Introduction
Prader-Willi syndrome (PWS; OMIM 176270) was first
reported by Prader et al (1) in 1956. As a complicated
neurodevelopmental genetic disorder, PWS classically presents with
severe hypotonia and feeding difficulties in the neonatal period,
sometimes with concurrent anorexia and a sucking deficit (2). Challenged by the difficulties in
early diagnosis, care and treatment, the majority of patients gain
excessive weight at three or four years of age, and begin to
exhibit signs of developmental delay and hypogonadism, eventually
leading to severe early obesity, intellectual disability, cognitive
impairment, genital hypoplasia and infertility (3,4). PWS
is characterized by a sporadic occurrence, which does not
discriminate among gender or race, with an estimated prevalence of
1 in 15,000–30,000 live births (5). An interstitial deletion of paternal
origin at 15q11.2-q13 may result in the absence of paternal
expression of imprinted genes located in this chromosomal region.
An imprinting defect (ID) or maternal uniparental disomy (UPD) of
chromosome 15 may also be an explanation for the pathogenic
mechanism of this disease (6). Due
to the severe complications and high mortality of PWS, a
multidisciplinary strategy is required to aid early diagnosis and
optimize clinical management in order to improve quality of life
and prognosis. Pediatricians worldwide are focusing effort on
prenatal diagnosis. However, some clinical aspects of PWS in
utero, including abnormal extremity positions, fetal
hypomobility and polyhydramnios, can only guide PWS diagnosis owing
to the lack of high specificity and sensitivity of these features.
To date, there have been few reports examining the prenatal
diagnosis of PWS using ultrasound or as an accidental finding by
cytogenetic molecular techniques (7–9). In
this present report, the case of an infant with PWS resulting from
complete maternal heterodisomy (hetero-UPD) of chromosome 15 is
presented. This case was not identified by fetal ultrasound
examination, chromosome karyotype analysis or chromosome microarray
(CMA) conducted during the pregnancy, but was identified
retrospectively by postnatal diagnosis of the syndrome according to
fetal ultrasound findings, postnatal clinical features and
molecular genetic investigations.
Patients and methods
Clinical history
The 1-day-old male proband was the second child of
healthy, non-consanguineous parents of 41 years of age. The family
history was unremarkable. The parents had a healthy daughter of 10
years of age. During the pregnancy, polyhydramnios and a lack of
fetal movements were noted. At 23 weeks and 6 days of gestation,
ultrasound screening showed normal fetal growth with no structural
abnormalities, but revealed polyhydramnios (amniotic fluid index,
25.2 cm) accompanied by reduced fetal movement. At 30 weeks and 6
days of gestation, polyhydramnios (amniotic fluid index, 29.0 cm)
and decreased fetal activity persisted. Fetal biometric
measurements confirmed that the fetal weight, estimated according
to the abdominal circumference, head circumference and femoral
length, was ~1,180 g, which was below the 10th percentile for this
gestational age. Considering the advanced maternal age and
polyhydramnios, interventional prenatal diagnosis was recommended
by obstetricians. With a thorough understanding of sampling
procedures, the potential risks and limitations of the test, the
couple agreed to amniocentesis at 24 weeks plus 2 days of gestation
for routine chromosome examination and CMA. Giemsa banded
chromosome analysis of amniotic fluid cells cultured in situ
and CMA revealed an apparently normal karyotype of 46, XY (data not
shown), and the pregnancy was continued. At 38 weeks plus 1 day of
gestation, the mother was hospitalized with fetal distress. An
elective caesarean section was performed on the day of admission
(October 2017) due to breech presentation. The caesarean was
performed in The Obstetrical Department of The First Affiliated
Hospital of Chongqing Medical University. The neonatal evaluation
recorded a birth weight of 2,680 g (5th percentile), a length of 47
cm (2.9th percentile), and a head circumference of 33.5 cm (20.7th
percentile), with an appearance, pulse, grimace, activity and
respiration score of 9, 10 and 10 at 1, 5 and 10 min (10,11),
respectively. The boy was admitted to the neonatal intensive care
unit directly after birth due to a severely poor suck and
hypotonia. Notable physical features included a narrow forehead,
up-slanted palpebral fissure, bilateral epicanthus, micrognathia,
wide spaced nipples, long slender fingers, hypopigmentation, a weak
cry and hypoplastic external genitalia (Fig. 1). A neurological exam showed
significant hypotonia. Gastric tube feeding was required due to the
poor sucking. Clinically, the physical features were indicative of
PWS. In view of the negative results of prenatal cytogenetic and
molecular analysis, rare types of PWS, including maternal
heterodisomy and ID were considered. On day 5 after birth, specific
genetic examinations were conducted. At 1 month of age, with a body
weight of 3,160 g (0.9th percentile), a length of 48.5 cm (0.1th
percentile) and a head circumference of 35.2 cm (8th percentile),
the boy was evaluated again by geneticists. Special feeding
techniques were still required due to the persistent difficulty in
mouth feeding and poor weight gain. On physical examination,
hypotonia remained and symptoms of upper respiratory infections,
including cough, fever and nasal discharge, were noticed. The boy
succumbed to from recurrent respiratory infections, hypoventilation
and food choking at 4 months of age.
Methylation status analysis
In order to confirm the diagnosis of PWS, a DNA
methylation test was conducted in which peripheral blood
lymphocytes were collected from 2 ml blood. To isolate genomic DNA,
the QIAamp DNA blood mini kit (cat. no. 51104; Qiagen GmbH) was
used according to the manufacturer's protocol. The DNA samples were
treated with the CpGenome™ Turbo Bisulfite Modification kit (cat.
no. S7820; Merck KGaA) following the manufacturer's protocol. The
DNA modified by bisulfite was amplified with primers specific to
the differentially methylated sites within exon 1 and the promoter
regions of the gene encoding small nuclear ribonucleoprotein
polypeptide N: Methylated-specific forward,
5′-TAAATAAGTACGTTTGCGCGGTC-3′ and reverse,
5′-AACCTTACCCGCTCCATCGCG-3′ amplified a 174-bp DNA region, and
non-methylated-specific forward, 5′-GTAGGTTGGTGTGTATGTTTAGGT-3′ and
reverse, 5′-ACATCAAACATCTCCAACAACCA-3′ were used to amplify a
100-bp DNA region. Methylation-specific (MS) PCR was carried out as
described in previous studies (12,13).
Amplification products were compared with appropriate positive
(patients with PWS) and negative (healthy patients) controls, using
agarose gel electrophoresis. The results showed two PCR products of
174 and 100 bp in unaffected individuals. By contrast, only the 174
bp product from the maternal allele was observed, with the absence
of the 100 bp product in this patient with PWS.
Chromosomal microarray analysis
Following salt-induced precipitation, DNA from
peripheral blood samples was genotyped using CytoScan HD array
(Affymetrix; Thermo Fisher Scientific, Inc.). In this experiment,
genotype calling, quality control and identification of copy-number
variation (CNV) were performed using Affymetrix Chromosome Analysis
Suite software (version 4.0; Affymetrix; Thermo Fisher Scientific,
Inc.), with various databases employed for evaluation of the array
data and analysis of genotype-phenotype correlations, including
OMIM (http://www.ncbi.nlm.nih.gov/omim), DECIPHER
(http://decipher.sanger.ac.uk/), DGV
(http://projects.tcag.ca/variation)
and ISCA (http://dbsearch.clinicalgenome.org/search/).
UPD classification with the
UPDtool
All CNVs were excluded before analysis to prevent
their potential interference with UPD detection. A UPD converter
tool (UPDtool; version 0.2) (14)
was used to transform the exported genotypes into the required
format, followed by the detection and classification of the UPD
with a UPD tool (http://www.uni-tuebingen.de/uni/thk/de/f-genomik-software.html).
Short tandem repeat (STR) linkage
analysis
The aforementioned abnormal findings were subject to
further verification, a multiplex-fluorescence-labeled STR assay
with DNA from the proband and the parents was performed. Using
microsatellite markers from chromosome 15 as the parameters for
linkage analysis, including D15S11, D15S646, D15S817, D15S128,
D15S1513, GABRB3, D15S822 [typical for PWS/Angelman syndrome (AS)
deletion region], D15S659 and FES proto-oncogene, tyrosine kinase
(distal region). The experiment was carried out according to the
procedure as described in a previous study (15). Data collection and allele sizing
were completed using GENEMAPPER software (version 2.2; Applied
Biosystems; Thermo Fisher Scientific, Inc.); two STR markers were
required as the minimum for identification of the abnormal
genotypes.
Results
Methylation specific PCR
Non-quantitative methylation-specific PCR for the
PWS/AS region demonstrated the absence of the paternally derived
allele, with only a single band from the maternal allele present
(Fig. 2), indicating a total loss
of paternal imprinting, typically associated with PWS.
 | Figure 2.Methylation specific-PCR showing that
the patient lacks the paternal 100 bp band. Lane 1, DNA ladder
marker; lane 2, father of the patient; lane 3, mother of the
patient; lane 4, patient; lane 5, Prader-Willi syndrome positive
control (deletion type); lane 6, negative control; lane 7, PWS
positive control (UPD type); lane 8, Angelman syndrome positive
control; lane 9, blank control; Lane 10, internal control
(non-methylated DNA). |
Chromosomal microarray analysis
For single nucleotide polymorphism (SNP)-based CMA,
a total of 276,527 markers were compared between the child and
parents. The SNP array analysis of the family did not identify any
copy number alterations genome-wide. However, trio analysis of SNP
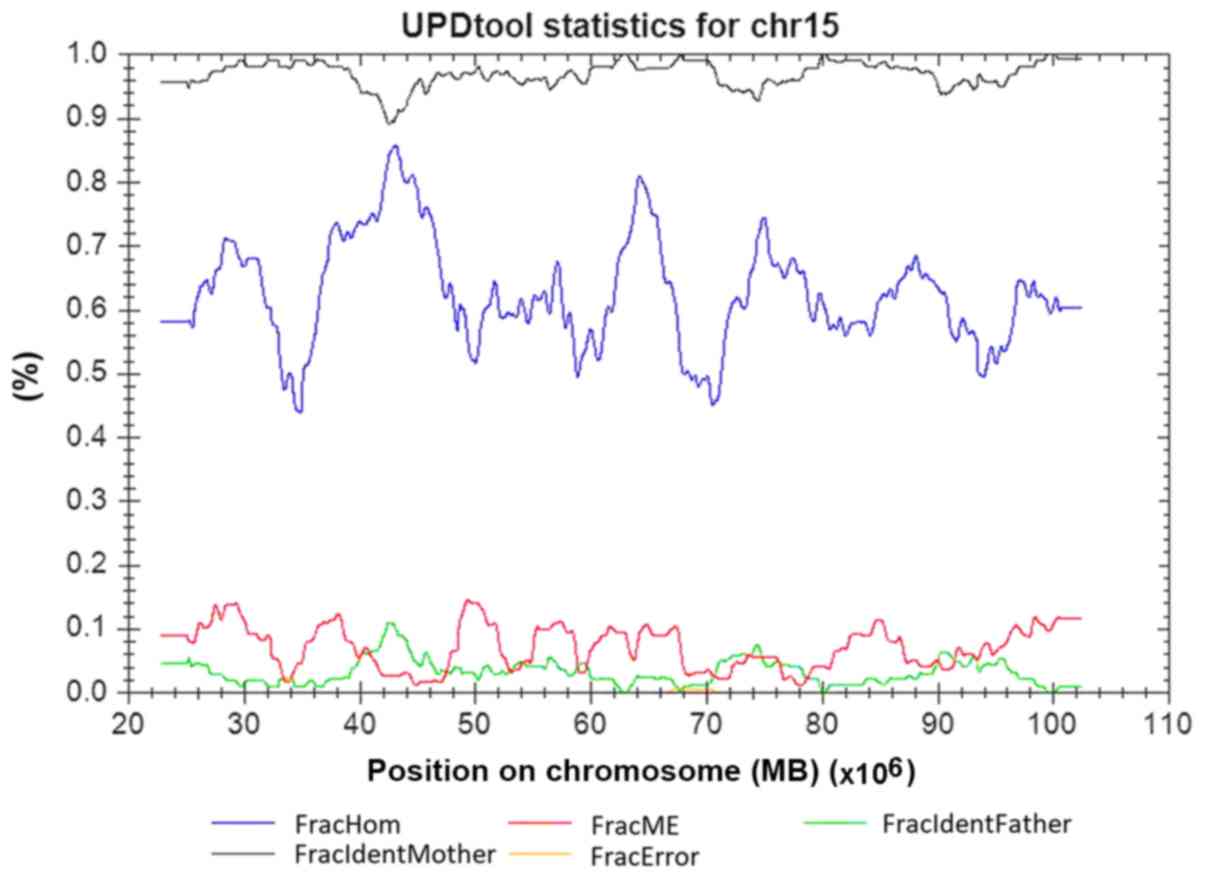
loci on chromosome 15 was consistent with uniparental inheritance,
and the classification of UPD using the UPDtool revealed a 100%
match in chromosome 15 between the child and the mother, which is
indicative of a maternal heterodisomy for the entire chromosome 15
(Fig. 3). No trisomy or monosomy
was found using the SNP array.
STR linkage analysis
Family linkage analysis based on STR analysis showed
four markers (D15S11, D15S1513, D15S822 and D15S659), mapping to
15q11.2q21.1, with only maternal alleles and the absence of a
paternal allele in this case. This result indicates maternal
heteroUPD of chromosome 15 (Fig.
4).
 | Figure 4.Schematic map of STR based linkage
analysis of the patient. Top panel, the PWS/AS critical region is
highlighted by a red box. In the middle panel, the locations of
nine STR loci alleles (D15S11, D15S646, D15S817, D15S128, D15S1513,
GABRB3, D15S822, D15S659, and FES) on 15q are shown as dots,
with the different colors denoting the different conditions of the
alleles. Seven STR loci alleles (D15S11, D15S646, D15S817, D15S128,
D15S1513, GABRB3, and D15S822) are located in the typical
PWS/AS critical region of BP1 to BP3, and the remaining two
(D15S659 and FES) are located in the distal region near the
telomere. At the bottom, four electrophoresis maps of STR loci
alleles (D15S11, D15S1513, D15S822, and D15S659) reveal that only
maternal alleles exist and no paternal allele is passed on to the
patient, indicating the maternal heteroUPD of chromosome 15. STR,
short tandem repeat; PWS, Prader-Willi syndrome; AS, Angelman
syndrome; BP, proximal breakpoint; F, father; M, mother; C,
child. |
In view of the clinical manifestation, karyotype,
MS-PCR, CMA and STR based linkage analysis, a diagnosis of PWS
resulting from complete maternal heteroUPD of chromosome 15 was
confirmed for this patient.
Discussion
As a sporadic genetic disorder with remarkable
developmental consequences, PWS is usually triggered by a paternal
deletion or maternal UPD of the chromosome region 15q11-q13. The
current genetic epidemiology is based on western populations,
according to which 15q11.2-q13 paternal deletion is responsible for
70–75%, maternal UPD for 25–28%, and ID for 2–5% of the cases of
PWS (16–18). However, studies of Asian
populations reveal a smaller proportion of PWS arising from
maternal UPD than has previously been recognized (5,19–21).
Furthermore, research on Western populations also revealed
significant discrepancies in both the genotypes and phenotypes in
cases of PWS arising from maternal UPD compared to those with
chromosomal region deletions, exhibiting fewer typical facial
phenotypes, a shorter course of gavage feeding, later onset of
hyperphagia, significantly higher birth weights and a lesser degree
of hypotonia (22). These milder
clinical manifestations among maternal UPD patients are usually
related to missed diagnosis, and thus should arouse vigilance among
pediatricians.
Widely accepted mechanisms for the development of
maternal UPD 15 are as follows: i) Trisomy rescue, triggered by the
loss of one copy of chromosome 15 in a trisomic fetus; ii) gamete
complementation, formed in a disomic egg fertilized by a nullisomic
sperm; and iii) post-zygotic duplication (23,24).
Trisomy rescue, the most commonly accepted mechanism, may give rise
to a number of outcomes in somatic cells mitosis, leading to
hetero-UPD (rescue), a normal cell (rescue) or trisomy (no or
incomplete rescue). Incomplete rescue of trisomy 15 may lead to the
development of mosaic mutations and chromosome rearrangements,
which are associated with cases of PWS with maternal UPD. In this
case, complete heterozygosity of the DNA markers implies
non-disjunction in the maternal meiosis II, which is also
consistent with studies demonstrating that the incidence of
chromosome 15 non-disjunction may exponentially increase with
maternal age, and that the children of older mothers have an
increased rate of maternal UPD for chromosome 15 compared to
mothers who are under 35 years of age at delivery (23,25).
Due to the severe complications associated with PWS,
it is important to clarify its features so that early diagnoses can
be made. At present, PWS is difficult to diagnose prenatally due to
a lack of precise and well-characterized fetal phenotypes, which,
otherwise, would have provided a basis for further molecular
genetic examinations. To date, only a few reports of PWS at the
prenatal stage have been published. Most of these reported
nonspecific prenatal signs indicative of PWS, and were accidentally
identified as trisomy 15 in a routine prenatal examination such as
chorionic villus sampling or amniotic fluid culture (26–28),
or as a retrospective discovery in postnatal diagnosis according to
fetal ultrasound findings and/or postnatal clinical features
(7–9,29–33).
Specific prenatal signs are required to allow
further molecular genetic examination for PWS. Studies have
identified a number of clinical features of the fetus as indicators
for PWS, including the abnormal position of feet and toes (9), polyhydramnios (9,31),
cerebral anomalies (33),
decreased fetal activity (9,34)
and hypoplasia of external genitalia (33,35),
with the hope that these features can facilitate an early prenatal
diagnosis of PWS. However, none of these are representative. In
2008, Bigi et al (9) issued
a report in which a possible fetal phenotype was delineated for the
first time, including abnormal extremity positions accompanied by
reduced fetal movement and polyhydramnios, which are also regarded
as informative in the diagnosis of PWS. Further studies followed
these, but there remains an absence of detailed prenatal
characteristics of fetuses with PWS. Therefore,
prenatally-diagnosed cases of PWS described in the literature have
been reviewed in the present report and the major characteristics
detailed in Table SI (9,26,28–41).
A previous report on a total of 28 prenatal cases with PWS suggests
that the phenotypes of the fetuses are variable despite the
similarities shared by most of the cases. Among the phenotypic
features commonly exhibited in prenatal individuals harboring PWS,
the top 10 are breech position (6/7, 85.7%), polyhydramnios (13/26,
50%), intrauterine growth restriction (IUGR) (10/26, 38.5%),
decreased fetal movement (8/26, 30.8%), facial dysmorphism (5/17,
29.4%), extended legs/feet with flexed toes (5/17, 29.4%), low
abdominal circumference (4/16, 25%), low femoral length (4/16,
25%), clenched hands (3/16, 18.8%), hypogonadism (3/16, 18.8%) and
low occipital frontal circumference (2/16, 12.5%). Some phenotypic
features presented in the present case are consistent with previous
reports, including polyhydramnios (9,30,33),
breech position (9), clenched
hands (33), peculiar position of
the extremities (9,31,33)
and diminished fetal movement (9).
Some of the features, including IUGR and facial dysmorphism, have
not been noted or summarized in previous studies to the best of our
knowledge. This phenotypic inconsistency was considered to be
associated with multiple factors, including the differences in the
deletion sizes and affected genes, and even the clinical experience
of the obstetricians and ultrasonologists. IUGR and facial
dysmorphism, which occur frequently in cases of PWS, should be
included as indicators for the prenatal diagnosis of PWS.
It should be noted that all the indicators have a
lack of specificity to allow a confirmed prenatal PWS diagnosis.
While a prenatal diagnosis solely relying on ultrasound findings
seems implausible, nevertheless, when fetal ultrasound examinations
detect these indicators, genetic analysis should be considered to
diagnose PWS.
A diagnosis of PWS is established in a proband
following a DNA methylation analysis demonstrating abnormal
parent-specific imprinting within the Prader-Willi critical region
on chromosome 15 in which the region demonstrates maternal-only
imprinting. Three molecular mechanisms that give rise to PWS
include paternal deletion, maternal UPD15 and ID. Optimizing a
diagnostic strategy requires clinicians to fully understand the
options available for testing for PWS, including the conditions to
use these methods, their technical superiorities and limitations,
and the cost of the testing (42).
For example, DNA methylation analysis is the only technique able to
diagnose PWS caused by the three aforementioned genetic mechanisms
and to differentiate PWS from AS in cases of deletion (43). Therefore, DNA methylation analysis
is regarded as the primary choice for differentiating PWS from
other confounders which cannot be identified using laboratory
information, before or after birth. Upon the diagnosis of PWS,
further testing is required to explore the underlying mechanism,
with the purpose of discriminating the individuals with a high
recurrence risk from the larger population with a very low
recurrence risk (deletions and maternal UPD). The genetic subtypes
and testing methods used in the diagnosis of Prader-Willi syndrome
are summarized in Table SII. As a
universally applied tool for CNV detection, CMA has substituted
karyotype analysis and some other specific tests as one of the
first choices for patients with developmental/intellectual
disorders, autism, congenital abnormalities and other dysmorphic
features (44–47). The Affymetrix CytoScan HD array has
great value in identifying human chromosomal aberrations due to its
broad genomic coverage, and is therefore deemed to be a reliable
method capable of specifically detecting 25- to 50-kb copy number
changes across the genome with allelic SNP call corroboration.
However, although CMA using the CytoScan HD has proved to be
successful in identifying maternal UPD patients, including all
iso-UPD cases and most of the iso/hetero-UPD cases, it is likely to
fail in the identification of complete hetero-UPD. In the case
reported in this present study, the diagnosis was missed in the
fetal ultrasound examination, chromosome karyotype analysis and CMA
conducted during the pregnancy, and identified retrospectively by
postnatal examinations according to fetal ultrasound findings,
postnatal clinical features, parent-child trio CMA-SNP array and
STR based linkage analysis (48).
As far as the present study is concerned, a failure
to diagnosis PWS occurred in prenatal ultrasound and routine
prenatal CMA examinations due to the technical limitations of these
approaches for detecting complete hetero-UPD. It was the postnatal
clinical features that allowed a diagnosis of PWS, which was
determined by trio CMA analysis and a
multiplex-fluorescence-labeled STR assay developed on the basis of
linkage analysis as a rapid and economic detection method for
chromosomal region deletion or maternal UPD15. It is now important
to determine which approach to the prenatal testing of PWS is
sufficient enough to make confirmed diagnoses. Certain clinical
features of the fetus, including anomalous extremity positions and
subdued fetal movement, combined with polyhydramnios, could be
considered indicators of the need for further testing to allow the
early prenatal diagnosis of PWS owing to the high potential risk of
PWS and the technical limitation of CMA. If clinical signs strongly
suggest PWS (abnormal ultrasonic results and advanced maternal
age), methylation and/or UPD analysis is highly recommendable.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was financially supported by
National Natural Science Foundation of China for Youth (grant no.
31501207) and Research Project of Traditional Chinese Medicine
Bureau of Guangdong Province (grant no. 20171030). We also would
like to appreciate the support from ‘111 program’ of Ministry of
Education P.R.C and State Administration of Foreign Experts Affairs
P.R.C.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YD and SL evaluated the patients clinically and
wrote the manuscript. JuL and JiL performed the prenatal ultrasonic
examinations and amniocentesis. QC and JYL worked on cell culture
of amniotic fluid and karyotype analysis. CL and HL undertook
molecular cytogenetic testing and bioinformatics analysis. HQ and
RL acted as the lead clinician in the study, counseled the parents,
and guided the writing of the manuscript.
Ethics approval and consent to
participate
The present work was approved by The Ethics
Committee of The First Affiliated Hospital of Chongqing Medical
University and The Ethics Committee of Guangdong Women and Children
Hospital. Written consent was given by the parents for full
photographs, clinical and laboratory studies. Blood samples used as
positive and negative controls were collected from patients with
PWS and healthy subjects, respectively. All subjects enrolled in
the present study signed a written informed consent. All the
procedures performed in the present study were in accordance with
the Declaration of Helsinki.
Patient consent for publication
Written informed consent was obtained from the
parents of all patients for the publication of all associated data
and images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Prader A, Labhart A and Willi H: Ein
Syndrom von Adipositas, Kleinwuchs, Kryptorchismus und Oligophrenie
nach myotonieartigem Zustand im Neugeborenalter. Schweir Med Wschr.
86:1260–1261. 1956.
|
|
2
|
Oiglane-Shlik E, Zordania R, Varendi H,
Antson A, Mägi ML, Tasa G, Bartsch O, Talvik T and Ounap K: The
neonatal phenotype of Prader-Willi syndrome. Am J Med Genet A.
140:1241–1244. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hirsch HJ, Eldar-Geva T, Bennaroch F,
Pollak Y and Gross-Tsur V: Sexual dichotomy of gonadal function in
Prader-Willi syndrome from early infancy through the fourth decade.
Hum Reprod. 30:2587–2596. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hartin SN, Hossain WA, Weisensel N and
Butler MG: Three siblings with Prader-Willi syndrome caused by
imprinting center microdeletions and review. Am J Med Genet A.
176:886–895. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ramsden SC, Clayton-Smith J, Birch R and
Buiting K: Practice guidelines for the molecular analysis of
Prader-Willi and Angelman syndromes. BMC Med Genet. 11:702010.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Horsthemke B and Wagstaff J: Mechanisms of
imprinting of the Prader-Willi/Angelman region. Am J Med Genet A
146A. 2041–2052. 2008. View Article : Google Scholar
|
|
7
|
Geysenbergh B, De Catte L and Vogels A:
Can fetal ultrasound result in prenatal diagnosis of Prader-Willi
syndrome? Genet Couns. 22:207–216. 2011.PubMed/NCBI
|
|
8
|
Whittington JE, Butler JV and Holland AJ:
Pre-, peri- and postnatal complications in Prader-Willi syndrome in
a UK sample. Early Hum Dev. 84:331–336. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bigi N, Faure JM, Coubes C, Puechberty J,
Lefort G, Sarda P and Blanchet P: Prader-Willi syndrome: Is there a
recognizable fetal phenotype? Prenat Diagn. 28:796–799. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Apgar V: A proposal for a new method of
evaluation of the newborn infant. Curr Res Anesth Analg.
32:260–267. 1953. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Butterfield J and Covey MJ: Practical
epigram of the Apgar score. JAMA. 181:3531962. View Article : Google Scholar
|
|
12
|
Hussain Askree S, Hjelm LN, Ali Pervaiz M,
Adam M, Bean LJ, Hedge M and Coffee B: Allelic dropout can cause
false-positive results for Prader-Willi and Angelman syndrome
testing. J Mol Diagn. 13:108–112. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kubota T, Das S, Christian SL, Baylin SB,
Herman JG and Ledbetter DH: Methylation-specific PCR simplifies
imprinting analysis. Nat Genet. 16:16–17. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Schroeder C, Sturm M, Dufke A,
Mau-Holzmann U, Eggermann T, Poths S, Riess O and Bonin M: UPDtool:
A tool for detection of iso- and heterodisomy in parent-child trios
using SNP microarrays. Bioinformatics. 29:1562–1564. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang K, Liu S, Feng B, Yang Y, Zhang H,
Dong R, Liu Y and Gai Z: Clinical application of an innovative
multiplex-fluorescent-labeled STRs assay for Prader-Willi syndrome
and angelman syndrome. PLoS One. 11:e01478242016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shaffer LG, Ledbetter DH and Lupski JR:
Molecular cytogenetics of contiguous gene syndromes: Mechanisms and
consequences of gene dosage imbalance. The metabolic and molecular
bases of inherited disease. Scriver CR, Beaudet AL, Sly WS and
Valle D: New York: McGraw-Hill; pp. 1291–1324. 2001
|
|
17
|
Jin DK: Systematic review of the clinical
and genetic aspects of Prader-Willi syndrome. Korean J Pediatr.
54:55–63. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cassidy SB and Driscoll DJ: Prader-Willi
syndrome. Eur J Hum Genet. 17:3–13. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hou JW and Wang TR: Prader-Willi syndrome:
Clinical and molecular cytogenetic investigations. J Formos Med
Assoc. 95:474–479. 1996.PubMed/NCBI
|
|
20
|
Kim HJ, Cho HJ, Jin DK, Kwon EK, Ki CS,
Kim JW and Kim SH: Genetic basis of Prader-Willi syndrome in Korea:
Less uniparental disomy than has been recognized? Clin Genet.
66:368–372. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lin HY, Lin SP, Chuang CK, Chen MR, Yen
JL, Lee YJ, Huang CY, Tsai LP, Niu DM, Chao MC and Kuo PL: Genotype
and phenotype in patients with Prader-Willi syndrome in Taiwan.
Acta Paediatr. 96:902–905. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cassidy SB, Forsythe M, Heeger S, Nicholls
RD, Schork N, Benn P and Schwartz S: Comparison of phenotype
between patients with Prader-Willi syndrome due to deletion 15q and
uniparental disomy 15. Am J Med Genet. 68:433–440. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fridman C and Koiffmann CP: Origin of
uniparental disomy 15 in patients with Prader-Willi or Angelman
syndrome. Am J Med Genet. 94:249–253. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kotzot D: Complex and segmental
uniparental disomy (UPD): Review and lessons from rare chromosomal
complements. J Med Genet. 38:497–507. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ginsburg C, Fokstuen S and Schinzel A: The
contribution of uniparental disomy to congenital development
defects in children born to mothers at advanced childbearing age.
Am J Med Genet. 95:454–460. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Morichon-Delvallez N, Mussat P, Dumez Y
and Vekemans M: Trisomy 15 in chorionic villi and Prader-Willi
syndrome at birth. Prenat Diagn. 13:307–308. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Christian SL, Smith AC, Macha M, Black SH,
Elder FF, Johnson JM, Resta RG, Surti U, Suslak L, Verp MS and
Ledbetter DH: Prenatal diagnosis of uniparental disomy 15 following
trisomy 15 mosaicism. Prenat Diagn. 16:323–332. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Roberts E, Stevenson K, Cole T, Redford DH
and Davison EV: Prospective prenatal diagnosis of Prader-Willi
syndrome due to maternal disomy for chromosome 15 following
trisomic zygote rescue. Prenat Diagn. 17:780–783. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hiroi H, Kozuma S, Hayashi N, Unno N,
Fujii T, Tsutsumi O, Okai T and Taketani Y: A fetus with
Prader-Willi syndrome showing normal diurnal rhythm and abnormal
ultradian rhythm on heart rate monitoring. Fetal Diagn Ther.
15:304–307. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fong BF and De Vries JI: Obstetric aspects
of the Prader-Willi syndrome. Ultrasound Obstet Gynecol.
21:389–392. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
L'Herminé AC, Aboura A, Brisset S, Cuisset
L, Castaigne V, Labrune P, Frydman R and Tachdjian G: Fetal
phenotype of Prader-Willi syndrome due to maternal disomy for
chromosome 15. Prenat Diagn. 23:938–943. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Haugen G, Rønnestad A and Kroken M:
Variations in fetal phenotype in Prader-Willi syndrome. Prenat
Diagn. 29:2942009. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Akiba Y, Ono M, Shirahashi M, Noda S,
Nishijima S, Amagata T, Kusano R, Fuke T, Hayashida S, Ikeda T, et
al: Polyhydramnios associated with Prader-Willi syndrome. J Obstet
Gynaecol. 35:752–753. 2015.PubMed/NCBI
|
|
34
|
Insoft RM, Hurvitz J, Estrella E and
Krishnamoorthy KS: Prader-Willi syndrome associated with fetal
goiter: A case report. Am J Perinatol. 16:29–31. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Le Bris-Quillevere MJ, Riviere D,
Pluchon-Riviere E, Chabaud JJ, Parent P, Volant A and Boog G:
Prenatal diagnosis of del(15)(q11q13). Prenat Diagn. 10:405–411.
1990. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bar C, Diene G, Molinas C, Bieth E, Casper
C and Tauber M: Early diagnosis and care is achieved but should be
improved in infants with Prader-Willi syndrome. Orphanet J Rare
Dis. 12:1182017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Liehr T, Brude E, Gillessen-Kaesbach G,
König R, Mrasek K, von Eggeling F and Starke H: Prader-Willi
syndrome with a karyotype 47,XY,+min(15)(pter->q11.1:) and
maternal UPD 15-case report plus review of similar cases. Eur J Med
Genet. 48:175–181. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Slater HR, Vaux C, Pertile M, Burgess T
and Petrovic V: Prenatal diagnosis of Prader-Willi syndrome using
PW71 methylation analysis-uniparental disomy and the significance
of residual trisomy 15. Prenat Diagn. 17:109–113. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Milunsky JM, Wyandt HE, Huang XL, Kang XZ,
Elias ER and Milunsky A: Trisomy 15 mosaicism and uniparental
disomy (UPD) in a liveborn infant. Am J Med Genet. 61:269–273.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cassidy SB, Lai LW, Erickson RP, Magnuson
L, Thomas E, Gendron R and Herrmann J: Trisomy 15 with loss of the
paternal 15 as a cause of Prader-Willi syndrome due to maternal
disomy. Am J Hum Genet. 51:701–708. 1992.PubMed/NCBI
|
|
41
|
Purvis-Smith SG, Saville T, Manass S, Yip
MY, Lam-Po-Tang PR, Duffy B, Johnston H, Leigh D and McDonald B:
Uniparental disomy 15 resulting from “correction” of an initial
trisomy 15. Am J Hum Genet. 50:1348–1350. 1992.PubMed/NCBI
|
|
42
|
Driscoll DJ, Miller JL, Schwartz S, et al:
Prader-Willi Syndrome. 1998 Oct 6 (Updated 2017 Dec 14). Adam MP,
Ardinger HH, Pagon RA, et al: GeneReviews® [Internet]
Seattle (WA): University of Washington, Seattle; 1993-2018,
https://www.ncbi.nlm.nih.gov/books/NBK1330/
|
|
43
|
Glenn CC, Driscoll DJ, Yang TP and
Nicholls RD: Genomic imprinting: Potential function and mechanisms
revealed by the Prader-Willi and Angelman syndromes. Mol Hum
Reprod. 3:321–332. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Miller DT, Adam MP, Aradhya S, Biesecker
LG, Brothman AR, Carter NP, Church DM, Crolla JA, Eichler EE, et
al: Consensus statement: Chromosomal microarray is a first-tier
clinical diagnostic test for individuals with developmental
disabilities or congenital anomalies. Am J Hum Genet. 86:749–764.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kearney HM, Thorland EC, Brown KK,
Quintero-Rivera F and South ST; Working Group of the American
College of Medical Genetics Laboratory Quality Assurance Committee,
: American College of Medical Genetics standards and guidelines for
interpretation and reporting of postnatal constitutional copy
number variants. Genet Med. 13:680–685. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hanemaaijer NM, Sikkema-Raddatz B, van der
Vries G, Dijkhuizen T, Hordijk R, van Essen AJ, Veenstra-Knol HE,
Kerstjens-Frederikse WS, Herkert JC, Gerkes EH, et al: Practical
guidelines for interpreting copy number gains detected by
high-resolution array in routine diagnostics. Eur J Hum Genet.
20:161–165. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
South ST, Lee C, Lamb AN, Higgins AW and
Kearney HM; Working Group for the American College of Medical
Genetics and Genomics Laboratory Quality Assurance Committee, :
ACMG Standards and Guidelines for constitutional cytogenomic
microarray analysis, including postnatal and prenatal applications:
Revision 2013. Genet Med. 15:901–909. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Tucker T, Schlade-Bartusiak K, Eydoux P,
Nelson TN and Brown L: Uniparental disomy: Can SNP array data be
used for diagnosis? Genet Med. 14:753–756. 2012. View Article : Google Scholar : PubMed/NCBI
|