Introduction
Atherosclerosis is a major disease that has a severe
effect on human health. Its morbidity and mortality rates have
demonstrated an increase in recent years (1,2).
Atherosclerosis develops due to impairment of molecular and
cellular activities, leading to the disruption of vascular
homeostasis (3). Currently, this
disease is primarily considered as a form of chronic inflammation,
which arises in response to lipid accumulation. Macrophages are
effector cells which stimulate vascular inflammatory reactions
throughout the pathological process (4). An increasing number of factors
reportedly participate in the inflammatory response in
atherosclerosis (5,6); however, the detailed mechanisms
underlying this process remain to be elucidated. Thus, an improved
understanding of the pathophysiology of atherosclerosis in addition
to the development of effective treatment methods are of great
importance.
Sirtuin (SIRT) 4 is a member of the sirtuin protein
family and is abundant in the heart, brain, kidney, liver and
skeletal muscles (7). The Sirtuin
family of proteins, from SIRT1 to SIRT7, serve key roles in the
prevention of atherosclerosis (8).
SIRT1 and SIRT6 inhibit foam cell formation and promote foam cell
egress to prevent atherogenesis (9,10).
SIRT2 may regulate macrophage polarization in order to inhibit
atherosclerotic plaque progression (11). SIRT4, initially reported as not
showing nicotinamide adenine dinucleotide (NAD)-dependent
deacetylase activity (12), is
localized in the mitochondrial matrix (13). It uses NAD to adenosine diphosphate
ribosylate glutamate dehydrogenase (GDH) and suppresses GDH
activity, limiting the generation of adenosine triphosphate
(14). A previous study suggested
that SIRT4 is a major regulator of lipid metabolism (15). Under nutrient-replete conditions,
SIRT4 acts to repress fatty acid oxidation and promote lipid
anabolism (16). In a previous
study, it was demonstrated that lipopolysaccharide (LPS) treatment
significantly decreased the expression of SIRT4 at mRNA and protein
levels in a dose-dependent manner (17). In colorectal cancer, SIRT4
suppressed the proliferation, migration and invasion of cancer
cells through the inhibition of glutamine metabolism via
upregulation of E-cadherin expression (18). SIRT4 overexpression protected
against diabetic nephropathy by preventing glucose-induced podocyte
apoptosis and production of reactive oxygen species (19). Our previous study indicated that
SIRT4 may inhibit inflammatory responses in human umbilical vein
endothelial cells (HUVECs) (20).
Oxidized low density lipoprotein (oxLDL) reportedly
induces atherosclerosis by triggering endothelial cell damage, and
promoting lipid accumulation and proinflammatory responses
(21). Nuclear factor (NF)-κB, a
multifunctional transcription regulator, serves an important role
in the inflammatory pathways of atherosclerosis, where expression
of NF-κB and its downstream genes can be regulated via the
phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt) signaling
pathway, which serves a vital role in the processes of cell
proliferation, apoptosis and inflammation (22,23).
The aim of the present study was to investigate the role of SIRT4
in the development of atherosclerosis. The results indicated that
oxLDL reduced the expression of SIRT4 in HUVECs. In addition, it
was identified that overexpression of SIRT4 may reverse
oxLDL-induced cell proliferation inhibition, rescue oxLDL-induced
apoptosis and attenuate the expression of pro-inflammatory
cytokines interleukin (IL)-1β, IL-6 and tumor necrosis factor
(TNF)-α induced by oxLDL, possibly via inhibition of the
PI3K/Akt/NF-κB signaling pathway.
Materials and methods
Cell culture and treatment
HUVECs were obtained from Capsugel, Morristown, NJ,
USA). The cells were cultured in Endothelial Growth Basal Medium
(EBM-2; Lonza Group, Ltd., Basel, Switzerland) supplemented with
growth factors according to the manufacturer's protocols. To
analyze the changes in SIRT4 expression in response to oxLDL
(Thermo Fisher Scientific, Inc., Waltham, MA, USA) at various
concentrations, cells were treated with oxLDL for 24 h at
concentrations of 0, 10, 50 and 100 µM at 37°C. In order to analyze
changes in SIRT4 expression in response to oxLDL treatment
following different time periods, cells were treated with 50 µM
oxLDL for 0, 12, 24 and 48 h at 37°C. Following treatment, cells
were harvested and assessed for changes in SIRT4 expression using
western blotting.
Stable overexpression of SIRT4 and
treatment
The complete open reading frame of SIRT4 was cloned
into the pLVX–IRES-ZsGreen1 (Clontech Laboratories, Inc., Mountain
View, CA, USA) plasmid (OV-SIRT4), using the empty vector as
negative control (-NC). pLVX-SIRT4 was generated by transiently
transfecting 293T cells (Beijing Zhongyuan Ltd., Beijing, China).
Lentiviral production, concentration and titration were performed
as follows: 293T cells (70–80% confluence) were seeded in 6-well
plates, and 1.5 µg pLVX–IRES-ZsGreen1 was transfected using the
Lipofectamine® 2000 (40 µl; Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. Then,
293T cells were cultured for 48 h at 37°C. Lentivirus particles
were directly collected and concentrated from the cell culture
medium at 48 h following transduction by multi-steps of
ultracentrifugation (50,000 × g at 4°C for 2 h). The titration
(transduction units, TU) and multiplicity of infection of
concentrated lentivirus particles were determined in 293T cells
grown in 96-well plates by serial dilutions. For infection
purposes, 2×105 HUVECs were divided into 2 groups and
subcultured in 6-well culture plates for 24 h prior to
transduction. For infection, the cell culture medium was removed
and cells were washed twice with phosphate-buffered saline (PBS).
Next, 0.5 ml of lentiviral suspension (1×108 IU/ml,
multiplicity of infection=100) containing 8 µg/ml Polybrene was
added to the cells. Cells were then incubated at 37°C overnight.
Vector suspension was then aspirated from the cells and the
transduced cells were added to 2 ml/flask of fresh growth medium.
Cells were then incubated at 37°C in a humidified atmosphere
containing 5% CO2. The growth medium was replaced after
24 h. After allowing the cells to incubate for 72 h at 37°C, HUVECs
cells were passaged twice per week with growth medium containing 5
µg/ml puromycin to select for cells expressing the transduced
vector. Positively screened cell lines were sub-cloned three times
using limiting dilution (diluent: EBM-2) and cultured in growth
medium containing puromycin for 1 month to generate stable cell
lines. HUVECs were infected by pLVX-SIRT4 and treated with 50 µM
oxLDL. Following incubation for 48 h at 37°C and 5% CO2,
HUVECs overexpressing SIRT4 and NC cells were harvested for western
blotting.
Western blotting
Following treatment or infection, cells were lysed
using a cell lysis buffer containing protease inhibitors [Tris-HCl
(pH 7.5) 50 mM, NaCl 250 mM, EDTA 10 mM, NP-40 0.5%, Leupeptin 10
µM, PMSF 1 mM and NaF 4 mM]. The protein concentration was measured
using a bicinchoninic acid protein assay kit (Thermo Fisher
Scientific, Inc.). Soluble lysate was mixed with loading buffer and
boiled for 5 min. Equal amounts of the protein samples (50 ng/lane)
were separated via 10% SDS-PAGE and transferred on to
polyvinylidene difluoride membranes. Membranes were blocked with
PBS, containing 10% non-fat dry milk, overnight at 4°C and
incubated with anti-SIRT4 antibody (1:1,000; cat. no. ab124521),
anti-GAPDH antibody (1:10,000; cat. no. 5174), anti-PI3K antibody
(1:1,500; cat. no. 4249), anti-Akt antibody (1:1,500; cat. no.
4691), anti-phosphorylated (p)-Akt antibody (1:500; cat. no. 4060),
anti-p-inhibitor of κBα (IκBα) antibody (1:500; cat. no. 2859),
anti-total (t)-IκBα antibody (1:1,000; cat. no. 4812), anti-p65
NF-κB antibody (1:500; cat. no. 8242) and anti-p-p65 NF-κB antibody
(1:500; cat. no. 3033) for 2 h at 25°C. Anti-SIRT4 was obtained
from Abcam (Cambridge, MA, USA); all other primary antibodies were
obtained from Cell Signaling Technology, Inc., (Danvers, MA, USA).
Membranes were washed with TBS-0.05% Tween 20 and incubated with
horseradish peroxidase-conjugated goat anti-rabbit immunoglobulin G
heavy and light chain secondary antibodies (1:10,000; cat. no.
7074; Cell Signaling Technology, Inc.) for 2 h at room temperature.
This process was followed by the development of protein bands for
visualization. Image-Pro Plus 6.0 software (Media Cybernetics,
Silver Spring, MD, USA) was used to quantify relative protein
densities. GAPDH was used as the loading control. Each experiment
was replicated three times.
Cell proliferation assay
Cell proliferation was assessed using a Cell
Counting Kit-8 assay (CCK-8; Beyotime Institute of Biotechnology,
Shanghai, China), according to the manufacturer's protocols. HUVECs
stably overexpressing SIRT4 and control cells were cultured in
96-well plates and treated with 50 µM oxLDL for 48 h at 37°C and 5%
CO2. Then, 10 µl of CCK-8 reagent was added to each well
and mixed gently; cells were then incubated at 37°C for 4 h. The
absorbance was evaluated at 450 nm using a microplate reader.
Survival rate=optical density (OD)450OV-SIRT4
group/OD450NC group. Each experiment was repeated three
times.
Apoptosis assay
Cell apoptosis was analyzed using flow cytometry.
HUVECs stably overexpressing SIRT4 and NC cells were cultured and
treated with 50 µM oxLDL for 48 h at 37°C and 5% CO2.
The cells were collected and the assay was performed using a
Propidium Iodide (PI)/Annexin V-fluorescein isothiocyanate (FITC)
apoptosis detection kit (Nanjing Keygen Biotech Co., Ltd., Nanjing,
China). Annexin V-FITC (5 µl) and PI (5 µl) were added, and cells
were incubated at 25°C in the dark for 15 min. Within 1 h, the
apoptotic cells was then assessed using a flow cytometer (BD
Biosciences, San Jose, CA, USA) and data were analyzed using FlowJo
version 10.07 software (FlowJo LLC, Ashland, OR, USA). The
apoptotic rate was defined as the percentage of cells in the upper
and lower right quadrants. Each experiment was repeated three
times.
Immunofluorescence
HUVECs stably overexpressing SIRT4 and NC cells
(4×105 cells/ml) were seeded into 24-well plates and
treated with 50 µM oxLDL for 48 h at 37°C and 5% CO2.
Following treatment, the cells were washed with PBS and then fixed
with 4% paraformaldehyde for 15 min at 37°C. The cell membranes
were then permeabilized using 0.3% Triton X-100 in PBS with 0.2%
(V/V) Tween-20 (PBST) on ice for 15 min, followed by blocking with
5% bovine serum albumin (Gibco; Thermo Fisher Scientific, Inc.) and
2.5% fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.) in
PBST and were then incubated for 2 h at 25°C with rabbit anti-NF-κB
p65 antibody (1:100; cat. no. 8242; Cell Signaling Technology,
Inc.). Next, cells were washed three times with PBST and incubated
with the Alexa Fluor® 594-conjugated secondary antibody
(1:200; cat. no. ab150084; Abcam) for 1 h at room temperature.
Following three washes with PBST, the cells were counterstained
with DAPI (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) for 5 min
at 25°C. The cells were then visualized and images captured via
fluorescence microscopy (magnification, ×400; Leica Microsystems
GmbH, Wetzlar, Germany).
Enzyme-linked immunosorbent assay
(ELISA) for cytokine detection
HUVECs stably overexpressing SIRT4 and NC cells
(4×105 cells/ml) were seeded into 24-well plates,
followed by treatment with 50 µM oxLDL for 48 h at 37°C and 5%
CO2. Cell-free supernatants were collected and used in
an assay for the detection of cytokines. The concentrations of the
cytokines IL-1β (E-EL-H0149c), IL-6 (E-EL-H0102c) and TNF-α
(E-EL-H0109c) were determined using commercially available ELISA
kits (eBioscience; Thermo Fisher Scientific, Inc.). The entire
procedure was performed according to the manufacturer's protocols.
Each experiment was replicated three times.
Statistical analysis
All statistical analyses were performed using SPSS
version 19.0 (IBM Corp., Armonk, NY, USA). Results are presented as
the mean ± standard deviation, and experiments were repeated in
triplicate. Student's t-test was performed for comparison between
two groups. Multiple groups were compared using one-way analysis of
variance, followed by a least significant difference post-hoc test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
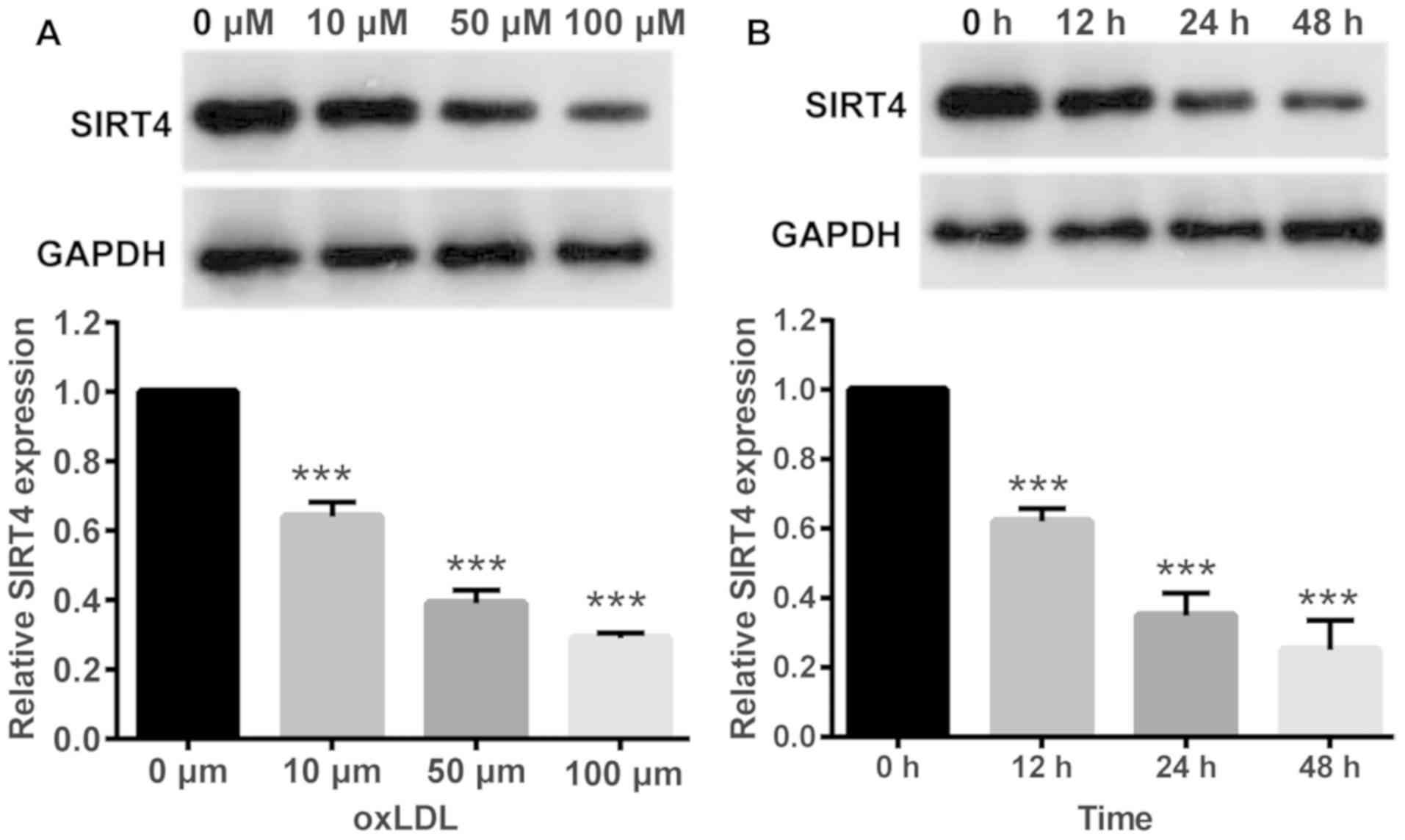
OxLDL reduces the expression of SIRT4
in HUVECs
In order to investigate changes in SIRT4 expression
in response to oxLDL, HUVECs were treated with oxLDL at various
concentrations. Western blotting results demonstrated that oxLDL
treatment significantly reduced SIRT4 expression in HUVECs,
compared with in control cells treated with 0 µM oxLDL. Higher
oxLDL concentrations resulted in lower SIRT4 expression levels
indicating a dose dependent association between oxLDL
concentrations and SIRT4 expression (Fig. 1A). Next, HUVECs were treated with
the same oxLDL concentration (50 µM) for different periods of time.
Western blotting results demonstrated that oxLDL treatment
significantly reduced SIRT4 expression in HUVECs compared with
control cells treated with 0 µM oxLDL, in a time dependent manner;
longer durations of treatment resulted in lower SIRT4 expression
levels (Fig. 1B).
Stably overexpressing SIRT4 in
HUVECs
Cells overexpressing SIRT4 were selected by
screening and treated with 50 µM oxLDL for 48 h. SIRT4 expression
was determined via western blotting. SIRT4 expression levels were
significantly higher in cells overexpressing SIRT4, compared with
the NC group following oxLDL treatment for 48 h (P<0.001;
Fig. 2). The results indicated
successful overexpression of SIRT4 in HUVECs.
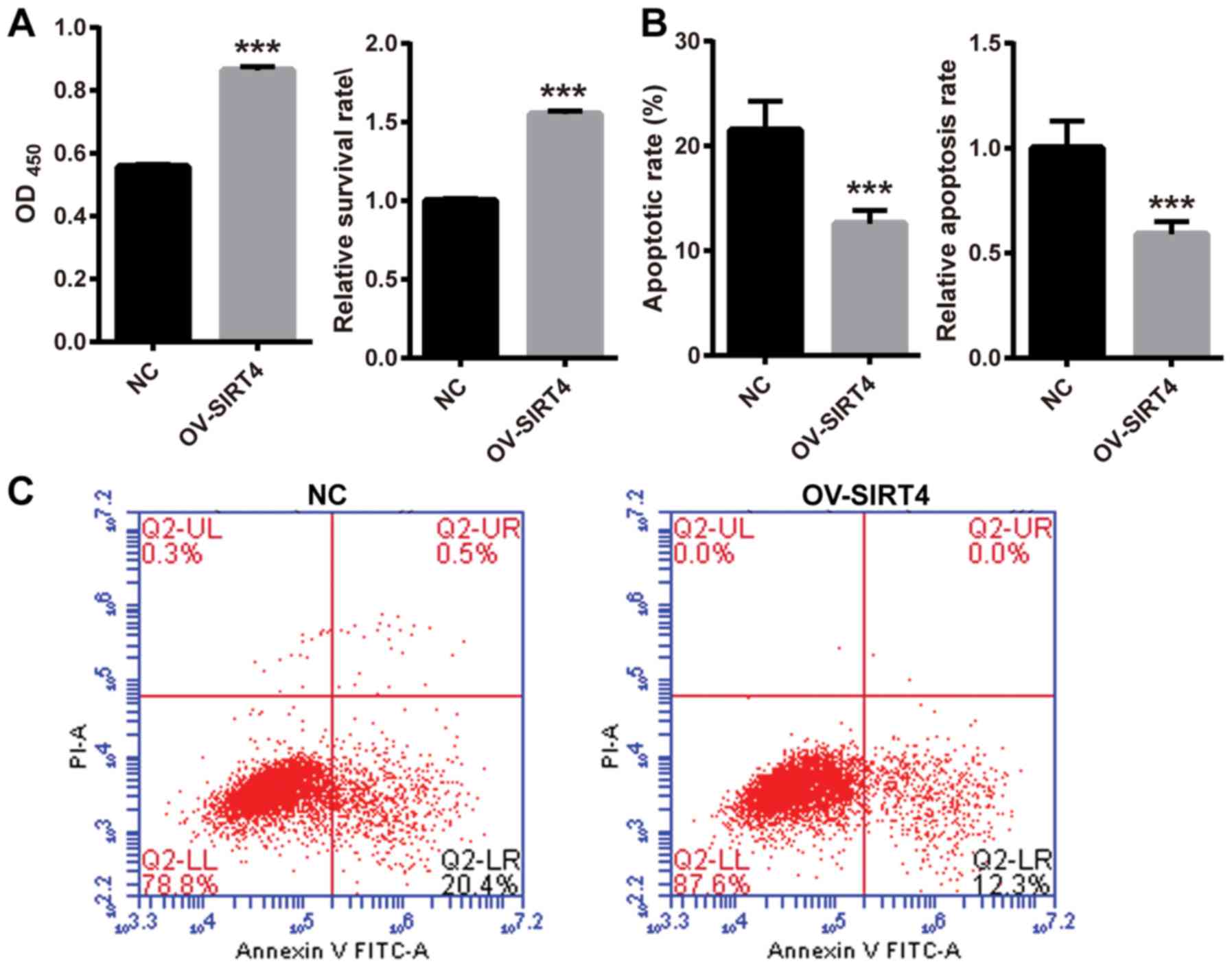
Overexpression of SIRT4 reverses
oxLDL-induced inhibition of cell proliferation
To investigate the effect of SIRT4 overexpression in
response to oxLDL on HUVEC proliferation, HUVECs overexpressing
SIRT4 and NC HUVECs were treated with 50 µM oxLDL for 48 h. Cell
proliferation was assessed using a CCK-8 assay. The survival rate
was significantly higher in HUVECs overexpressing SIRT4, compared
with NC HUVECs following oxLDL treatment (Fig. 3A). The results indicated that SIRT4
overexpression may reverse oxLDL-induced inhibition of cell
proliferation. In order to explore the effects of SIRT4
overexpression in response to oxLDL on HUVEC apoptosis, HUVECs
overexpressing SIRT4 and NC HUVECs were treated with 50 µM oxLDL
for 48 h. Cell apoptosis was examined via flow cytometry. Following
oxLDL treatment, the apoptotic rate significantly decreased in
HUVECs overexpressing SIRT4 compared with in NC HUVECs (Fig. 3B and C). These results indicated
that overexpression of SIRT4 may inhibit oxLDL-induced
apoptosis.
Overexpression of SIRT4 inhibits the
PI3K/Akt/NF-κB signaling pathway
In order to elucidate the role of SIRT4 in
regulating the PI3K/Akt/NF-κB signaling pathway, HUVECs
overexpressing SIRT4 and NC HUVECs were treated with 50 µM oxLDL
for 48 h. Western blotting analysis was conducted to quantify
protein expression associated with the PI3K/Akt/NF-κB signaling
pathway. Compared with the NC, SIRT4 overexpression suppressed the
PI3K/Akt/NF-κB pathway in HUVECs by inhibiting PI3K expression
(Fig. 4A and B). It also
significantly inhibited p-Akt, p-IκBα and p-P65 NF-κB expression
compared with the control. P65 NF-κB nuclear translocation was
analyzed via immunofluorescence. Immunofluorescence analysis
revealed that P65 NF-κB was largely expressed in the nucleus, while
a small quantity was expressed in the cytoplasm of cells in the
oxLDL + NC treatment group. In contrast, in the oxLDL + OV-SIRT4
treatment group, P65 NF-κB was largely expressed in the cytoplasm,
while a small quantity was expressed in the nucleus. These results
suggest that OV-SIRT4 may inhibit P65 NF-κB nuclear translocation
(Fig. 4C).
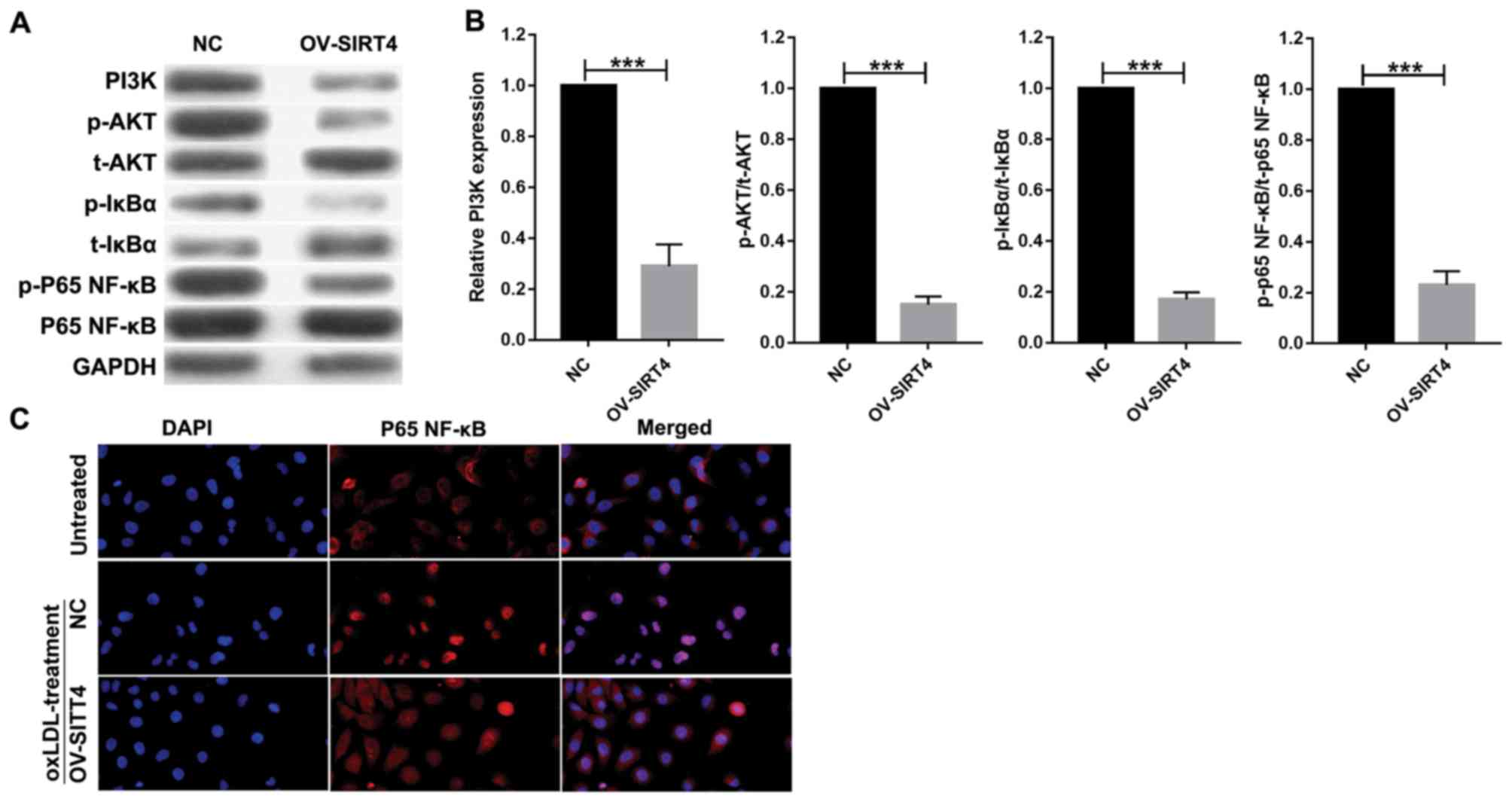 | Figure 4.PI3K/Akt/NF-κB signaling pathway,
activated by oxLDL, was suppressed by SIRT4 overexpression in
HUVECs. (A and B) Expression of PI3K, p-Akt, t-Akt, p-IκBα, t-IκBα,
p-p65 NF-κB, p65 NF-κB proteins were detected by western blotting
following 50 µM oxLDL treatment at 48 h in overexpressed
SIRT4-HUVECs and NC-HUVECs. Each experiment was replicated three
times (***P<0.001 vs. NC). (C) P65 NF-κB nuclear translocation
was examined via immunofluorescence in overexpressed SIRT4-HUVECs,
NC-HUVECs and HUVECs (magnification, ×400). HUVECs, human umbilical
vein endothelial cells; IκBα, inhibitor of κBα; NF, nuclear factor;
NF, nuclear factor, NC, negative control; oxLDL, oxidized low
density lipoprotein; PI3K, phosphoinositide 3-kinase; Akt, protein
kinase B; SIRT4, sirtuin 4; p-, phosphorylated; t-, total. |
Overexpression of SIRT4 attenuates the
expression of inflammatory cytokines induced by oxLDL
In order to further confirm the effect of SIRT4
overexpression on endothelial activation in response to
inflammatory cytokines, HUVECs overexpressing SIRT4 and NC HUVECs
were treated with 50 µM oxLDL for 48 h. The expression of IL-1β,
IL-6 and TNF-α were assessed using ELISA. Compared with NC HUVECs,
the expression levels of IL-1β, IL-6 and TNF-α were significantly
decreased in HUVECs overexpressing SIRT4 (Fig. 5). The results indicated that SIRT4
overexpression attenuated the expression levels of IL-1β, IL-6 and
TNF-α induced at the protein level by oxLDL.
Discussion
Previous studies have indicated that endothelial
dysfunction may serve a vital role during the initial stages of
atherosclerosis (6,24). Although much progress has been made
in studies of pathophysiology and clinical aspects of
atherosclerosis, the detailed mechanism underlying this disease
remains to be elucidated (25,26).
OxLDL reportedly induces endothelial cell injury and dysfunction
(27). In the present study,
HUVECs were stimulated by oxLDL to mimic the conditions of damage
and inflammation of endothelial cells. The results demonstrated
that oxLDL may reduce SIRT4 expression in HUVECs in a dose- and
time-dependent manner. SIRT4 is a member of the sirtuin protein
family which is able to improve vascular smooth muscle and
endothelial cell injury induced by lipid deposition, oxidative
stress and inflammation, thus opposing atherogenesis (28). In a previous study, it was
demonstrated that LPS treatment significantly reduced SIRT4
expression at the mRNA and protein levels in a dose-dependent
manner (16). In the present
study, it was demonstrated that oxLDL treatment inhibited SIRT4
expression in a dose-dependent manner in HUVECs, suggesting that
SIRT4 may be critically involved in oxLD L-induced endothelial
dysfunction. In diabetic nephropathy, SIRT4 overexpression
increased mouse podocyte cell proliferation and suppressed
apoptosis, which was accompanied with an increase in mitochondrial
membrane potential and decreased production of reactive oxygen
species (18). The results of the
present study demonstrated that overexpression of SIRT4 may reverse
oxLDL-induced cell proliferation inhibition and rescue
oxLDL-induced apoptosis. These results indicated that SIRT4
overexpression may enhance the survival of HUVECs in an
oxLDL-induced inflammatory environment.
It has been reported that the PI3K/Akt pathway may
be implicated in the regulation of a number of inflammatory
diseases, including atherosclerosis (29). In addition, it was previously
demonstrated that activation of the PI3K/Akt pathway was also
involved in the activation of NF-κB via the phosphorylation of IκBα
protein (30). The NF-κB complex
is localized and inactivated in the cytoplasm by binding to the
inhibitory protein IκB (31).
Phosphorylation of IκBα protein results in the dissociation of IκBα
from NF-κB, allowing NF-κB to freely migrate into the nucleus and
activate transcription (32). The
current study identified that SIRT4 overexpression suppressed PI3K,
p-Akt, p-IκBα and p-P65 NF-κB expression. Furthermore,
immunofluorescence analysis revealed that p65 NF-κB, which is
localized in the nucleus, was significantly decreased in HUVECs
with SIRT4-overexpression. These results suggested that SIRT4 may
inhibit the PI3K/Akt/NF-κB pathway. NF-κB activity has been
demonstrated to be essential for the production of pro-inflammatory
cytokines in atherosclerosis. Inflammation contributes to AS
development from plaque onset to plaque rupture (33). Inflammatory cytokines promote the
recruitment of monocytes to endothelial cells and induce to foam
cell formation (34). A previous
study demonstrated that SIRT4 suppressed NF-κB nuclear
translocation, and inhibited IL-1β, IL-6 and IL-8 expression in
HUVECs (19). The findings of the
present study demonstrated that SIRT4 overexpression significantly
decreased the levels of IL-1β, IL-6 and TNF-α induced by oxLDL. The
results of the present study indicated that SIRT4 overexpression
attenuated the expression of IL-1β, IL-6 and TNF-α induced by oxLDL
by suppressing the PI3K/Akt/NF-κB pathway, which improves the
inflammatory environment induced by oxLDL and enhances the survival
of HUVECs.
The present study involved several limitations.
First, all experiments were performed using one cell line and,
therefore, it is possible that the conclusions are specific to this
cell line only and may not apply to atherosclerosis per se.
Thus, further validation of these findings using more cell and
animal models are required in subsequent studies. Additionally, the
potential pathways that mediate the effects of oxLDL on SIRT4 and
the effects of SIRT4 on the PI3K/Akt pathway remain to be
elucidated. Whether SIRT4 attenuates oxLDL-induced HUVEC apoptosis
via the PI3K/Akt/NF-κB signaling pathway requires further
investigation.
In conclusion, the present study provided notable
evidence suggesting that SIRT4 overexpression enhanced HUVEC
survival, suppressed the PI3K/Akt/NF-κB signaling pathway and
inhibited inflammatory cytokine expression induced by oxLDL. These
results may serve as a theoretical basis to provide novel insight
into the treatment of atherosclerosis.
Acknowledgements
Not applicable.
Funding
This study was supported by the grant from Jiangxi
Province Science Foundation for Youths (grant no. 20161BAB215257)
and the Science and Technology Program by the Health and Family
Planning Commission of Jiangxi Province (grant no. 20162304).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
YT, SY and HL conceived and designed the present
study. MC, YW and JX developed the methodology. YT, SY, MC, YW and
JX completed the experiment and collected the data. YT, SY and HL
analyzed and interpreted the data. YT and SY drafted the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Heuslein JL, Meisner JK, Li X, Song J,
Vincentelli H, Leiphart RJ, Ames EG, Blackman BR, Blackman BR and
Price RJ: Mechanisms of amplified arteriogenesis in collateral
artery segments exposed to reversed flow direction. Arterioscler
Thromb Vasc Biol. 35:2354–2365. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tousoulis D, Oikonomou E, Economou EK,
Crea F and Kaski JC: Inflammatory cytokines in atherosclerosis:
Current therapeutic approaches. Eur Heart J. 37:1723–1732. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Park KH and Park WJ: Endothelial
dysfunction: Clinical implications in cardiovascular disease and
therapeutic approaches. J Korean Med Sci. 30:1213–1225. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tuttolomondo A, Di Raimondo D, Pecoraro R,
Arnao V, Pinto A and Licata G: Atherosclerosis as an inflammatory
disease. Curr Pharm Des. 18:4266–4288. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chistiakov DA, Melnichenko AA, Grechko AV,
Myasoedova VA and Orekhov AN: Potential of anti-inflammatory agents
for treatment of atherosclerosis. Exp Mol Pathol. 104:114–124.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ren K, Lu YJ, Mo ZC, -Liu X, Tang ZL,
Jiang Y, Peng XS, Li L, Zhang QH and Yi GH: ApoA-I/SR-BI modulates
S1P/S1PR2-mediated inflammation through the PI3K/Akt signaling
pathway in HUVECs. J Physiol Biochem. 73:287–296. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pirinen E, Lo Sasso G and Auwerx J:
Mitochondrial sirtuins and metabolic homeostasis. Best Pract Res
Clin Endocrinol Metab. 26:759–770. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kane AE and Sinclair DA: Sirtuins and
NAD(+) in the development and treatment of metabolic and
cardiovascular diseases. Circ Res. 123:868–885. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zheng H, Fu Y, Huang Y, Zheng X, Yu W and
Wang W: mTOR signaling promotes foam cell formation and inhibits
foam cell egress through suppressing the SIRT1 signaling pathway.
Mol Med Rep. 16:3315–3323. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
D'Onofrio N, Servillo L and Balestrieri
ML: SIRT1 and SIRT6 signaling pathways in cardiovascular disease
protection. Antioxid Redox Signal. 28:711–732. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang B, Ma Y and Xiang C: SIRT2 decreases
atherosclerotic plaque formation in low-density lipoprotein
receptor-deficient mice by modulating macrophage polarization.
Biomed Pharmacother. 97:1238–1242. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Haigis MC, Mostoslavsky R, Haigis KM,
Fahie K, Christodoulou DC, Murphy AJ, Valenzuela DM, Yancopoulos
GD, Karow M, Blander G, et al: SIRT4 inhibits glutamate
dehydrogenase and opposes the effects of calorie restriction in
pancreatic beta cells. Cell. 126:941–954. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Komlos D, Mann KD, Zhuo Y, Ricupero CL,
Hart RP, Liu AY and Firestein BL: Glutamate dehydrogenase 1 and
SIRT4 regulate glial development. Glia. 61:394–408. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Michishita E, Park JY, Burneskis JM,
Barrett JC and Horikawa I: Evolutionarily conserved and
nonconserved cellular localizations and functions of human SIRT
proteins. Mol Biol Cell. 16:4623–4635. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Laurent G, de Boer VC, Finley LW, Sweeney
M, Lu H, Schug TT, Cen Y, Jeong SM, Li X, Sauve AA and Haigis MC:
SIRT4 represses peroxisome proliferator-activated receptor α
activity to suppress hepatic fat oxidation. Mol Cell Biol.
33:4552–4561. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Laurent G, German NJ, Saha AK, de Boer VC,
Davies M, Koves TR, Dephoure N, Fischer F, Boanca G, Vaitheesvaran
B, et al: SIRT4 coordinates the balance between lipid synthesis and
catabolism by repressing malonyl CoA decarboxylase. Mol Cell.
50:686–698. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Qiu Y, Lai H, Huang Y, Hong L, Wang H and
Tao Y: Effect of shear force on SIRT4 in LPS-injured human
umbilical vein endothelial cells. Int J Clin Exp Pathol.
9:4921–4930. 2016.
|
|
18
|
Miyo M, Yamamoto H, Konno M, Colvin H,
Nishida N, Koseki J, Kawamoto K, Ogawa H, Hamabe A, Uemura M, et
al: Tumour-suppressive function of SIRT4 in human colorectal
cancer. Br J Cancer. 113:492–499. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shi JX, Wang QJ, Li H and Huang Q: SIRT4
overexpression protects against diabetic nephropathy by inhibiting
podocyte apoptosis. Exp Ther Med. 13:342–348. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tao Y, Huang C, Huang Y, Hong L, Wang H,
Zhou Z and Qiu Y: SIRT4 suppresses inflammatory responses in human
umbilical vein endothelial cells. Cardiovasc Toxicol. 15:217–223.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hasanally D, Edel A, Chaudhary R and
Ravandi A: Identification of oxidized phosphatidylinositols present
in OxLDL and human atherosclerotic plaque. Lipids. 52:11–26. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kang Q, Liu W, Liu H and Zhou M: Effect of
compound chuanxiong capsule on inflammatory reaction and
PI3K/Akt/NF-κB signaling pathway in atherosclerosis. Evid Based
Complement Alternat Med. 2015:5845962015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Schlegel N, Leweke R, Meir M, Germer CT
and Waschke J: Role of NF-κB activation in LPS-induced endothelial
barrier breakdown. Histochem Cell Biol. 138:627–641. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu Z, Wang J, Huang X, Li Z and Liu P:
Deletion of sirtuin 6 accelerates endothelial dysfunction and
atherosclerosis in apolipoprotein E-deficient mice. Transl Res.
172:18–29.e2. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jackson SW, Scharping NE, Jacobs HM, Wang
S, Chait A and Rawlings DJ: Cutting Edge: BAFF overexpression
reduces atherosclerosis via TACI-Dependent B cell activation. J
Immunol. 197:4529–4534. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Min J, Weitian Z, Peng C, Yan P, Bo Z, Yan
W, Yun B and Xukai W: Correlation between insulin-induced estrogen
receptor methylation and atherosclerosis. Cardiovasc Diabetol.
15:1562016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Di Pietro N, Formoso G and Pandolfi A:
Physiology and pathophysiology of oxLDL uptake by vascular wall
cells in atherosclerosis. Vascul Pharmacol. 84:1–7. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sosnowska B, Mazidi M, Penson P,
Gluba-Brzózka A, Rysz J and Banach M: The sirtuin family members
SIRT1, SIRT3 and SIRT6: Their role in vascular biology and
atherogenesis. Atherosclerosis. 265:275–282. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Garat CV, Crossno JT Jr, Sullivan TM,
Reusch JE and Klemm DJ: Inhibition of phosphatidylinositol
3-kinase/Akt signaling attenuates hypoxia-induced pulmonary artery
remodeling and suppresses CREB depletion in arterial smooth muscle
cells. J Cardiovasc Pharmacol. 62:539–548. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shen T, Yang Z, Cheng X, Xiao Y, Yu K, Cai
X, Xia C and Li Y: CXCL8 induces epithelial-mesenchymal transition
in colon cancer cells via the PI3K/Akt/NF-κB signaling pathway.
Oncol Rep. 37:2095–2100. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Napetschnig J and Wu H: Molecular basis of
NF-κB signaling. Annu Rev Biophys. 42:443–468. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hayden MS and Ghosh S: NF-κB, the first
quarter-century: remarkable progress and outstanding questions.
Genes Dev. 26:203–234. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Onat D, Brillon D, Colombo PC and Schmidt
AM: Human vascular endothelial cells: A model system for studying
vascular inflammation in diabetes and atherosclerosis. Curr Diab
Rep. 11:193–202. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zernecke A and Weber C: Inflammatory
mediators in atherosclerotic vascular disease. Basic Res Cardiol.
100:93–101. 2005. View Article : Google Scholar : PubMed/NCBI
|