Introduction
Chronic rhinosinusitis (CRS) is characterized by
inflammation of the nose and the sinus mucosa, and has a morbidity
rate of approximately 14% (1). CRS
is one of the most common conditions associated with upper airway
illness and severely affects patients' quality of life. According
to the European Position Paper on Rhinosinusitis and Nasal Polyps
(EPOS), CRS can be classified into the following two categories:
CRS without nasal polyposis (CRSsNP) and CRS with nasal polyposis
(CRSwNP) (2,3).
Although CRSsNP is more prevalent, CRSwNP accounts
for ~20% of all CRS cases. CRSwNP, which is often accompanied by
asthma, fungal rhinosinusitis and aspirin-exacerbated respiratory
disease, is considered more difficult to treat compared with CRSsNP
(4). The disease often requires a
combination of surgical and medical treatments. However, CRSwNP
often recurs even after therapy. Despite numerous studies in this
field, the pathophysiological mechanisms of human CRS with nasal
polyps (CRSwNP) remain to be fully elucidated (5,6).
Development of human diseases can proceed through
the accumulation of different genetic alterations affecting the
structure and function of the genome. Combined analyses of
molecular data at multiple levels, including DNA copy-number
alteration, and mRNA, long non-coding RNA (lncRNA) and miRNA
expression, can clarify biological functions and pathways
dysregulated in various diseases (7). The rapid development of
high-throughput sequencing technology has provided a more
comprehensive and efficient analytical method for the study of the
pathogenesis of CRSwNP. In our study, 994 differentially expressed
mRNAs [false discovery rate (FDR) <0.05] and 265 differentially
expressed lncRNAs (P<0.05) were identified. By constructing an
lncRNA-mRNA interaction network, and performing GO functional and
KEGG pathway enrichment analyses of target genes regulated by
lncRNAs, this study aimed to understand the mechanism of CRSwNP and
screen potential lncRNAs and mRNAs for determining the prognosis of
CRSwNP patients.
Materials and methods
Eligible CRSwNP gene expression
profiles
Gene Expression Omnibus database (GEO, www.ncbi.nlm.nih.gov/geo) is the largest
available database of high-throughput gene expression data and we
selected GEO datasets containing gene expression profiles of
patients with CRSwNP (7). The
following search key words were used: ‘rhinosinusitis’ [All Fields]
AND ‘Homo sapiens’[porgn] AND ‘gse’ [Filter]. The study types were
limited to ‘profiling by array’ and the following inclusion
criteria were used: (1) The
selected dataset must contain genome-wide mRNA transcriptome data;
(2) Data must originate from
CRSwNP nasal polyp tissue (NP) samples and normal control nasal
mucosa tissue samples; (3) Both
normalized and raw datasets were considered. According to these
requirements, two CRSwNP datasets [GSE36830 (case: Normal
ratio=6:6) and GSE72713 (case: Normal ratio=6:3)] were
obtained.
Identification of differentially
expressed mRNAs and lncRNAs in CRSwNP
We performed background correction for the raw data.
Using metaMA package analysis (version 3.1.2; Guille mette Marot),
P-values of differentially expressed genes were calculated by
moderated t-tests (Limma version 3.30.13) for each dataset and
combined by the inverse normal method. The adopted standard for
determination of DEGs was P<0.05, and all datasets had the same
direction of differential expression. Finally, the differentially
expressed mRNAs and lncRNAs between case and normal control samples
were identified (8,9).
Functional annotation
We performed Kyoto Encyclopedia of Genes and Genomes
(KEGG) pathway enrichment analysis and Gene Ontology (GO)
classification according to the GO categories of molecular function
(MF), biological process (BP) and cellular component, to identify
the enriched functions and pathways of DEGs, using the online
software GeneCoDis 3 (genecodis.cnb.csic.es/analysis) (10). We set FDR<0.05 as the threshold
of statistical significance. A total of 58 mRNAs were subjected to
GO and KEGG enrichment analyses using the R language (GSEABase
package), P-value <0.01 (P<0.01 is considered to indicate a
statistically significant difference).
Protein-protein interaction (PPI)
network construction in CRSwNP
To further research the biological functions of
DEGs, we constructed a PPI network based on the top 100 DEGs in
CRSwNP using Biological General Repository for Interaction Datasets
(BioGRID; thebiogrid.org) and Cytoscape software
version 3.5.0 (11,12). Based on the existing protein
interaction data included in the BioGRID database, Cytoscape was
used to search for common and regulatory mRNAs with opposite
effects in case vs. normal samples (13). After removing non-differentially
expressed genes, a protein interaction network was mapped. Such a
network consists of nodes and edges. The nodes in the network
represent the proteins and edges represent the interactions between
them (14).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) confirmation
Based on the results of the integrated
high-throughput transcriptome data analysis, five DEGs and one
DElncRNA with higher degrees between case and normal control groups
were identified and subsequently screened as candidate genes. The
sequences of the primers used to detect the differentially
expressed genes/lncRNA and the endogenous control were listed in
Table I. Blood samples from
patients with CRSwNP and healthy individuals from the First
People's Hospital of Jining (Jining, China) were collected on Nov
24th, 2017. All individuals provided signed informed consent for
use of their samples in this present study. The present study was
approved by the Ethics Committee of the First People's Hospital of
Jining [committee's reference no. 2016(012)]. RNA samples were used
to verify the differential expression of candidate genes in disease
and control groups using RT-qPCR (15). Blood samples were collected from 3
patients with CRSwNP (cases 1–3) and 3NCs and were frozen at −80°C
within 2 h. We performed RNA isolation using TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
after thawing the frozen samples at room temperature. We generated
cDNA from 1 µg extracted RNA using SuperScript® III
Reverse Transcriptase (Invitrogen; Thermo Fisher Scientific, Inc.).
qPCR reactions were performed in an ABI 7500 real-time PCR system
with Power SYBR® Green PCR Master Mix (Applied
Biosystems; Thermo Fisher Scientific, Inc.). We analyzed relative
gene expression using the 2−ΔΔCq method (16). We selected the human 18srRNA as an
endogenous control for mRNA expression analysis (17). The thermocycling conditions were as
follows: 95°C for 15 min, followed by 40 cycles of 95°C for 10 sec,
55°C for 30 sec and 72°C for 32 sec, and then 95°C for 15 sec, 60°C
for 60 sec and 95°C for 15 sec.
 | Table I.Sequence of primers. |
Table I.
Sequence of primers.
| Name | Sequence (5′-3′) | Size of product
(bp) |
|---|
| GAPDH-F (endogenous
control) |
GGAGCGAGATCCCTCCAAAAT | 197 |
| GAPDH-R (endogenous
control) |
GGCTGTTGTCATACTTCTCATGG |
|
| CCL18-F |
AGCATCATGAAGGGCCTTGC | 209 |
| CCL18-R |
TGCCGGCCTCTCTTGGTTAG |
|
| CCL8-F |
TGGAGAGCTACACAAGAATCACC | 133 |
| CCL8-R |
TGGTCCAGATGCTTCATGGAA |
|
| CUL4B-F |
TTTACAACCCAGGGATTCGGC | 154 |
| CUL4B-R |
GGATTCCTCAGCCATCTTCGC |
|
| NEDD4L-F |
CTGGGAAATGAGGATAGCGCC | 191 |
| NEDD4L-R |
AAAACGTTCGGCCATCCAAGT |
|
| GALNT7-F |
GGTTCATCTTACGCAGTTTGCT | 140 |
| GALNT7-R |
GGGCATGGGGTCATTGACA |
|
| RP11-798M19.6-F |
AAAGTTTTGGGAAGCTGGCAAG | 192 |
| RP11-798M19.6-R |
GGAGAAAGCAATCAGGGCACA |
|
Statistical analysis
The metaMA package in R version 3.1.2 (cran.r-project.org/web/packages/metaMA/index.html;
Guille mette Marot) was used to combine data from multiple
microarray datasets and obtain the individual P-values in the
present study. DEGs in CRSwNP compared with normal control were
identified with P<0.05. Values were presented as the mean ±
standard deviation. P<0.05 was considered to indicate a
statistically significant difference.
Results
Differential expression analysis of
genes in CRSwNP
The probes corresponding to multiple genes were
removed and average values of expression were calculated for genes
corresponding to multiple probes. Subsequently, lncRNAs included in
the GSE72713 dataset were screened out to obtain 1,747 lncRNAs
(1483 were newly identified and 264 were previously known). A total
of 15,996 mRNAs were overlapping between the two datasets. Compared
with the normal control group, 994 DEmRNAs in CRSwNP were
identified with P<0.05, among which, 354 genes were upregulated
and 640 genes were downregulated. The top 20 most significantly up-
or downregulated genes were listed in Table II. The top 100 DEmRNAs were
included in the cluster analysis. The heatmap was shown in Fig. 1.
| Table II.Top 20 differentially expressed
mRNAs. |
Table II.
Top 20 differentially expressed
mRNAs.
| ID | Symbol | Combined.ES | P-value | FDR | Up/down |
|---|
| 8564 | KMO | 3.53 |
5.66×10−8 |
4.14×10−4 | Up |
| 28959 | TMEM176B | 3.26 |
1.54×10−7 |
4.64×10−4 | Up |
| 3426 | CFI | 3.59 |
1.60×10−7 |
4.64×10−4 | Up |
| 2206 | MS4A2 | 3.27 |
2.51×10−7 |
5.75×10−4 | Up |
| 23406 | COTL1 | 3.25 |
4.93×10−7 |
7.82×10−4 | Up |
| 2207 | FCER1G | 3.06 |
5.38×10−7 |
7.82×10−4 | Up |
| 6355 | CCL8 | 3.20 |
6.33×10−7 |
8.44×10−4 | Up |
| 85329 | LGALS12 | 2.89 |
1.13×10−6 |
1.32×10−3 | Up |
| 6275 | S100A4 | 2.89 |
1.15×10−6 |
1.32×10−3 | Up |
| 151258 | SLC38A11 | 2.81 |
1.40×10−6 |
1.49×10−3 | Up |
| 342574 | KRT27 | −3.99 |
1.13×10−8 |
1.81×10−4 | Down |
| 140597 | TCEAL2 | −3.80 |
7.76×10−8 |
4.14×10−4 | Down |
| 372 | ARCN1 | −3.34 |
1.74×10−7 |
4.64×10−4 | Down |
| 54682 | MANSC1 | −3.22 |
3.36×10−7 |
6.72×10−4 | Down |
| 5523 | PPP2R3A | −3.06 |
4.88×10−7 |
7.82×10−4 | Down |
| 64816 | CYP3A43 | −2.97 |
1.72×10−6 |
1.63×10−3 | Down |
| 138046 | RALYL | −2.76 |
1.78×10−6 |
1.63×10−3 | Down |
| 6663 | SOX10 | −2.75 |
1.83×10−6 |
1.63×10−3 | Down |
| 56994 | CHPT1 | −2.73 |
2.19×10−6 |
1.65×10−3 | Down |
| 81621 | KAZALD1 | −2.96 |
2.23×10−6 |
1.65×10−3 | Down |
Limma package was used for differential expression
analysis. Using the standard P<0.05, a total of 265
differentially expressed lncRNAs were obtained, including 56
upregulated and 209 downregulated genes. The top 20 significant
differences in expression of lncRNA are shown in Table III. The differential expression
of lncRNAs was presented using heat map in Fig. 2.
| Table III.Top 20 differentially expressed long
non-coding RNAs. |
Table III.
Top 20 differentially expressed long
non-coding RNAs.
| ID | Symbol | Log2FC | P-value | FDR | Up/down |
|---|
| XLOC_003006 | – | 6.63 |
4.19×10−7 |
1.83×10−4 | Up |
| XLOC_016248 | – | 5.91 |
4.63×10−6 |
1.62×10−3 | Up |
|
ENSG00000253339.1 | RP11-434I12.3 | 4.48 |
8.09×10−4 |
8.83×10−2 | Up |
| XLOC_017561 | – | 2.46 |
9.52×10−4 |
9.23×10−2 | Up |
| XLOC_011814 | – | 2.44 |
2.05×10−3 |
1.43×10−1 | Up |
| XLOC_018649 | – | 3.99 |
2.32×10−3 |
1.43×10−1 | Up |
| XLOC_015500 | – | 5.85 |
2.39×10−3 |
1.43×10−1 | Up |
|
ENSG00000248810.1 | RP11-362F19.1 | 1.50 |
2.82×10−3 |
1.43×10−1 | Up |
| XLOC_018891 | – | 1.87 |
4.59×10−3 |
1.78×10−1 | Up |
| XLOC_000122 | – | 5.61 |
5.16×10−3 |
1.88×10−1 | Up |
| XLOC_010540 | – | −9.00 |
1.79×10−9 |
3.13×10−6 | Down |
| XLOC_010305 | – | −6.89 |
7.56×10−9 |
6.60×10−6 | Down |
|
ENSG00000250360.1 | CTD-2089N3.1 | −5.53 |
8.81×10−8 |
5.13×10−5 | Down |
| XLOC_018529 | – | −2.83 |
6.32×10−5 |
1.84×10−2 | Down |
| XLOC_025155 | – | −2.98 |
7.93×10−5 |
1.98×10−2 | Down |
| XLOC_005882 | – | −4.35 |
1.39×10−4 |
3.04×10−2 | Down |
| XLOC_018024 | – | −2.16 |
2.09×10−4 |
4.06×10−2 | Down |
| XLOC_015712 | – | −2.44 |
3.15×10−4 |
5.50×10−2 | Down |
| XLOC_019396 | – | −1.70 |
3.72×10−4 |
5.91×10−2 | Down |
|
ENSG00000181123.4 | RP4-539M6.14 | −1.83 |
5.77×10−4 |
7.80×10−2 | Down |
Functional annotation
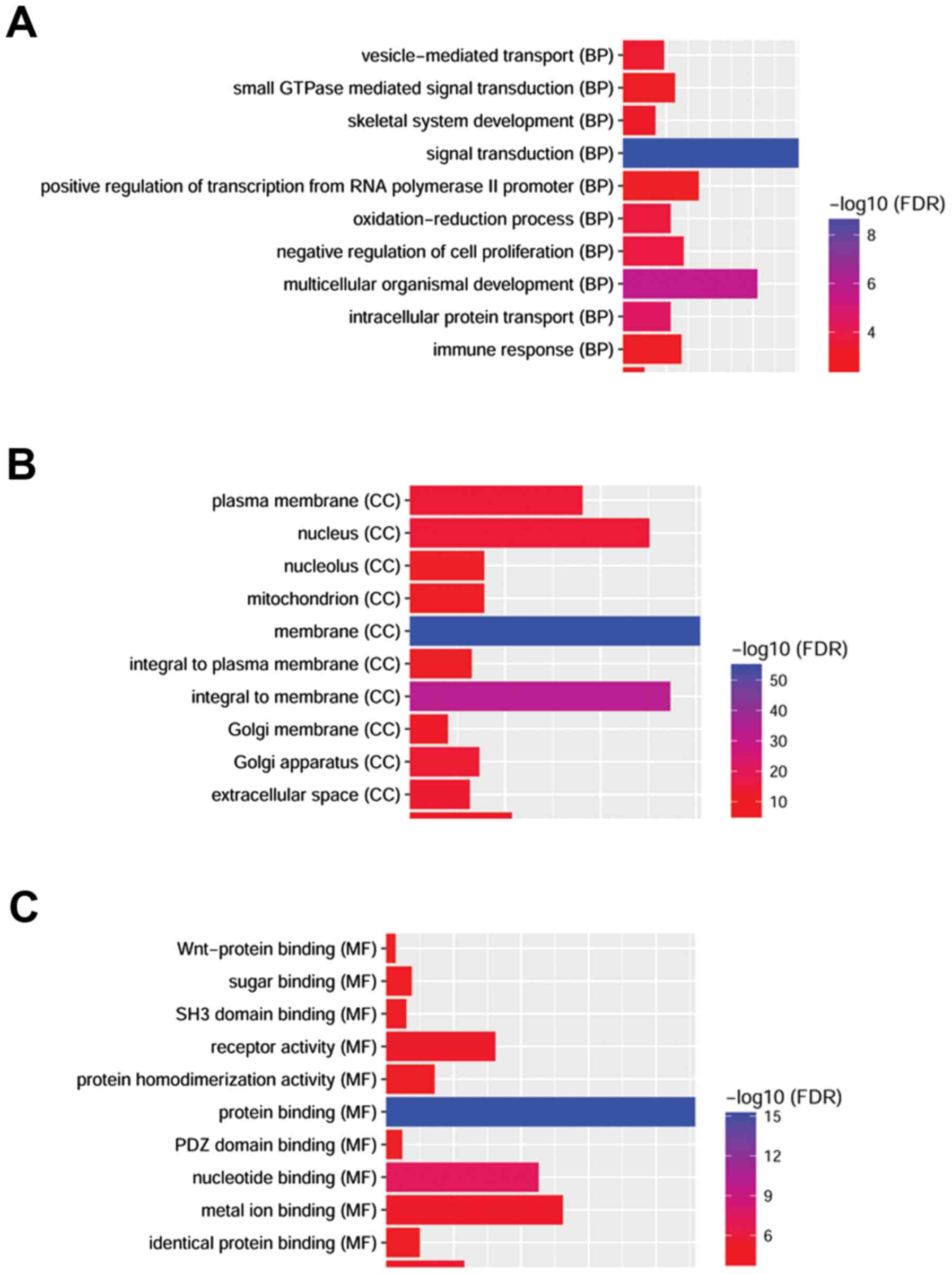
As demonstrated in Fig.
3, several GO categories were enriched among the upregulated
and downregulated DEmRNAs. ‘Signal transduction’ (including 81
genes, FDR=1.18×10−9) and ‘multicellular organismal
development’ (including 62 genes, FDR=1.55×10−6) were
the most significantly enriched BP; the top ranked cellular
component (CC) GO categories were ‘membrane’ (including 304 genes,
FDR=4.87×1056) and ‘cytoplasm’ (including 305 genes,
FDR=4.87×1056); and ‘protein binding’ (including 229
genes, FDR=3.94×1016) was the most significantly
enriched MF. The most significantly enriched BP of upregulated and
downregulated DEGs was ‘signal transduction’ and the most
significantly enriched MF was ‘protein binding’. The most
significantly enriched cellular component (CC) was ‘membrane’.
Several pathways were significantly enriched after the KEGG pathway
enrichment analysis (FDR<0.05, Fig.
4), including ‘cytokine-cytokine receptor interaction’
(including 26 genes, FDR=3.94×1016) and ‘cell adhesion
molecules’ (CAMs) (including 17 genes,
FDR=1.28×10−5).
Ppi network of differentially
expressed mRNAs
Based on the existing protein interaction data
included in the BioGRID database, Cytoscape was used to search the
top 100 upregulated and top 100 downregulated differentially
expressed mRNAs. After removing the non-differentially expressed
genes from analysis, the protein interaction network map was drawn
(Fig. 5). A total of 144 nodes and
123 edges were identified. Among them, the genes with the highest
degree were CUL4B (degree=20), COPS5 (degree=8), NEDD4L (degree=8),
ITGA4 (degree=5), CSF1R (degree=5), EPRS (degree=5), HSP90AA1
(degree=5).
DElncRNAs, and GO and KEGG analyses of
mRNAs in CRSwNP
Twenty-six pairs formed by lncRNAs and their
neighboring genes (including 23 lncRNAs and 25 mRNAs) were searched
to identify differentially expressed genes upstream and downstream
of lncRNAs, as shown in Table IV.
GO and KEGG enrichment analyses of the differentially expressed
genes were performed using GeneCoDis3
(genecodis.cnb.csic.es/analysis). KEGG enrichment results were
obtained using an FDR cut off of 0.05 and are shown in Table V.
| Table IV.Differentially expressed long
non-coding RNAs and adjacent mRNAs. |
Table IV.
Differentially expressed long
non-coding RNAs and adjacent mRNAs.
|
| Long non-coding
RNA | mRNA |
|---|
|
|
|
|
|---|
| ID | Symbol | Start-100 kb | End+100 kb | Symbol | Start | End |
|---|
|
ENSG00000204380.2 | PKP4-AS1 | 159,435,168 | 159,691,330 | PKP4 | 159,313,476 | 159,539,391 |
|
ENSG00000268001.1 | CARD8-AS1 | 48,658,932 | 48,861,456 | EMP3 | 48,824,766 | 48,833,810 |
|
ENSG00000272870.1 | RP11-798M19.6 | 174,185,145 | 174,390,966 | GALNT7 | 174,089,904 | 174,245,118 |
| XLOC_001856 | – | 60,327,121 | 60,534,026 | HOOK1 | 60,280,458 | 60,342,050 |
| XLOC_002179 | – | 153,430,979 | 153,633,083 | S100A2 | 153,533,584 | 153,540,366 |
| XLOC_002179 | – | 153,430,979 | 153,633,083 | S100A4 | 153,516,089 | 153,522,612 |
| XLOC_003728 | – | 115,617,190 | 115,820,554 | NHLRC2 | 115,614,420 | 115,676,953 |
| XLOC_004001 | – | 34,532,537 | 34,734,403 | EHF | 34,642,640 | 34,682,604 |
| XLOC_005757 | – | 108,928,967 | 109,130,444 | SELPLG | 109,016,053 | 109,027,735 |
| XLOC_005757 | – | 108,928,967 | 109,130,444 | CORO1C | 109,038,885 | 109,125,372 |
| XLOC_006494 | – | 118,455,546 | 118,673,787 | PEBP1 | 118,573,663 | 118,583,389 |
| XLOC_009628 | – | 20,654,577 | 20,875,447 | ACSM3 | 20,621,565 | 20,808,903 |
| XLOC_010280 | – | 34,304,118 | 34,520,635 | CCL18 | 34,391,640 | 34,399,392 |
| XLOC_010305 | – | 37,141,750 | 37,342,737 | PLXDC1 | 37,219,556 | 37,310,647 |
| XLOC_011271 | – | 72,625,994 | 72,827,011 | CD300LF | 72,690,450 | 72,709,117 |
| XLOC_013363 | – | 58,726,344 | 58,938,866 | RPS5 | 58,897,767 | 58,906,173 |
| XLOC_013363 | – | 58,726,344 | 58,938,866 | ZSCAN22 | 58,838,385 | 58,853,698 |
| XLOC_014821 | – | 98,570,692 | 98,801,316 | VWA3B | 98,703,579 | 98,929,762 |
| XLOC_016476 | – | 38,383,272 | 38,584,377 | SOX10 | 38,366,693 | 38,383,429 |
| XLOC_018137 | – | 128,110,058 | 128,311,374 | GATA2 | 128,198,270 | 128,212,028 |
| XLOC_018236 | – | 151,046,653 | 151,250,581 | P2RY13 | 151,044,100 | 151,047,336 |
| XLOC_019500 | – | 174,101,606 | 174,311,598 | GALNT7 | 174,089,904 | 174,245,118 |
| XLOC_019578 | – | 1,831,467 | 2,036,070 | IRX4 | 1,877,527 | 1,887,350 |
| XLOC_024517 | – | 134,039,147 | 134,241,605 | WISP1 | 134,203,282 | 134,242,587 |
| XLOC_025491 | – | 113,925,958 | 114,147,241 | OR2K2 | 114,089,763 | 114,092,463 |
| XLOC_026071 | – | 139,765,127 | 139,967,176 | CDR1 | 139,864,570 | 139,867,036 |
| Table V.Kyoto Encyclopedia of Genes and
Genomes of differentially expressed mRNAs adjacent to
differentially expressed long non-coding RNA. |
Table V.
Kyoto Encyclopedia of Genes and
Genomes of differentially expressed mRNAs adjacent to
differentially expressed long non-coding RNA.
| ID | Term | FDR | Count | Symbols |
|---|
| 00512 | Mucin type O-Glycan
biosynthesis |
3.59×10−10 | 1 | GALNT7 |
| 00650 | Butanoate
metabolism |
3.59×10−10 | 1 | ACSM3 |
| 05150 | Staphylococcus
aureus infection |
5.43×10−7 | 1 | SELPLG |
| 03010 | Ribosome |
4.46×10−4 | 1 | RPS5 |
| 04514 | CAMs |
4.86×10−3 | 1 | SELPLG |
| 04062 | Chemokine signaling
pathway |
2.86×10−2 | 1 | CCL18 |
RT-qPCR confirmation
To validate the results of the integrated analysis,
the expression levels of five genes including (CCL18, GALNT7, CCL8,
CUL4B, NEDD4L and the lncRNA RP11-798M19.6 were selected and
verified by RT-qPCR. In Fig. 6,
with the exception of CCL8, NEDD4L and GALNT7, the expression of
the other 3 selected genes detected using RT-qPCR was consistent
with the results of our integrated analysis.
Discussion
Although many studies have found that CRSwNP has
various inflammatory phenotypes, drug sensitivities and prognoses,
the detailed molecular mechanisms remain unclear (18). Since effective treatment is
lacking, finding novel treatment strategies for CRS has long been a
research objective. In our study, 994 DEmRNAs (354 upregulated and
640 downregulated) and 265 DElncRNAs (56 upregulated and 209
downregulated) were identified. Based on the results of integrated
high-throughput transcriptome data analysis and a literature
search, we selected five differentially expressed genes (CCL18,
GALNT7, CCL8, CUL4B, NEDD4L) and one lncRNA (RP11-798M19.6) and the
expression of these candidate genes was verified by RT-qPCR in
disease and control groups. Expression profiles of CCL8, CUL4B and
RP11-798M19.6 were consistent with the results of high-throughput
sequencing. However, the RT-qPCR results for CCL18, NEDD4L and
GALNT7 were inconsistent with our integrated analysis, which may be
due to the small sample size.
GO and KEGG analyses were performed to identify the
biological functions enriched in the DEmRNAs. Following the
analysis of the dysregulated mRNAs, we found that two central
characteristics of CRSwNP were significantly enriched. These
included the inflammatory and immune response, such as
‘cytokine-cytokine receptor interaction’
(FDR=3.94×1016), and regional microenvironment changes
in nasal polyps, such as ‘signal transduction’ and ‘protein
binding’. We found that the most significantly enriched BP of
upregulated and downregulated DEGs was ‘signal transduction’ and
the most significantly enriched MF was ‘protein binding’. The most
significantly enriched cellular component (CC) was ‘membrane’.
Several pathways were demonstrated to be significantly enriched
following the KEGG pathway enrichment analysis (FDR<0.05,
Fig. 3), including
‘cytokine-cytokine receptor interaction’
(FDR=3.94×1016), and ‘cell adhesion molecules (CAMs)’
(FDR=1.28×10−5). KEGG analysis of DEmRNAs adjacent to
DElncRNAs, revealed that DEmRNAs were enriched in ‘mucin type
O-Glycan biosynthesis (GALNT7)’ and ‘chemokine signaling pathway
(CCL18)’. Chemokine receptors are potential targets for the
treatment of many inflammatory diseases (19–21).
Collectively these findings indicate that CCL18, closely related to
chemokine signaling, is involved in CRSwNP.
CCL18, a 7.8 kDa protein composed of 69 amino acids,
is known as pulmonary and activation-regulated chemokine (PARC)
(22). Peterson et al
(22) found that compared with
normal control uncinate tissue (UT), CCL18 mRNA was significantly
increased in NP (P<0.001) and UT (P<0.05) from patients with
CRSwNP, but not in UT from patients with CRSsNP. And the author
guessed that overproduction of CCL18 might contribute to the
pathogenesis of CRSwNP (23). In
our study, CCL18 was upregulated in patients with CRSwNP and the
adjacent lncRNA XLOC_010280 was also upregulated. Wang et al
(24) also found an important
lncRNA, XLOC_010280, which was highly correlated with CCL18 in
cis prediction of functions of lncRNAs. In our study,
differentially expressed CCL18 was enriched in CCL18, and
XLOC_010280 was determined as the adjacent lncRNA of CCL18. CCL18
and XLOC_010280 were both upregulated in patients with CRSwNP. We
may conclude that lncRNA XLOC_010280 upregulates the mRNA
expression of CCL18, and causes the inflammatory disease.
As part of the acetylgalactosaminyltransferase
family, N-acetylgalactosaminyltransferase 7 (GALNT7) acts as a
glycosyltransferase in protein O-GlcNAcylation. A previous study
demonstrated that GGalNAc-transferase-7 (GALNT7) was involved in
the regulation of cell proliferation and was related to
tumorigenesis. miR-494 and GALNT7-small interfering RNA (siRNA)
have been shown to inhibit tumor growth in nude mice (25). In our study, GALNT7 was upregulated
in patients with CRSwNP, and its adjacent lncRNA RP11-798M19.6 was
downregulated in these patients. Besides, in the KEGG analysis, the
mRNA of GALNT7 was enriched in ‘mucin type O-Glycan biosynthesis’,
which may be associated with CRSwNP.
In conclusion, the present study provides further
insight into the molecular aspects of CRSwNP, suggesting new
molecular signatures and new targets for application as specific
biomarkers. In particular, our findings suggest that
inflammation-related genes and cell proliferation-associated genes
may be factors indicating poor prognosis of CRSwNP.
Acknowledgements
The authors would like to thank Beijing Medintell
Bioinformatic Technology Co., Ltd. (Beijing, China) for assisting
with the in high-throughput sequencing and data analysis.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ML and YL supervised the research program, drafted
the manuscript and revised it critically for important intellectual
content. PG and JA participated in the acquisition, analysis and
interpretation of data. CG contributed to the acquisition and
analysis of data. FL and YL carried out the data analysis. ML and
YL gave final approval of the version to be published. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
The present study has been approved by the Ethics
Committee of The First People's Hospital of Jining.
Patient consent for publication
All individuals provided signed informed consent for
the use of their samples in the present study.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Beule A: Epidemiology of chronic
rhinosinusitis, selected risk factors, comorbidities, and economic
burden. GMS Curr Top Otorhinolaryngol Head Neck Surg.
14:Doc112015.PubMed/NCBI
|
|
2
|
Ma Z, Shen Y, Zeng Q, Liu J, Yang L, Fu R
and Hu G: miR-150-5p regulates EGR2 to promote the development of
chronic rhinosinusitis via the DC-Th axis. Int Immunopharmacol.
54:188–197. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bohman A, Juodakis J, Oscarsson M, Bacelis
J, Bende M and Torinsson Naluai Å: A family-based genome-wide
association study of chronic rhinosinusitis with nasal polyps
implicates several genes in the disease pathogenesis. PLoS One.
12:e01852442017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chaaban MR, Walsh EM and Woodworth BA:
Epidemiology and differential diagnosis of nasal polyps. Am J
Rhinol Allergy. 27:473–478. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lin H, Li Z, Lin D, Zheng C and Zhang W:
Role of NLRP3 inflammasome in eosinophilic and non-eosinophilic
chronic rhinosinusitis with nasal polyps. Inflammation.
39:2045–2052. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kim JY, Kim DK, Yu MS, Cha MJ, Yu SL and
Kang J: Role of epigenetics in the pathogenesis of chronic
rhinosinusitis with nasal polyps. Mol Med Rep. 17:1219–1227.
2018.PubMed/NCBI
|
|
7
|
Cava C, Bertoli G and Castiglioni I:
Integrating genetics and epigenetics in breast cancer: Biological
insights, experimental, computational methods and therapeutic
potential. BMC Syst Biol. 9:622015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Saris CG, Horvath S, van Vught PW, van Es
MA, Blauw HM, Fuller TF, Langfelder P, DeYoung J, Wokke JH, Veldink
JH, et al: Weighted gene co-expression network analysis of the
peripheral blood from Amyotrophic Lateral Sclerosis patients. BMC
Genomics. 10:4052009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li WB, Zhou J, Xu L, Su XL, Liu Q and Pang
H: Identification of genes associated with papillary thyroid
carcinoma (PTC) for diagnosis by integrated analysis. Horm Metab
Res. 48:226–231. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Huang QX, Cui JY, Ma H, Jia XM, Huang FL
and Jiang LX: Screening of potential biomarkers for
cholangiocarcinoma by integrated analysis of microarray data sets.
Cancer Gene Ther. 23:48–53. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang L, Zhang J, Yang G, Wu D, Jiang L,
Wen Z and Li M: Investigating the concordance of gene ontology
terms reveals the intra- and inter-platform reproducibility of
enrichment analysis. BMC Bioinformatics. 14:1432013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yang L, Feng S and Yang Y: Identification
of transcription factors (TFs) and targets involved in the
cholangiocarcinoma (CCA) by integrated analysis. Cancer Gene Ther.
23:439–445. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang H, Zhang C, Feng R, Zhang H, Gao M
and Ye L: Investigating the microRNA-mRNA regulatory network in
acute myeloid leukemia. Oncol Lett. 14:3981–3988. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang F, Wang R, Li Q, Qu X, Hao Y, Yang J,
Zhao H, Wang Q, Li G, Zhang F, et al: A transcriptome profile in
hepatocellular carcinomas based on integrated analysis of
microarray studies. Diagn Pathol. 12:42017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang J, Qu D, An J, Yuan G and Liu Y:
Integrated microarray analysis provided novel insights to the
pathogenesis of glaucoma. Mol Med Rep. 16:8735–8746. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wu T, Wu HD, Xu ZX, Han F, Zhang BQ, Sun J
and Hu SJ: Abnormal expression of long non-coding RNAs in
myocardial infarction. Heart Vessels. 32:1253–1261. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhao B, Wang M, Xu J, Li M and Yu Y:
Identification of pathogenic genes and upstream regulators in
age-related macular degeneration. BMC Ophthalmol. 17:1022017.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Okada N, Nakayama T, Asaka D, Inoue N,
Tsurumoto T, Takaishi S, Otori N, Kojima H, Matsuda A, Oboki K, et
al: Distinct gene expression profiles and regulation networks of
nasal polyps in eosinophilic and non-eosinophilic chronic
rhinosinusitis. Int Forum Allergy Rhinol. 8:592–604. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Charo IF and Ransohoff RM: The many roles
of chemokines and chemokine receptors in inflammation. N Engl J
Med. 354:610–621. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Viola A and Luster AD: Chemokines and
their receptors: Drug targets in immunity and inflammation. Annu
Rev Pharmacol Toxicol. 48:171–197. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Peterson S, Poposki JA, Nagarkar DR,
Chustz RT, Peters AT, Suh LA, Carter R, Norton J, Harris KE,
Grammer LC, et al: Increased expression of CC chemokine ligand 18
in patients with chronic rhinosinusitis with nasal polyps. J
Allergy Clin Immunol. 129:119–127.e1-9. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hieshima K, Imai T, Baba M, Shoudai K,
Ishizuka K, Nakagawa T, Tsuruta J, Takeya M, Sakaki Y, Takatsuki K,
et al: A novel human CC chemokine PARC that is most homologous to
macrophage-inflammatory protein-1 alpha/LD78 alpha and chemotactic
for T lymphocytes, but not for monocytes. J Immunol. 159:1140–1149.
1997.PubMed/NCBI
|
|
24
|
Wang W, Gao Z, Wang H, Li T, He W, Lv W
and Zhang J: Transcriptome analysis reveals distinct gene
expression profiles in eosinophilic and noneosinophilic chronic
rhinosinusitis with nasal polyps. Sci Rep. 6:266042016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nie GH, Luo L, Duan HF, Li XQ, Yin MJ, Li
Z and Zhang W: GALNT7, a target of miR-494, participates in the
oncogenesis of nasopharyngeal carcinoma. Tumour Biol. 37:4559–4567.
2016. View Article : Google Scholar : PubMed/NCBI
|