisplatin
treatment. Furthermore, there was a notable decrease in miR-29a
expression in cisplatin-resistant A549rCDDP cells compared to their
A549 cell counterpart. Therefore, it was suggested that miR-29a may
promote the sensitivity of NSCLC cells to cisplatin, and loss of
miR-29a may be responsible for cisplatin resistance in NSCLC.
REV3L is the catalytic subunit of DNA polymerase ζ,
which is involved in translesional DNA synthesis (14). The role of REV3L in cancer
progression is controversial. For example, low expression of REV3L
was observed in colon carcinomas compared with normal tissues,
which suggested a role as a tumor suppressor (29). By contrast, another study reported
that REV3L depletion induced cell growth arrest in cancer cells of
different origins, and identified REV3L as an oncogene (30). As for chemotherapy resistance,
upregulation of REV3L was previously demonstrated to serve a
crucial role in many cancer types, including NSCLC, through
regulation of DNA repair (16,31,32).
In the present study, an elevated expression level of REV3L was
observed in A549rCDDP cells. In addition, REV3L was predicted and
confirmed to be directly regulated by miR-29a. Silencing of REV3L
or overexpression of miR-29a inhibited cell growth and increased
accumulation of cells in the G2/M phase in cells co-treated with
cisplatin. This was consistent with a previous study that
demonstrated that depletion of REV3L led to cumulative DNA damage,
and resulted in inhibition of cell proliferation and G2/M arrest
(30). Furthermore, results from
the present study indicated that decreased miR-29a expression may
contribute to the elevation of REV3L expression in A549rCDDP cells,
and overexpression of miR-29a may reverse cisplatin resistance and
induce growth arrest and apoptosis in A549rCDDP cells treated with
cisplatin. Therefore, miR-29a/REV3L may promote the development of
cisplatin resistance in NSCLC cells. The results of the present
study provided further validation that REV3L depletion may amend
cisplatin-based chemotherapy, and revealed that miR-29a may target
REV3L to enhance cisplatin sensitivity of NSCLC cells.
In conclusion, the present study demonstrated that
miR-29a positively regulates the sensitivity of NSCLC cells to
cisplatin via direct suppression of REV3L expression.
Downregulation of miR-29a led to cisplatin resistance in NSCLC
cells and may be a promising prognostic tool and a target for the
treatment of patients with NSCLC.
Acknowledgements
Not applicable.
Funding
This present work is supported by The Research
Program on the Application of Public Welfare Technology in Zhejiang
(grand no. 2016C33224), The Natural Science Foundation of Anhui
(grant no. 1608085QH215) and The Medical Scientific Program of
Shanghai Health and Family Planning Commission (grant no.
201540163).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XC, HZ and WY performed the experiments and analyzed
the data. YC analyzed the data. MC analyzed the data, collected the
funding and prepared the manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Zhejiang Cancer Hospital, and written informed consent
was obtained from each patient prior to surgery and enrolment in
the study.
Patient consent for publication
Patients consented to the publication of their
clinical data.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Riaz SP, Lüchtenborg M, Coupland VH,
Spicer J, Peake MD and Møller H: Trends in incidence of small cell
lung cancer and all lung cancer. Lung Cancer. 75:280–284. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Toschi L, Cappuzzo F and Janne PA:
Evolution and future perspectives in the treatment of locally
advanced non-small cell lung cancer. Ann Oncol. 18 Suppl
9:ix150–ix155. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gu L, Deng JZ, Roy S and Hammond PT: A
combination RNAi-chemotherapy layer-by-layer nanoparticle for
systemic targeting of KRAS/P53 with cisplatin to treat non-small
cell lung cancer. Clin Cancer Res. 23:7312–7323. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Barr MP, Gray SG, Hoffmann AC, Hilger RA,
Thomale J, O'Flaherty JD, Fennell DA, Richard D, O'Leary JJ and
O'Byrne KJ: Generation and characterisation of cisplatin-resistant
non-small cell lung cancer cell lines displaying a stem-like
signature. PLoS One. 8:e541932013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
statistics, 2014. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lin S and Gregory RI: MicroRNA biogenesis
pathways in cancer. Nat Rev Cancer. 15:321–333. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pan JY, Sun CC, Bi ZY, Chen ZL, Li SJ, Li
QQ, Wang YX, Bi YY and Li DJ: miR-206/133b Cluster: A Weapon
against lung cancer? Mol Ther Nucleic Acids. 8:442–449. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ni J, Bucci J, Chang L, Malouf D, Graham P
and Li Y: Targeting MicroRNAs in prostate cancer radiotherapy.
Theranostics. 7:3243–3259. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ayers D and Vandesompele J: Influence of
microRNAs and long non-coding RNAs in cancer chemoresistance.
Genes. 8(pii): E952017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pal MK, Jaiswar SP, Dwivedi VN, Tripathi
AK, Dwivedi A and Sankhwar P: MicroRNA: A new and promising
potential biomarker for diagnosis and prognosis of ovarian cancer.
Cancer Biol Med. 12:328–341. 2015.PubMed/NCBI
|
|
13
|
Yu PN, Yan MD, Lai HC, Huang RL, Chou YC,
Lin WC, Yeh LT and Lin YW: Downregulation of miR-29 contributes to
cisplatin resistance of ovarian cancer cells. Int J Cancer.
134:542–551. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lawrence CW and Hinkle DC: DNA polymerase
zeta and the control of DNA damage induced mutagenesis in
eukaryotes. Cancer Surv. 28:21–31. 1996.PubMed/NCBI
|
|
15
|
Zhang S, Chen H, Zhao X, Cao J, Tong J, Lu
J, Wu W, Shen H, Wei Q and Lu D: REV3L 3′UTR 460 T>C
polymorphism in microRNA target sites contributes to lung cancer
susceptibility. Oncogene. 32:242–250. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wu F, Lin X, Okuda T and Howell SB: DNA
polymerase zeta regulates cisplatin cytotoxicity, mutagenicity and
the rate of development of cisplatin resistance. Cancer Res.
64:8029–8035. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2-ΔΔCT method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yang L, Shi T, Liu F, Ren C, Wang Z, Li Y,
Tu X, Yang G and Cheng X: REV3L, a promising target in regulating
the chemosensitivity of cervical cancer cells. PLoS One.
10:e01203342015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hu Y, Hong Y, Xu Y, Liu P, Guo DH and Chen
Y: Inhibition of the JAK/STAT pathway with ruxolitinib overcomes
cisplatin resistance in non-small-cell lung cancer NSCLC.
Apoptosis. 19:1627–1636. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
He J, Yu JJ, Xu Q, Wang L, Zheng JZ, Liu
LZ and Jiang BH: Downregulation of ATG14 by EGR1-MIR152 sensitizes
ovarian cancer cells to cisplatin-induced apoptosis by inhibiting
cyto-protective autophagy. Autophagy. 11:373–384. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li J, Wang Y, Song Y, Fu Z and Yu W:
miR-27a regulates cisplatin resistance and metastasis by targeting
RKIP in human lung adenocarcinoma cells. Mol Cancer. 13:1932014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wu DW, Wu TC, Wu JY, Cheng YW, Chen YC,
Lee MC, Chen CY and Lee H: Phosphorylation of paxillin confers
cisplatin resistance in non-small cell lung cancer via activating
ERK-mediated Bcl-2 expression. Oncogene. 33:4385–4395. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dehghanzadeh R, Jadidi-Niaragh F, Gharibi
T and Yousefi M: MicroRNA-induced drug resistance in gastric
cancer. Biomed Pharmacother. 74:191–199. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zou J, Yin F, Wang Q, Zhang W and Li L:
Analysis of microarray-identified genes and microRNAs associated
with drug resistance in ovarian cancer. Int J Clin Exp Pathol.
8:6847–6858. 2015.PubMed/NCBI
|
|
25
|
Dong Z, Zhong Z, Yang L, Wang S and Gong
Z: MicroRNA-31 inhibits cisplatin-induced apoptosis in non-small
cell lung cancer cells by regulating the drug transporter ABCB9.
Cancer Lett. 343:249–257. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ma Y, Li X, Cheng S, Wei W and Li Y:
MicroRNA-106a confers cisplatin resistance in non-small cell lung
cancer A549 cells by targeting adenosine triphosphatase-binding
cassette A1. Mol Med Rep. 11:625–632. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Muniyappa MK, Dowling P, Henry M, Meleady
P, Doolan P, Gammell P, Clynes M and Barron N: MiRNA-29a regulates
the expression of numerous proteins and reduces the invasiveness
and proliferation of human carcinoma cell lines. Eur J Cancer.
45:3104–3118. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hu Z, Cui Y, Zhou Y, Zhou K, Qiao X, Li C
and Wang S: MicroRNA-29a plays a suppressive role in non-small cell
lung cancer cells via targeting LASP1. Onco Targets Ther.
9:6999–7009. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Brondello JM, Pillaire MJ, Rodriguez C,
Gourraud PA, Selves J, Cazaux C and Piette J: Novel evidences for a
tumor suppressor role of Rev3, the catalytic subunit of Pol zeta.
Oncogene. 27:6093–6101. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Knobel PA, Kotov IN, Felley-Bosco E,
Stahel RA and Marti TM: Inhibition of REV3 expression induces
persistent DNA damage and growth arrest in cancer cells. Neoplasia.
13:961–970. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang H, Zhang SY, Wang S, Lu J, Wu W, Weng
L, Chen D, Zhang Y, Lu Z, Yang J, et al: REV3L confers
chemoresistance to cisplatin in human gliomas: the potential of its
RNAi for synergistic therapy. Neuro Oncol. 11:790–802. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang W, Sheng W, Yu C, Cao J, Zhou J, Wu
J, Zhang H and Zhang S: REV3L modulates cisplatin sensitivity of
non-small cell lung cancer H1299 cells. Oncol Rep. 34:1460–1468.
2015. View Article : Google Scholar : PubMed/NCBI
|