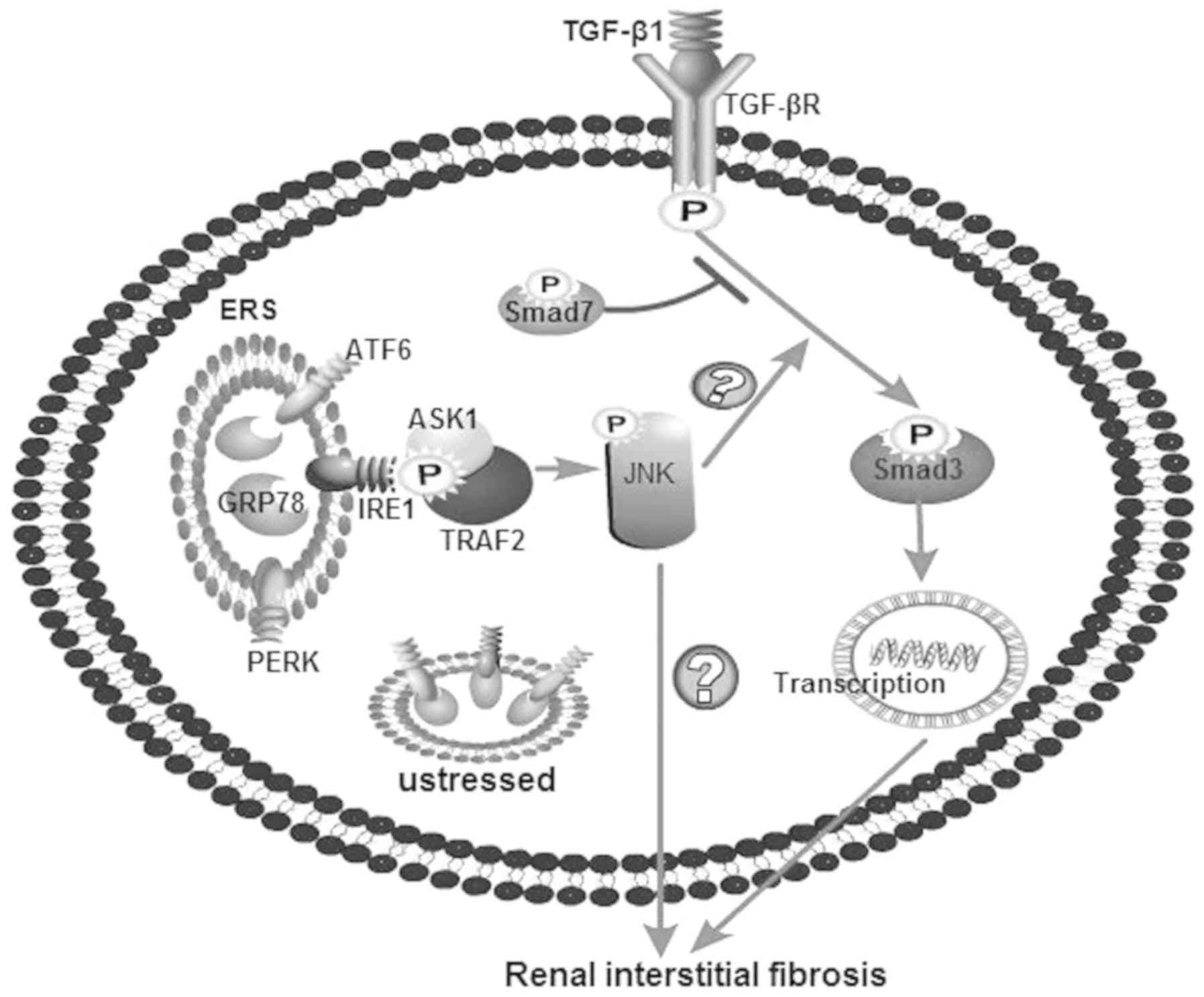
Introduction
The prevalence of chronic kidney disease (CKD) is
currently increasing worldwide, making this disease a rising global
health concern (1). CKD can result
in end-stage renal disease, which is associated with a number of
complications, including mineral and bone disorders, anemia,
cognitive decline and cardiovascular disease (2). However, no cure exists for CKD at
present, and thus the available treatments are primarily aimed at
halting or delaying disease progression. Renal interstitial
fibrosis serves a key role in the process of progressive renal
injury and is a common characteristic of CKD (3). Therefore, inhibition of renal
interstitial fibrosis is of great significance in CKD therapy. A
great number of studies have reported that excessive endoplasmic
reticulum stress (ERS) participated in the development of renal
interstitial fibrosis (4–6); however, the exact mechanism is not
fully clear.
The endoplasmic reticulum (ER) is an organelle that
is responsible for the transmembrane, secretory and ER luminal
protein synthesis (7). A
disruption in ER proteostasis may occur under tissue fibrosis,
leading to an imbalance between the protein folding demand and
capacity (8). Glucose-regulated
protein 78 (GRP78) is a central regulator of ER homeostasis, and is
commonly used as a biomarker for ERS. Under increased ERS, the
unfolded protein response triggers the dissociation of GRP78 from
three known transmembrane sensors in the ER, including the protein
kinase R-like ER kinase, activating transcription factor 6 and
inositol-requiring enzyme I (IRE1). Among them, IRE-1 interacts
with the TNF receptor-associated factor 2 (TRAF2) and attracts
apoptosis signal-regulating kinase-1 (ASK1) to form
IRE1-TRAF2-ASK1, which subsequently activates c-Jun N-terminal
kinase (JNK) (9,10).
The JNK family of kinases, also known as
stress-activated mitogen-activated protein kinases, includes three
distinct members, namely: JNK1, JNK2 and JNK3. The JNK1 and JNK2
isoforms are ubiquitously expressed in the majority of tissues,
including the kidney, while JNK3 expression is restricted to
nervous system tissues (11).
Previous studies have suggested that disorder of the JNK signaling
pathway serves a pivotal role in several diseases, such as lung
fibrosis, human fibrosarcoma and renal fibrosis (12–14).
In addition, it has been reported that JNK was able to mediate the
fibronectin and connective tissue growth factor (CTGF) synthesis,
suggesting that JNK was involved in tissue fibrosis (12,13).
A study conducted by Ma et al (14) demonstrated that CC-401, which is a
specific JNK inhibitor, suppressed JNK signaling and significantly
reduced renal fibrosis in rats with obstructed kidney, indicating
that JNK signaling served a pathogenic role in renal fibrosis. It
has also been reported that SP600125, another JNK inhibitor, was
able to effectively prevent the transforming growth factor
(TGF)-β1-induced phosphorylation of Smad3, and the changes in
E-cadherin, α-smooth muscle actin (α-SMA) and collagen I
expression, which suggested that JNK possibly mediated peritoneal
fibrosis through the TGF-β/Smad3 pathway (15). A recent study revealed that
4-phenylbutyrate (4-PBA), a specific inhibitor of ERS, evidently
attenuated JNK phosphorylation and TGF-β-induced profibrogenic
CTGF, collagen I protein expression in renal tubular NRK-52E cells
(16).
Therefore, it can be hypothesized that ESR may
mediate renal interstitial fibrosis through the JNK/TGF-β/Smad3
pathway (Fig. 1). In the present
study, the expression level of GRP78 was initially examined in
human renal tissue. Next, different pathways were inhibited using
chemical agents in human tubular HK-2 cells in order to investigate
the possible mechanism of ERS-mediated renal interstitial
fibrosis.
Materials and methods
Reagents
Tunicamycin (TM; cat. no. T7765) and the 4-PBA (an
ERS inhibitor; cat. no. SML0309) were obtained from Sigma-Aldrich
(Merck KGaA, Darmstadt, Germany). SP600125 (a JNK inhibitor, cat.
no. ab120065) and antibody against phosphorylated (p)-Smad3 (cat.
no. ab52903) were obtained from Abcam (Cambridge, UK). The antibody
against p-JNK (cat. no. 4668s) was purchased from Cell Signaling
Technology, Inc. (Danvers, MA, USA), while antibodies targeting
GRP78 (cat. no. 11587-1-AP), TGF-β1 (cat. no. 18978-1-AP), CTGF
(cat. no. 23936-1-AP), α-SMA (cat. no. 14395-1-AP) and β-actin
(cat. no. 20536-1-AP) were acquired from ProteinTech Group, Inc.
(Wuhan, China).
Patients and tissues
A total of 6 patients (3 male and 3 female; age,
29–60 years old) with asymptomatic hematuria, and/or proteinuria
(patients without edema, hypertension and/or renal injury but with
more than 3 erythrocytes per high-power field by the urine sediment
and/or mild proteinuria of less than 1 g/day of protein) or chronic
kidney disease (17), were
identified in a retrospective review of renal biopsies received at
the Suzhou Municipal Hospital (Suzhou, China) from June 2014 to May
2015. Patients with other systemic diseases (including diabetes,
heart disease, pulmonary fibrosis, liver fibrosis and
neurodegenerative diseases) or disease complications (including
infection, anemia and hypoproteinemia) were excluded via
examination of clinical history, physical examination and
laboratory test results. Paraffin-embedded renal biopsy specimens
from these patients were conserved at room temperature. With
consent from the patients, paraffin-embedded specimens were sliced
into 4 µm sections for immunohistochemical evaluation.
Sample classification and
immunohistochemical staining
The six paraffin-embedded specimens, including three
specimens of minor glomerular lesion (with asymptomatic hematuria
and/or proteinuria, regarded as control group) and three of severe
renal interstitial fibrosis (CKD), were evaluated via
immunohistochemical staining. According to the World Health
Organization (18), glomerular
minor lesions are regarded as kidney tissue without evident
interstitial fibrosis, while severe renal interstitial fibrosis is
defined as kidney tissue with >50% of interstitial fibrosis. The
estimated glomerular filtration rates[calculated according to the
CKD-EPI formula (19)] of the
three patients with glomerular minor lesion were 123.5, 90.1 and
96.7 ml/min, while the respective rates of patients with severe
renal interstitial fibrosis were 54.6, 36.7 and 73.6 ml/min.
Paraffin-embedded 4 µm sections were analyzed by
immunohistochemical staining. Briefly, the sections were
rehydrated, and antigen retrieval was performed with heated
citrate. Sections were incubated with 3% H2O2
at room temperature for 15 min to remove endogenous peroxidase
activity. Immunohistochemical staining was performed using primary
antibodies against GRP78 (1:50) and p-JNK (1:20) overnight at 4°C.
Following incubation with a reaction enhancer (OriGene
Technologies, Inc., Rockville, MD, USA) at 37°C for 20 min,
sections incubated with horseradish peroxidase labeled goat
anti-rabbit immunoglobulin G (IgG) polymer (cat. no. PV9001,
OriGene Technologies, Inc.) at 37°C for 30 min. The signals were
developed with DAB Peroxidase Substrate kit (Vector Laboratories,
Ltd., Burlingame, CA, USA) at room temperature for 3 min. Brown
staining represents the GRP78 and p-JNK expression. Intensity of
immunohistochemistry staining was assessed as follows: 0, negative;
1, faint yellow; 2, light brown; and 3, dark brown. The
immunohistochemistry staining range was classified as: 1 (0–25%
staining of the section); 2 (26–50% staining of the section); 3
(51–75% staining of the section); and 4 (76–100% staining of the
section). The final immunohistochemistry staining score was the sum
of the intensity scores and staining range scores, and was graded
from 1 to 7. All immunohistochemical analyses were repeated at
least three times, and representative images are presented.
Cell culture and treatment
All cell experiments were performed using HK-2
cells, a human proximal tubular cell line (supplied by the Second
Xiangya Hospital of Central South University, Changsha, China).
HK-2 cells were maintained in Dulbecco's modified Eagle's
medium/F12 (cat. no. SH30023.01; HyClone; GE Healthcare Life
Sciences, Logan, Utah, USA) supplemented with 10% fetal bovine
serum (cat. no. P303-3302, PAN-Biotech GmbH, Aidenbach, Germany),
100 µg/ml streptomycin and 100 U/ml penicillin at 37°C in 5%
CO2. The media were changed every 3 days until
confluence was reached. Cells were growth-arrested in serum-free
medium for 24 h prior to use in experiments. To determine whether
ESR inhibition alleviated the activation of JNK and TGF-β/Smad3
pathway, HK-2 cells were incubated with TM (0.2 µM) for 24 h and
then treated with 4-PBA (1.0 mM) for 2 h. Subsequently, to
determine whether JNK signaling inhibition alleviated the
activation of TGF-β/Smad3 pathway, HK-2 cells were incubated with
TM (0.2 µM) for 24 h and then treated with SP600125 (10 µM) for 1
h. The selection of TM, 4-PBA and SP600125 concentrations was based
on previous studies and preliminary experiments (20–22).
Western blotting
After treatment, cells were harvested, rinsed with
PBS and lysed on ice in RIPA buffer (Applygen Technologies, Inc.,
Beijing, China) supplied with protease inhibitors (Merck KGaA). The
protein concentrations were measured using a bicinchoninic acid
assay kit. Equal amount of protein (50 µg total protein) from whole
cell lysates was separated by 10% SDS-PAGE and then transferred to
nylon membranes. The membranes were blocked in a TBS solution with
5% skimmed milk containing 0.1% Tween-20 at 37°C for 1 h. Next, the
membranes were incubated with primary antibody against GRP78
(1:1,000), p-JNK (1:1,000), TGF-β1 (1:500), p-Smad3 (1:2,000), CTGF
(1:500), α-SMA (1:1,000) and β-actin (1:4,000) overnight at 4°C,
followed by incubation with horseradish peroxidase-conjugated
secondary antibody goat anti-rabbit IgG (cat. no. SA00001-2) or
goat anti-mouse IgG (cat. no. SA00001-1; both at 1:3,000;
ProteinTech Group, Inc.) at 37°C for 45 min. Chemiluminescent
detection was performed using an enhanced chemiluminescence
substrate kit (Pierce; Thermo Fisher Scientific, Inc., Waltham, MA,
USA). Quantification of the band intensities was conducted using
Quantity One version 4.6.2 (Bio-Rad Laboratories, Inc., Hercules,
CA, USA), and the intensity was normalized to that of β-actin for
standardization.
Statistical analysis
SPSS software (version 17.0; SPSS, Inc., Chicago,
IL, USA) was used for data analysis. The experimental results are
expressed as the mean ± standard deviation. Analysis between groups
was performed using one-way analysis of variance followed by Tukey
post hoc tests for multiple comparisons for normally distributed
quantitative data. Categorical data was compared using Mann-Whitney
U tests. P<0.05 was considered to indicate a statistically
significant difference.
Results
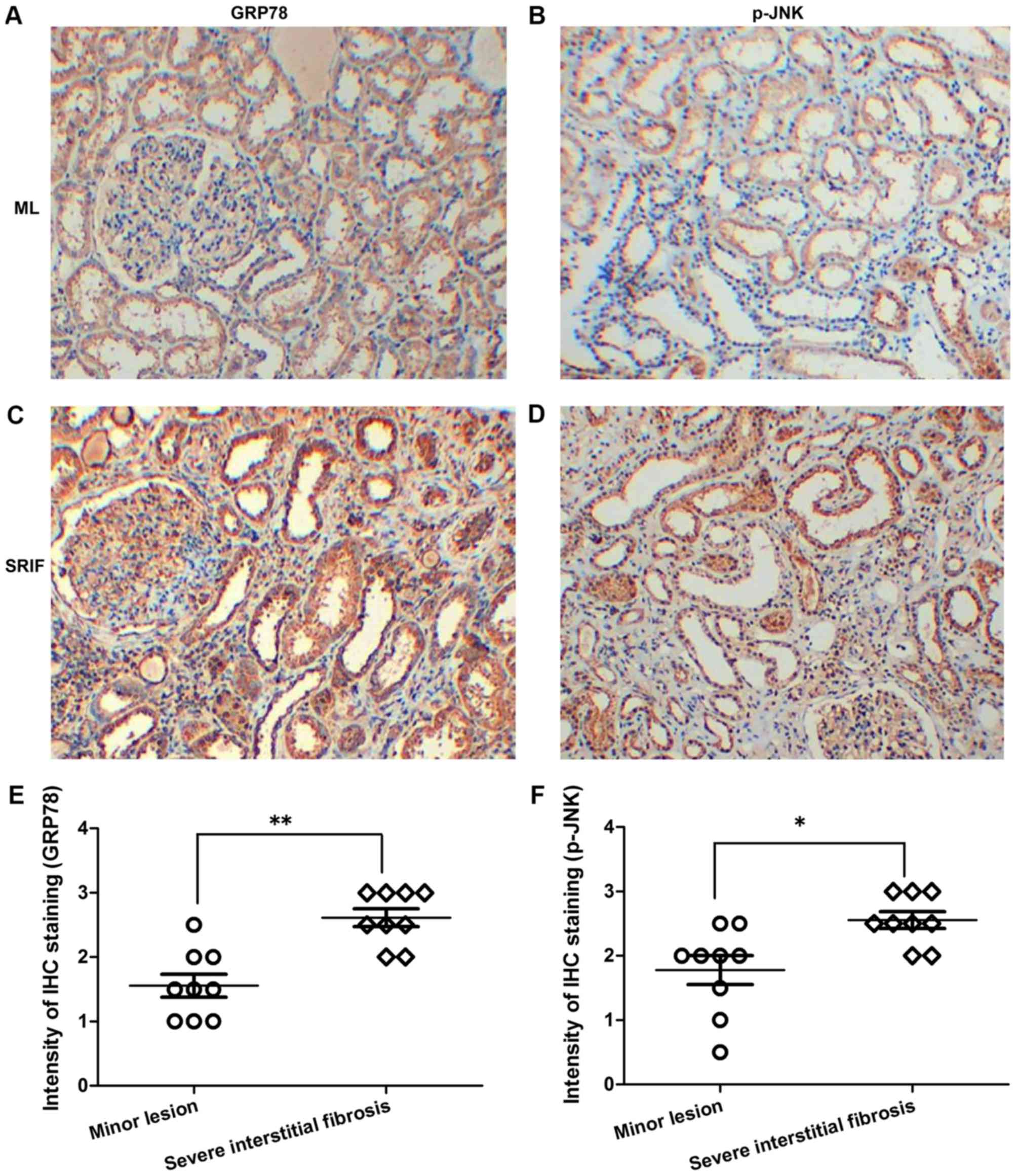
Expression levels of GRP78 and p-JNK
in renal tissues of CKD patients are increased
To investigate the association between ERS and renal
interstitial fibrosis, the expression levels of GRP78 and p-JNK
were analyzed in human renal tissues of different lesion degrees by
immunohistochemical staining. The results revealed evident
expression of GRP78 and p-JNK in the renal tissues of CKD patients.
In addition, GRP78 and p-JNK expression levels were significantly
higher in renal tissues with severe interstitial fibrosis compared
with those withglomerularminor lesions (P<0.01 and P<0.05 for
each protein, respectively; Fig.
2), which verified that ERS potentially accelerates the
progression of renal interstitial fibrosis.
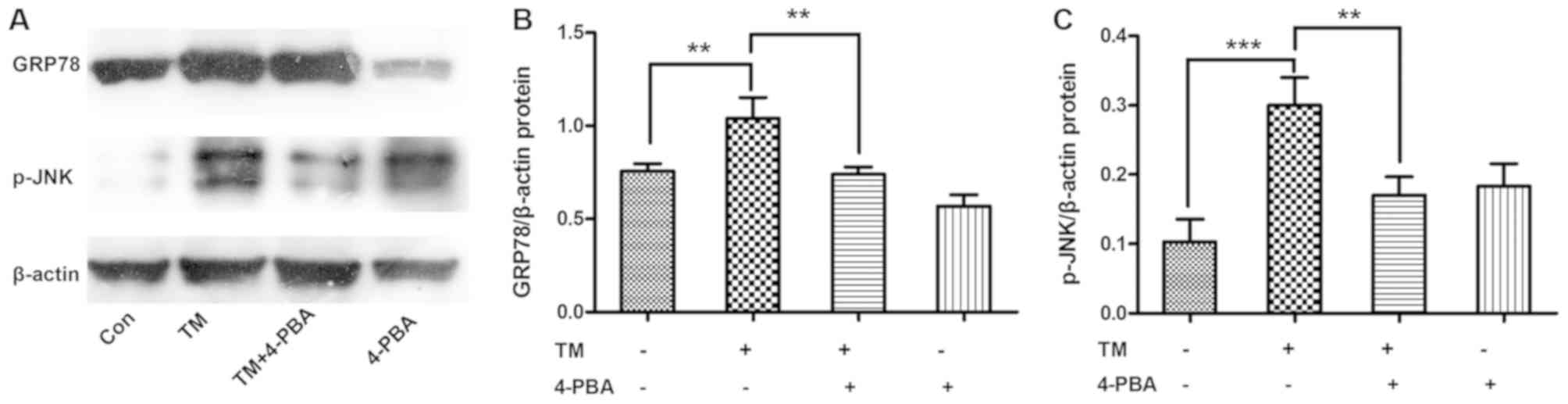
ESR inhibition alleviates the
expression levels of GRP78 and p-JNK in HK-2 cells
To determine whether ESR inhibition alleviated the
activation of JNK, HK-2 cells were incubated with the ESR chemical
inducer TM (0.2 µM) for 24 h and then treated with the ESR chemical
inhibitor 4-PBA (1.0 mM) for 2 h. The results demonstrated that
incubation with TM significantly increased the expression levels of
GRP78 and p-JNK proteins compared with the untreated cells
(P<0.01 and P<0.001, respectively). However, co-treatment
with 4-PBA markedly ameliorated the TM-induced, ERS-associated
expression of GRP78 and p-JNK proteins in HK-2 cells (both
P<0.01; Fig. 3).
ESR inhibition alleviates the
expression of TGF-β/Smad3 pathway-associated proteins in HK-2
cells
To determine whether ESR inhibition alleviated the
activation of the TGF-β/Smad3 pathway, HK-2 cells were incubated
with the ESR chemical inducer TM (0.2 µM) for 24 h and then treated
with the ESR chemical inhibitor 4-PBA (1.0 mM) for 2 h. The results
revealed that TM incubation increased the expression levels of
TGF-β/Smad3 signals, including TGF-β1, p-Smad3, CTGF and α-SMA
protein levels (P<0.01 and P<0.001). By contrast, 4-PBA
co-treatment evidently ameliorated the TM-induced expression levels
of TGF-β/Smad3 pathway-associated proteins in HK-2 cells (P<0.01
and P<0.001; Fig. 4).
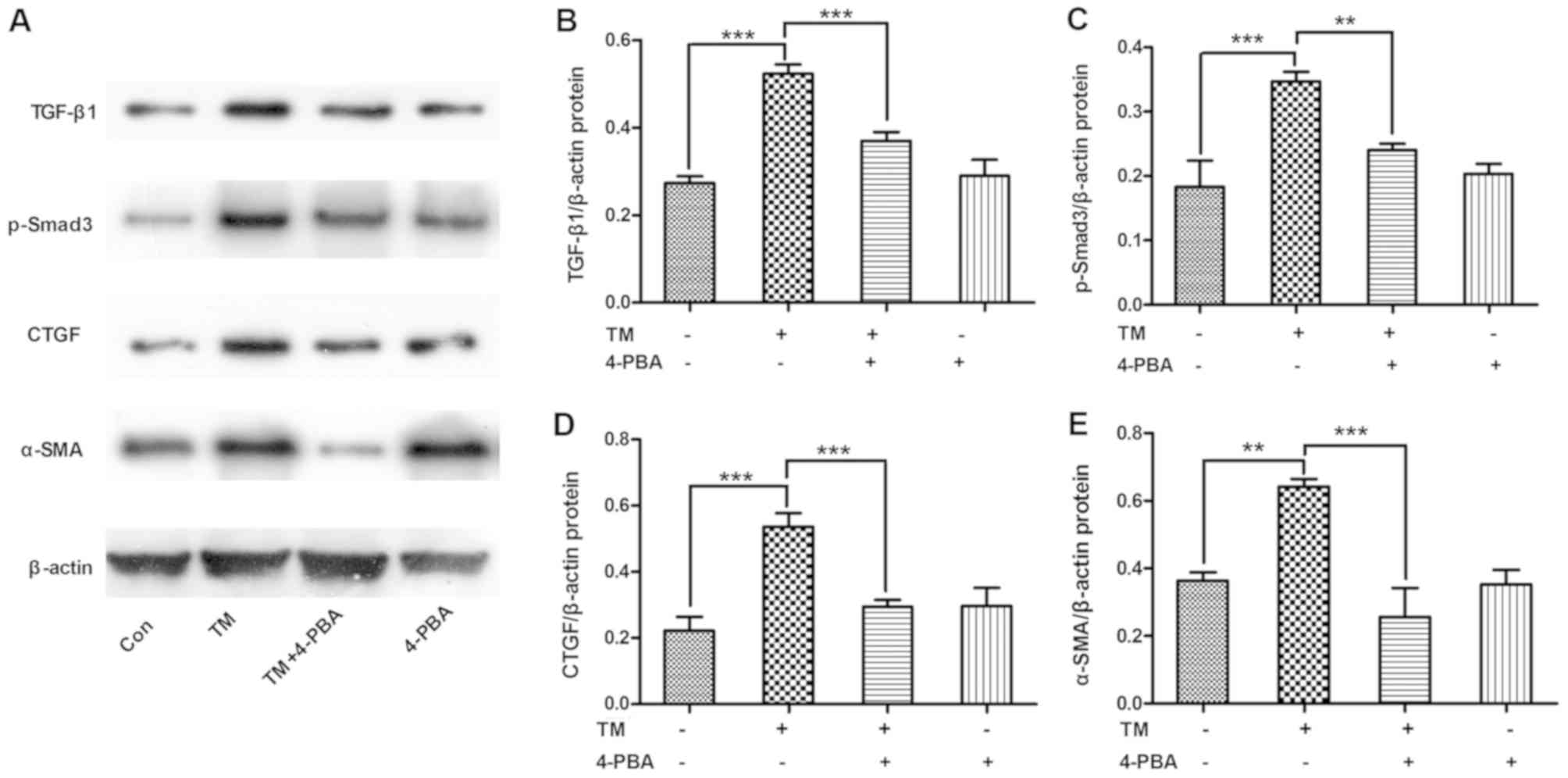 | Figure 4.ESR inhibition alleviated the
expression of TGF-β/Smad3 pathway proteins in HK-2 cells. HK-2
cells were incubated with the ESR chemical inducer TM (0.2 µM) for
24 h and then treated with the ESR chemical inhibitor 4-PBA (1.0
mM) for 2 h. (A) Western blot analysis of cell lysates for TGF-β1,
p-Smad3, CTGF and α-SMA proteins, with blots reprobed for β-actin.
(B) TGF-β1, (C) p-Smad3, (D) CTGF and (E) α-SMA protein expression
levels relative to β-actin, with pooled data are shown. Data are
presented as the mean ± standard deviation, and were assessed by
analysis of variance and Tukey post-hoc test (n=3). **P<0.01 and
***P<0.001. ERS, endoplasmic reticulum stress; TM, tunicamycin;
4-PBA, 4-phenylbutyrate; TGF-β1, transforming growth factor-β1;
p-Smad3, phosphorylated Smad3; CTGF, connective tissue growth
factor; α-SMA, α-smooth muscle actin. |
Inhibition of JNK signaling alleviates
the expression levels of TGF-β/Smad3 pathway-associated proteins in
HK-2 cells
To determine whether JNK signaling inhibition
alleviated the activation of TGF-β/Smad3 pathway, HK-2 cells were
incubated with the ESR chemical inducer TM (0.2 µM) for 24 h and
then treated with the JNK pathway chemical inhibitor SP600125 (10
µM) for 1 h. The data indicated that SP600125 treatment
significantly decreased the TM-induced p-JNK expression (P<0.01)
and the expression levels of TGF-β/Smad3-associated proteins,
including TGF-β1 (P<0.01), p-Smad3 (P<0.05), CTGF (P<0.01)
and α-SMA (P<0.01), in HK-2 cells (Fig. 5).
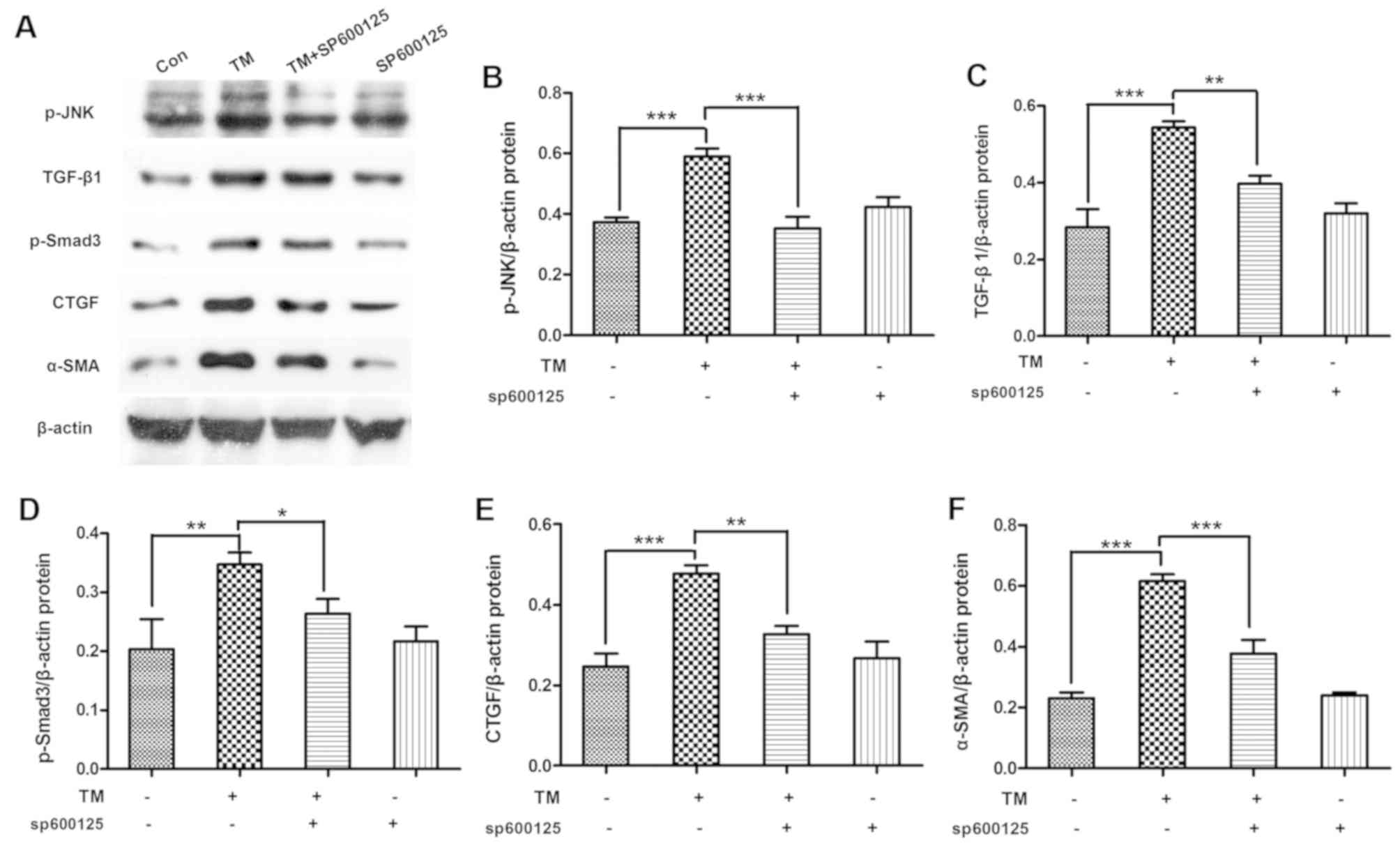 | Figure 5.Inhibition of JNK signal alleviated
TGF-β/Smad3 pathway expression in HK-2 cells. HK-2 cells were
incubated with the ESR chemical inducer TM (0.2 µM) for 24 h and
then treated with the JNK pathway chemical inhibitor SP600125 (10
µM) for 1 h. (A) Western blot analysis of cell lysates for p-JNK,
TGF-β1, p-Smad3, CTGF and α-SMA, with blots reprobed for β-actin.
(B) p-JNK, (C) TGF-β1, (D) p-Smad3, (E) CTGF and (F) α-SMA protein
expression graphs relative to β-actin, with pooled data are shown.
Data are presented as the mean ± standard deviation, and were
assessed by analysis of variance and Tukey post-hoc test (n=3).
*P<0.05, **P<0.01 and ***P<0.001. JNK, c-Jun N-terminal
kinase; TGF-β, transforming growth factor-β; ERS, endoplasmic
reticulum stress; TM, tunicamycin; p-, phosphorylated; CTGF,
connective tissue growth factor; α-SMA, α-smooth muscle actin. |
Discussion
Renal tubulointerstitial fibrosis is the common and
final pathologic change of the kidney in end-stage renal disease
(23). Inhibition of renal
interstitial fibrosis is of great significance in CKD therapy.
Previous cell and animal experiments have suggested that ERS was
implicated in the development of renal interstitial fibrosis
(4,16). In the present study,
immunohistochemical staining revealed that GRP78 was evidently
expressed in the renal tissues of CKD patients. In addition, higher
GRP78 expression was detected in renal tissue with severe
interstitial fibrosis as compared with that of minor glomerular
lesion tissues, indicating the potentially key role of ERS in the
renal interstitial fibrosis process, consistent with the
observations of previous studies (4,16).
TGF-β is regarded as a central mediator of renal
interstitial fibrosis (3,24,25),
and its upregulation occurs in nearly all types of CKD. TGF-β acts
on downstream signaling through Smad phosphorylation. Smad proteins
are highly conserved transcription factors that are central to
signal transduction pathways and mediate numerous effects
associated with the TGF-β superfamily signaling pathway (26). Among them, Smad3 is a key mediator
in renal fibrosis, whereas Smad2 and Smad7 exhibit renal-protective
properties (27,28). In addition, CTGF is considered to
be determinant of progressive renal fibrosis and a downstream
mediator of TGF-β1 signaling in the fibrosis process (29,30).
During the fibrosis process, epithelial-mesenchymal transition,
characterized by the loss of cell adhesion markers and the de
novo expression of mesenchymal markers such as α-SMA, is
considered to be an indicator of interstitial fibrosis (31). In the present study, TM was used to
activate ERS in HK-2 cells, which resulted in increased expression
levels of TGF-β1, p-Smad3, CTGF and α-SMA. In contrast, treatment
with 4-PBA to inhibit ERS led to decreased expression levels of
these proteins, which further indicated that ERS may have served a
key role in the development of renal interstitial fibrosis.
Currently, it remains unclear how ERS induces renal
interstitial fibrosis. Previous research has shown that ERS
mediated tubular cell apoptosis that then resulted in renal
interstitial fibrosis (4,16). Additionally, a number of other
studies revealed that JNK participates in the development of tissue
fibrosis, including in lung, liver and ovarian tissues, among
others (13,32,33).
Based on previous animal experiments, Ma et al (14) reported that JNK signaling served a
pathogenic role in renal interstitial fibrosis. Consistent with
previous studies, the data of the current study revealed that p-JNK
was evidently expressed in human renal tissues obtained from CKD
patients, and its expression level was correlated with the
interstitial fibrosis degree, further suggesting the potential role
of JNK signaling in renal interstitial fibrosis.
Understanding the mediation of renal interstitial
fibrosis by JNK, a downstream signaling molecule of the ERS, is
also of great significance. A study by Liu et al (15) reported that blocking the activation
of JNK with SP600125 resulted in the ineffective inhibition of
TGF-β1-induced phosphorylation of Smad3, suppressed the
TGF-β1-induced upregulation of α-SMA and collagen I, and prevented
the TGF-β1-induced downregulation of E-cadherin expression in rat
peritoneal mesothelial cells. These observations suggested that JNK
possibly mediated peritoneal fibrosis through the TGF-β/Smad3
pathway (15). According to the
present study, the expression levels of p-JNK, TGF-β1, p-Smad3,
CTGF and α-SMA in HK-2 cells were increased when ERS was activated
by TM. However, their expression levels were decreased when the
cells were subsequently treated with 4-PBA, which suggested that
ERS potentially activated TGF-β/Smad3 signaling through JNK pathway
regulation, which may have resulted in renal interstitial fibrosis.
In addition, JNK blocking with SP600125 resulted in downregulation
of TGF-β1, p-Smad3, CTGF and α-SMA levels in HK-2 cells, which
further verified the aforementioned hypothesis.
A potential limitation of the present study is the
absence of normalization of phosphorylated proteins to the total
proteins. Nevertheless, the upregulation of p-JNK is indicative of
the potential role of the JNK pathway in ERS-induced renal
interstitial fibrosis.
In conclusion, the present study demonstrated that
ERS possibly mediated renal interstitial fibrosis through the
JNK/TGF-β/Smad3 pathway, which may provide a useful insight for CKD
therapy. However, due to limitations in the methods of the current
study, the exact mechanism of ERS-mediated renal interstitial
fibrosis remains to be further clarified by in vivo studies
through genetic methods, such as transfection with small
interfering RNA or gene knockout.
Acknowledgements
Not applicable.
Funding
This study was supported by a grant from the Program
of Ke Jiao Xing Wei of Suzhou City (grant no. kjxw2015021) and the
Program of Key Discipline of Suzhou City (grant no.
Szxk201807).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
HC and YL were responsible for the experimental
design. YL and PS performed the majority of the experiments and
drafted the manuscript. YZ and LT participated in sample
classification and immunohistochemical staining. YL and LT assisted
with analyzing the experimental results. HC, YZ and PS revised the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The described experiments were approved by the
Scientific and Ethics Committee of the Affiliated Suzhou Hospital
of Nanjing Medical University (Suzhou, China; permit no. KL901006)
prior to conducting the study. All patients provided written
informed consent prior to participation.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Jha V, Garcia-Garcia G, Iseki K, Li Z,
Naicker S, Plattner B, Saran R, Wang AY and Yang CW: Chronic kidney
disease: Global dimension and perspectives. Lancet. 382:260–272.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Collister D, Ferguson T, Komenda P and
Tangri N: The patterns, risk factors, and prediction of progression
in chronic kidney disease: A narrative review. Semin Nephrol.
36:273–282. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Boor P, Ostendorf T and Floege J: Renal
fibrosis: Novel insights into mechanisms and therapeutic targets.
Nat Rev Nephrol. 6:643–656. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chiang CK, Hsu SP, Wu CT, Huang JW, Cheng
HT, Chang YW, Hung KY, Wu KD and Liu SH: Endoplasmic reticulum
stress implicated in the development of renal fibrosis. Mol Med.
17:1295–1305. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Piret SE, Olinger E, Reed AAC, Nesbit MA,
Hough TA, Bentley L, Devuyst O, Cox RD and Thakker RV: A mouse
model for inherited renal fibrosis associated with endoplasmic
reticulum stress. Dis Model Mech. 10:773–786. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ke B, Zhu N, Luo F, Xu Y and Fang X:
Targeted inhibition of endoplasmic reticulum stress: New hope for
renal fibrosis (Review). Mol Med Rep. 16:1014–1020. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dickhout JG and Krepinsky JC: Endoplasmic
reticulum stress and renal disease. Antioxid Redox Signal.
11:2341–2352. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Inagi R, Ishimoto Y and Nangaku M:
Proteostasis in endoplasmic reticulum-new mechanisms in kidney
disease. Nat Rev Nephrol. 10:369–378. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lee H, Noh JY, Oh Y, Kim Y, Chang JW,
Chung CW, Lee ST, Kim M, Ryu H and Jung YK: IRE1 plays an essential
role in ER stress-mediated aggregation of mutant hunting in via the
inhibition of autophagy flux. Hum Mol Genet. 21:101–114. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zheng QY, Li PP, Jin FS, Yao C, Zhang GH,
Zang T and Ai X: Ursolic acid induces ER stress response to
activate ASK1-JNK signaling and induce apoptosis in human bladder
cancer T24 cells. Cell Signal. 25:206–213. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Davis RJ: Signal transduction by the JNK
group of MAP kinases. Cell. 103:239–252. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hocevar BA, Brown TL and Howe PH: TGF-beta
induces fibronectin synthesis through a c-Jun N-terminal
kinase-dependent, Smad4-independent pathway. EMBO J. 18:1345–1356.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Utsugi M, Dobashi K, Ishizuka T, Masubuchi
K, Shimizu Y, Nakazawa T and Mori M: C-Jun-NH2-terminal kinase
mediates expression of connective tissue growth factor induced by
transforming growth factor-beta1 in human lung fibroblasts. Am J
Respir Cell Mol Biol. 28:754–761. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ma FY, Flanc RS, Tesch GH, Han Y, Atkins
RC, Bennett BL, Friedman GC, Fan JH and Nikolic-Paterson DJ: A
pathogenic role for c-Jun amino-terminal kinase signaling in renal
fibrosis and tubular cell apoptosis. J Am Soc Nephrol. 18:472–484.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu Q, Mao H, Nie J, Chen W, Yang Q, Dong
X and Yu X: Transforming growth factor {beta}1 induces
epithelial-mesenchymal transition by activating the JNK-Smad3
pathway in rat peritoneal mesothelial cells. Perit Dial Int. 3
(Suppl 28):S88–S95. 2008.
|
|
16
|
Liu SH, Yang CC, Chan DC, Wu CT, Chen LP,
Huang JW, Hung KY and Chiang CK: Chemical chaperon 4-phenylbutyrate
protects against the endoplasmic reticulum stress-mediated renal
fibrosis in vivo and in vitro. Oncotarget. 7:22116–22127.
2016.PubMed/NCBI
|
|
17
|
National Kidney Foundation: K/DOQI
clinical practice guidelinesfor chronic kidney disease: Evaluation,
classification, and stratification. Am J Kidney Dis. 39 (Suppl
1):S1–S266. 2002.PubMed/NCBI
|
|
18
|
Churg J, Bernstein J and Glassock RJ:
Renal Disease: Classification and atlas of glomerular diseases.
2nd. Igaku-Shoin Ltd.; Tokyo: 1995
|
|
19
|
Levey AS, Stevens LA, Schmid CH, Zhang YL,
Castro AF III, Feldman HI, Kusek JW, Eggers P, Van Lente F, Greene
T, et al: A new equation to estimate glomerular filtration rate.
Ann Intern Med. 150:604–612. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chang JW, Kim H, Baek CH, Lee RB, Yang WS
and Lee SK: Up-regulation of SIRT1 reduces endoplasmic reticulum
stress and renal fibrosis. Nephron. 133:116–128. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhu S, Wang Y, Jin J, Guan C, Li M, Xi C,
Ouyang Z, Chen M, Qiu Y, Huang M and Huang Z: Endoplasmic reticulum
stress mediates aristolochic acid I-induced apoptosis in human
renal proximal tubular epithelial cells. Toxicol In Vitro.
26:663–671. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hirai Y, Iyoda M, Shibata T, Kuno Y,
Kawaguchi M, Hizawa N, Matsumoto K, Wada Y, Kokubu F and Akizawa T:
IL-17A stimulates granulocyte colony-stimulating factor production
via ERK1/2 but not p38 or JNK in human renal proximal tubular
epithelial cells. Am J Physiol Renal Physiol. 302:F244–F250. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hewitson TD: Renal tubulointerstitial
fibrosis: Common but never simple. Am J Physiol Renal Physiol.
296:F1239–F1244. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang Q, Usinger W, Nichols B, Gray J, Xu
L, Seeley TW, Brenner M, Guo G, Zhang W, Oliver N, et al:
Cooperative interaction of CTGF and TGF-β in animal models of
fibrotic disease. Fibrogenesis Tissue Repair. 4:42011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yeh YC, Wei WC, Wang YK, Lin SC, Sung JM
and Tang MJ: Transforming growth factor-{beta}1 induces
Smad3-dependent {beta}1 integrin gene expression in
epithelial-to-mesenchymal transition during chronic
tubulointerstitial fibrosis. Am J Pathol. 177:1743–1754. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Meng XM, Huang XR, Xiao J, Chung AC, Qin
W, Chen HY and Lan HY: Disruption of Smad4 impairs TGF-β/Smad3 and
Smad7 transcriptional regulation during renal inflammation and
fibrosis in vivo and in vitro. Kidney Int. 81:266–279. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lan HY: Diverse roles of TGF-β/Smads in
renal fibrosis and inflammation. Int J Biol Sci. 7:1056–1067. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Meng XM, Huang XR, Chung AC, Qin W, Shao
X, Igarashi P, Ju W, Bottinger EP and Lan HY: Smad2 protects
against TGF-beta/Smad3-mediated renal fibrosis. J Am Soc Nephrol.
21:1477–1487. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Grotendorst GR: Connective tissue growth
factor: A mediator of TGF-beta action on fibroblasts. Cytokine
Growth Factor Rev. 8:171–179. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yokoi H, Sugawara A, Mukoyama M, Mori K,
Makino H, Suganami T, Nagae T, Yahata K, Fujinaga Y, Tanaka I and
Nakao K: Role of connective tissue growth factor in profibrotic
action of transforming growth factor-beta: A potential target for
preventing renal fibrosis. Am J Kidney Dis 38 (4 Suppl 1).
S134–S138. 2001. View Article : Google Scholar
|
|
31
|
Grande MT and Lopez-Novoa JM: Fibroblast
activation and myofibroblast generation in obstructive nephropathy.
Nat Rev Nephrol. 5:319–328. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhao X, Fu J, Xu A, Yu L, Zhu J, Dai R, Su
B, Luo T, Li N, Qin W, et al: Gankyrin drives malignant
transformation of chronic liver damage-mediated fibrosis via the
Rac1/JNK pathway. Cell Death Dis. 6:e17512015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bulut G, Kurdoglu Z, Dönmez YB, Kurdoglu M
and Erten R: Effects of jnk inhibitor on inflammation and fibrosis
in the ovary tissue of a rat model of polycystic ovary syndrome.
Int J Clin Exp Pathol. 8:8774–8785. 2015.PubMed/NCBI
|