Introduction
Head and neck squamous cell carcinoma (HNSCC) refers
to a group of malignancies that originates in the oral cavity,
oropharynx, larynx, or hypopharynx (1,2). It
is the seventh most common cancer worldwide with an annual
incidence of more than half a million (3). The average 5-year overall survival
(OS) of patients with HNSCC is between 42–64% (4). Given the intrinsic heterogeneity of
this disease, the identification of prognostic gene signatures is
of a particular interest to improve HNSCC diagnosis.
DNA methylation patterns are largely altered in
cancer cells as compared with normal cells (5). Epigenetic gene silencing caused by
DNA methylation has been widely accepted as an important mechanism
of tumorigenesis (6). Studies have
revealed the aberrant methylation of multiple genes in HNSCC
(7,8). Kostareli et al demonstrated
and confirmed a human papilloma virus (HPV)-related prognostic
methylation score for patients with HNSCC (9,10).
Moreover, a recent study identified an HPV infection-related
epigenetic signature consisting of five CpGs as survival predictors
in HPV-positive HNSCC (11).
Despite these marked findings, there is a lack of a reliable
methylation prognostic signature for risk stratification in
patients with HNSCC.
Similar to surgery, radiotherapy alone may be used
for HNSCC treatment at early stages. For patients with HNSCC in
middle and late stages, radiotherapy is generally implemented in
combination with surgical excision that may decrease local
recurrence and improve tumor control and the survival of patients
(12). DNA methylation is known to
play a critical role in the resistance of tumors to radiotherapy
(13). Epigenetic silencing of
tumor suppressor genes by methylation was revealed to be associated
with radio-resistance of oral squamous cell carcinoma and poor
outcome of patients (14). Given
the close associations between DNA methylation and radiotherapy,
herein we focused on the identification of a prognostic methylation
signature from radiotherapy-related differentially methylated CpG
sites based on the methylation data obtained from 388 patients with
HNSCC from The Cancer Genome Atlas (TCGA) database. The prognostic
robustness of this methylation signature was assessed in a training
set as well as a validation set.
Materials and methods
Retrieval of public data
The DNA methylation profile of 580 patients with
HNSCC was retrieved from TCGA data portal (https://tcga-data.nci.nih.gov/tcga/) based on the
Illumina Infinium Human Methylation 450 BeadChip platform. Of these
patients, 388 with corresponding clinical data concerning
radiotherapy and survival were selected as a training cohort. The
GSE75537 (15) dataset downloaded
from the National Center for Biotechnology Information Gene
Expression Omnibus repositories (http://www.ncbi.nlm.nih.gov/geo/) contained 108
samples of oral tongue squamous cell carcinomas, and 53 of these
samples with available survival information were selected as a
validation cohort.
Differential methylation analyses
between favorable and poor prognostic samples
Poor prognostic samples (patients not receiving
radiotherapy with an OS of 12 months or shorter) and favorable
prognostic samples (patients receiving radiotherapy with an OS of
48 months or longer) were selected from the training cohort. The
acquired methylated sites were annotated based on the platform
annotation information, and only those in the CpGs sites were
retained. The genes with differentially methylated CpG sites (DMGs)
between the two groups were selected using the limma package
(16) (version 3.34.7, http://bioconductor.org/packages/release/bioc/html/limma.html)
with significant cut-off values of false discovery rate (FDR)
<0.05 and |log2 fold change (FC)|>0.1.
Selection of co-methylation
modules
A weighted gene co-methylation network with all
methylated CpGs obtained from the training set was constructed
using the Weighted Gene Co-expression Network Analysis (WGCNA)
software (17,18) (https://cran.r-project.org/web/packages/WGCNA/index.html).
To achieve scale-free topology, a soft-thresholding power of β=٥
with a scale-free R2 value of 0.9 for calculating
adjacency, was selected. The genes with similar methylation levels
were grouped into the same module. The modules with minSize=100 and
cutHeight=0.95 were identified by dynamic tree cut algorithm using
dynamicTreeCut version 1.63 (https://cran.r-project.org/web/packages/dynamicTreeCut/index.html).
Enrichment analysis of DMGs was carried out in each identified
module with a hypergeometric-based test (19). The modules with P<0.05 and fold
enrichment >1 were selected as DMG-enriched modules, which were
subjected to gene ontology (GO) (20) functional enrichment analysis using
the DAVID (https://david.ncifcrf.gov/)
bioinformatics online tool (21).
Correlation analysis of methylation
and expression data
Methylation and expression data of the DMGs were
focused on in these selected DMG-enriched modules. Using
methylation data and the matched mRNA expression data, overall
methylation levels of all DMGs and their overall gene expression
levels were correlated by calculating Pearson correlation
coefficient (CC) using the cor.test function (https://stat.ethz.ch/R-manual/R-devel/library/stats/html/cor.test.html).
Correlation between methylation levels and expression levels was
explored for every individual DMG. As a result, the genes with a
negative CC value were selected for further analysis.
Statistical analysis for survival
Based on the survival information of patients in the
training set, a univariate Cox regression analysis was performed to
evaluate the association of the aforementioned genes which had a
negative CC with prognosis. The significant genes with a log-rank
P<0.05 were regarded as prognosis-related genes, which were used
as input for a L1 penalized (LASSO) Cox-proportional hazard (PH)
model (22) to identify the
optimal panel of prognostic genes using the penalized package
(https://cran.r-project.org/web/packages/penalized/) of
R language (version 3.4.1). Cox-PH coefficients and methylation
levels of these prognostic genes were combined to construct the
following prognostic model:
Risk
score=∑coefgenexMethylationgene
where, Coefgene represents the Cox-PH
coefficient of an individual gene; and Methylationgene
represents the methylation level of an individual gene.
A risk score was assigned to each sample in the
training cohort. With a median methylation risk score as the
cutoff, the training set was dichotomized into a high-risk group
and a low-risk group. Survival probabilities of the two groups were
analyzed by Kaplan-Meier estimates (23) using survival package of R language.
P-values from the log-rank test suggested significance of the
prognostic model. Specificity and sensitivity of this model were
assessed by the area under the receiver operating characteristic
(ROC) curve (AUC) analysis. Prognostic performance of the
methylation signature was assessed in the validation cohort.
Results
Identification of radiotherapy-related
DMGs
Clinical and demographic data of the training and
validation cohorts are presented in Table I. Based on the aforementioned
criterion of sample classification, the training cohort had 30 poor
prognostic samples and 27 favorable prognostic samples. Between the
poor and favorable prognostic samples, a total of 976 DMGs
associated with radiotherapy (3.43×10−8 < Pnominal
<6.807×10−4, FDR <0.05) were obtained by
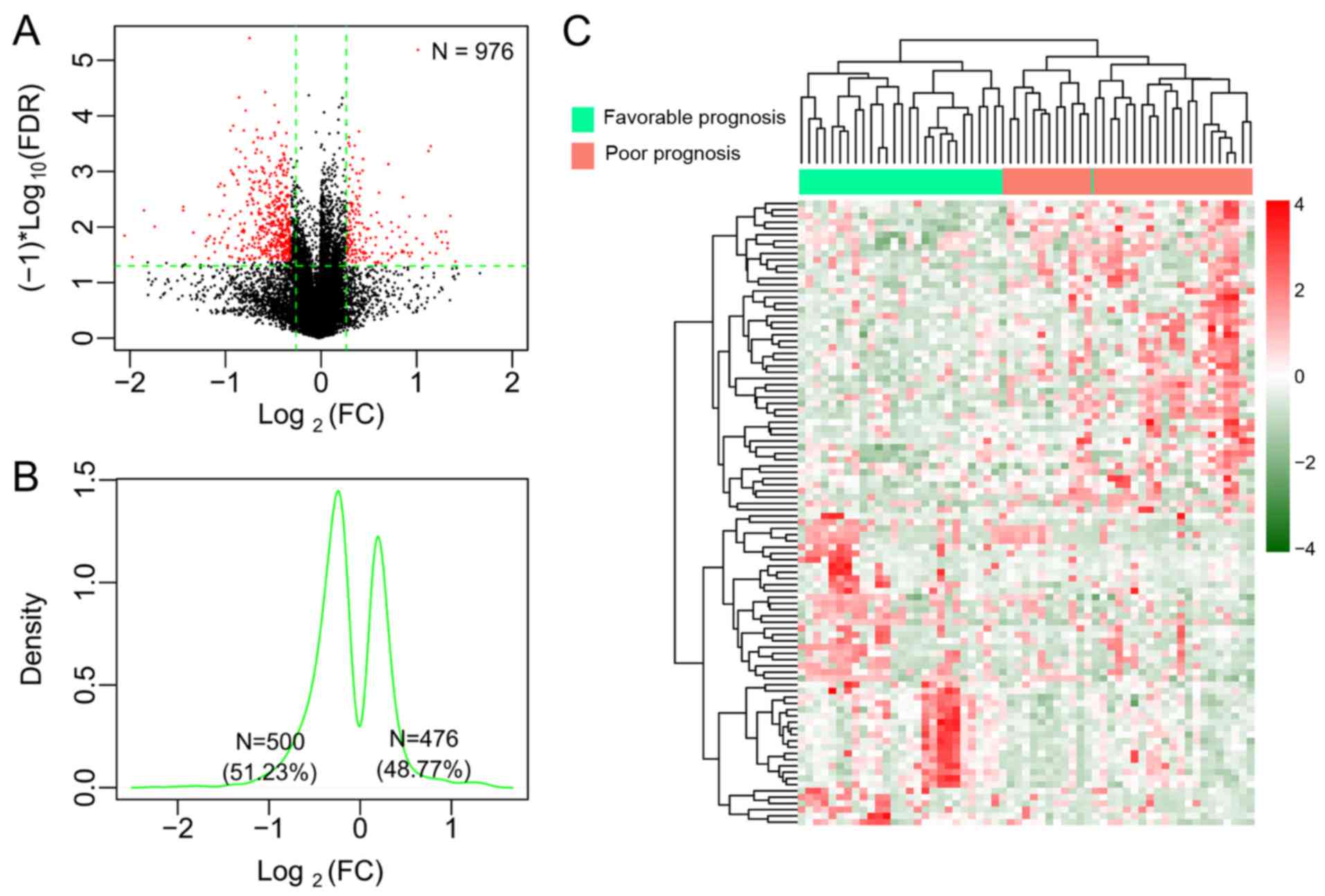
differential methylation analysis (Fig. 1A). Among these DMGs, 476 (48.77%)
were hypermethylated and 500, hypomethylated (Fig. 1B). The two-way hierarchical
clustering analysis based on methylation levels of the top 100 DMGs
revealed the difference in the DNA methylation pattern between the
favorable prognostic samples and the poor prognostic samples
(Fig. 1C). With regard to the CpG
sites in the 976 DMGs, 192 DMGs were located in the TSS region, 220
in the gene body region, 30 in the first exon, 53 in the
5′-untranslated regions (UTRs), 35 in the 3′-UTRs, and 446 in the
promoters. All significant DMGs were ranked according to the FDR
value. As a consequence, the top 20 DMGs with significance were
selected (Table II).
 | Table I.Clinical covariates of patients in
the training set and the validation set. |
Table I.
Clinical covariates of patients in
the training set and the validation set.
| Clinical
covariates | Training set
(n=388) | Validation set
(n=53) |
|---|
| Age (mean ± SD,
years) | 60.81±11.66 | 49.36±13.47 |
| Sex
(male/female) | 288/100 | 42/11 |
| Death
(dead/alive/-) | 117/271 | 15/38 |
| OS time (mean ± SD,
months) | 26.43±26.21 | 30.46±26.98 |
| Table II.Top 20 DMGs between favorable and
poor prognostic samples. |
Table II.
Top 20 DMGs between favorable and
poor prognostic samples.
| ID | Chr | Position | Gene | Location | β-favorable | β-poor | Effect | Pnominal | FDR |
|---|
| cg26934993 | chr17 | 37528248 | KAT2A | TSS200 | 0.3667 | 0.6062 | −0.7253 |
5.43×10−8 |
3.980×10−6 |
| cg26370886 | chr19 | 53930240 | RASIP1 | Body | 0.3655 | 0.5390 | −0.5604 |
5.12×10−7 |
3.750×10−5 |
| cg24351916 | chr3 | 39427639 | RPSA | Body | 0.7753 | 0.8339 | −0.1051 |
5.82×10−7 |
4.270×10−5 |
| cg09938490 | chr15 | 41876101 | SERINC4 | Body | 0.8672 | 0.7346 | 0.2394 |
8.47×10−7 |
6.210×10−5 |
| cg08069338 | chr6 | 160127888 | SNORA29 | TSS1500 | 0.8294 | 0.7166 | 0.2110 |
9.56×10−7 |
7.010×10−5 |
| cg27211576 | chr15 | 72880218 | CSK | Body | 0.6376 | 0.5534 | 0.2045 |
1.60×10−6 |
1.171×10−4 |
| cg26264697 | chr19 | 3529064 | HMG20B | Body | 0.4428 | 0.5928 | −0.4210 |
1.78×10−6 |
1.307×10−4 |
| cg27266479 | chr1 | 9217469 | H6PD | Promoter | 0.1574 | 0.1290 | 0.2868 |
1.95×10−6 |
1.432×10−4 |
| cg21657521 | chr19 | 7650447 | C19orf59 | TSS1500 | 0.0907 | 0.1250 | −0.4637 |
2.75×10−6 |
2.017×10−4 |
| cg05657416 | chr6 | 27213771 | HIST1H4I | TSS1500 | 0.8716 | 0.8073 | 0.1106 |
2.86×10−6 |
2.099×10−4 |
| cg21205305 | chr19 | 54884437 | C19orf76 | Promoter | 0.2889 | 0.3970 | −0.4583 |
3.09×10−6 |
2.263×10−4 |
| cg27537591 | chr10 | 116687888 | TRUB1 | Promoter | 0.0501 | 0.0402 | 0.3154 |
3.30×10−6 |
2.422×10−4 |
| cg25739003 | chr11 | 62097836 | EEF1G | Promoter | 0.0751 | 0.0606 | 0.3092 |
4.23×10−6 |
3.100×10−4 |
| cg26740494 | chr1 | 1556994 | MMP23B | TSS1500 | 0.4795 | 0.6275 | −0.3881 |
4.68×10−6 |
3.434×10−4 |
| cg26222042 | chr5 | 31567957 | C5orf22 | Promoter | 0.0580 | 0.0447 | 0.3762 |
4.68×10−6 |
3.434×10−4 |
| cg26615259 | chr1 | 154977769 | MRPL24 | Promoter | 0.0558 | 0.0447 | 0.3208 |
4.75×10−6 |
3.483×10−4 |
| cg27085584 | chr5 | 61735043 | DIMT1L | Promoter | 0.0392 | 0.0496 | −0.3411 |
4.85×10−6 |
3.555×10−4 |
| cg27535410 | chr19 | 797354 | PRTN3 | Body | 0.8207 | 0.9326 | −0.1844 |
5.20×10−6 |
3.812×10−4 |
| cg27065374 | chrX | 67976906 | EFNB1 | Body | 0.5659 | 0.4863 | 0.2185 |
5.20×10−6 |
3.816×10−4 |
| cg22961457 | chr20 | 61840754 | SLC2A4RG | 3′UTR | 0.2397 | 0.4364 | −0.8641 |
5.41×10−6 |
3.965×10−4 |
WGCNA network analysis and key module
identification
To evaluate the correlation between all methylated
CpGs obtained from the training dataset, a weighted gene
co-methylation network was applied to these methylated CpGs. Twelve
modules of co-methylated genes were identified, wherein CC varied
from 0.514 to 0.772 (mean value=0.642, Table III and Fig. 2A). Other CpGs that exhibited no
significant correlation with methylation levels were grouped into a
grey module. The enrichment of DMGs was evaluated in these modules.
As revealed in Fig. 2B and C,
yellow-green, magenta, purple, and turquoise modules had 44, 92, 45
and 113 DMGs, respectively, and each of these met the criteria of
fold enrichment >1 and P<0.05. Thus, the four DMG-enriched
modules were subjected to GO enrichment analysis. All DMGs in the
four modules were predominately linked to protein-DNA complex
assembly, nucleosome organization, chromatin assembly, and the cell
cycle process (Table IV).
| Table III.WGCNA network analysis identified
gene modules with co-methylated CpG sites. |
Table III.
WGCNA network analysis identified
gene modules with co-methylated CpG sites.
| Module color | Count of CpGs | Correlation | Pcorr | Count of DM
CpGs | Enrichment fold
(95% CI) | Phyper |
|---|
| Black | 301 | 0.702 |
7.01×10−32 | 30 | 1.033
(0.679–1.521) |
8.43×10−1 |
| Blue | 562 | 0.514 |
1.70×10−6 | 38 | 0.701
(0.486–0.985) |
4.01×10−2 |
| Brown | 469 | 0.689 |
6.68×10−11 | 28 | 0.619
(0.403–0.915) |
1.31×10−2 |
| Green | 397 | 0.561 |
1.33×10−7 | 46 | 1.201
(0.856–1.652) |
2.63×10−1 |
| Green-yellow | 196 | 0.687 |
2.14×10−33 | 44 | 2.327
(1.622–3.277) |
5.59×10−6 |
| Grey | 2,623 | 0.215 |
8.56×10−2 | 180 | 0.711
(0.596–0.846) |
7.10×10−5 |
| Magenta | 221 | 0.642 |
7.05×10−10 | 92 | 4.314
(3.301–5.604) |
2.20×10−16 |
| Pink | 248 | 0.565 |
1.59×10−4 | 2 | 0.084
(0.010–0.306) |
9.75×10−8 |
| Purple | 208 | 0.687 |
2.71×10−23 | 45 | 2.243
(1.571–3.142) |
1.00×10−5 |
| Red | 324 | 0.701 |
5.87×10−4 | 13 | 0.416
(0.218–0.727) |
6.86×10−4 |
| Tan | 182 | 0.772 |
4.53×10−26 | 16 | 0.911
(0.507–1.532) |
8.99×10−1 |
| Turquoise | 649 | 0.645 |
1.47×10−27 | 113 | 1.805
(1.442–2.245) |
2.96×10−7 |
| Yellow | 431 | 0.538 |
1.66×10−15 | 10 | 0.241
(0.114–0.449) |
4.86×10−8 |
| Table IV.Significantly enriched GO terms for
genes with differentially methylated CpGs in four important gene
modules. |
Table IV.
Significantly enriched GO terms for
genes with differentially methylated CpGs in four important gene
modules.
| GO term | Count of genes | P-value | Genes |
|---|
| Protein-DNA complex
assembly | 8 |
2.71×10−4 | HIST2H2AA3,
HIST4H4, HIST1H2AG, HIST1H2BL, CENPA, HIST1H2BG, HIST1H3A,
HIST1H2AH, MIS12 |
| Nucleosome
organization | 8 |
3.10×10−4 | HIST2H2AA3,
HIST4H4, HIST1H2AG, HIST1H2BL, CENPA, HIST1H2BG, HIST1H3A, SUPT16H,
HIST1H2AH |
| Protein
folding | 10 |
8.25×10−4 | GRPEL1, CRYAA,
PFDN5, CCT8, C19ORF2, CCT3, CCT6A, DNAJC2, CLPX, PIN1 |
| Nucleosome
assembly | 7 |
1.10×10−3 | HIST2H2AA3,
HIST4H4, HIST1H2AG, HIST1H2BL, CENPA, HIST1H2BG, HIST1H3A,
HIST1H2AH |
| DNA packaging | 8 |
1.23×10−3 | HIST2H2AA3, CHMP1A,
HIST4H4, HIST1H2AG, HIST1H2BL, CENPA, HIST1H2BG, HIST1H3A,
HIST1H2AH |
| Chromatin
assembly | 7 |
1.32×10−3 | HIST2H2AA3,
HIST4H4, HIST1H2AG, HIST1H2BL, CENPA, HIST1H2BG, HIST1H3A,
HIST1H2AH |
| Chromatin assembly
or disassembly | 8 |
1.97×10−3 | HIST2H2AA3,
HIST4H4, HIST1H2AG, HIST1H2BL, CENPA, HIST1H2BG, HIST1H3A, SUPT16H,
HIST1H2AH |
| Translation | 13 |
2.31×10−3 | MRPL24, EIF4G1,
RPSA, MRPS16, MRPL27, RARS, EIF2S2, RPL35, MARS2, EIF5A, DPH1,
RPL10A, MRPL34 |
| Chromosome
organization | 16 |
3.19×10−3 | KAT2A, HIST2H2AA3,
HIST4H4, HIST1H2AG, HIST1H2BG, NDC80, LIG4, MIS12, C20ORF20, KDM1A,
CHMP1A, HIST1H2BL, CENPA, HIST1H3A, SUPT16H, BRE, HIST1H2AH |
| Cell cycle | 21 |
5.67×10−3 | CCNT2, MAD1L1,
CRYAA, NDC80, PMF1, PBK, LIG4, TACC3, ESCO2, UHMK1, MIS12, PIN1,
CHMP1A, PSMB6, CENPA, PSMA3, SKA2, RAD51L3, MAPK7, KPNA2,
DNAJC2 |
| DNA metabolic
process | 15 |
1.11×10−2 | NEIL3, SMC5, PPT1,
LIG4, ESCO2, PCNA, SUPT16H, PSIP1, BRE, DDB2, RAD51L3, KPNA2,
DNAJC2, APEX1, DUT |
| Cell cycle
process | 16 |
1.24×10−2 | MAD1L1, CRYAA,
NDC80, PMF1, PBK, TACC3, UHMK1, MIS12, CHMP1A, PSMB6, CENPA, PSMA3,
SKA2, RAD51L3, KPNA2, DNAJC2 |
| Mitotic cell
cycle | 12 |
1.44×10−2 | MAD1L1, CHMP1A,
PSMB6, CENPA, PSMA3, NDC80, SKA2, PMF1, PBK, DNAJC2, KPNA2,
MIS12 |
| Response to DNA
damage stimulus | 12 |
1.52×10−2 | NEIL3, DDB2, BRE,
SUPT16H, SMC5, PCNA, AATF, RAD51L3, LIG4, ATMIN, APEX1, ESCO2 |
| Chromatin
organization | 12 |
1.67×10−2 | KAT2A, KDM1A,
HIST2H2AA3, HIST4H4, HIST1H2AG, HIST1H2BL, CENPA, HIST1H2BG,
HIST1H3A, BRE, SUPT16H, HIST1H2AH, C20ORF20 |
| M phase | 11 |
1.68×10−2 | MAD1L1, CHMP1A,
CRYAA, NDC80, SKA2, RAD51L3, PMF1, PBK, TACC3, KPNA2, MIS12 |
Identification and validation of a
four-gene prognostic methylation signature
In correlation analysis, an inverse correlation was
evident between overall methylation and overall expression of all
DMGs in the four DMG-enriched modules (CC=−0.5051,
P=2.20×10−16; Fig. 3).
In addition, an inverse correlation was also observed between
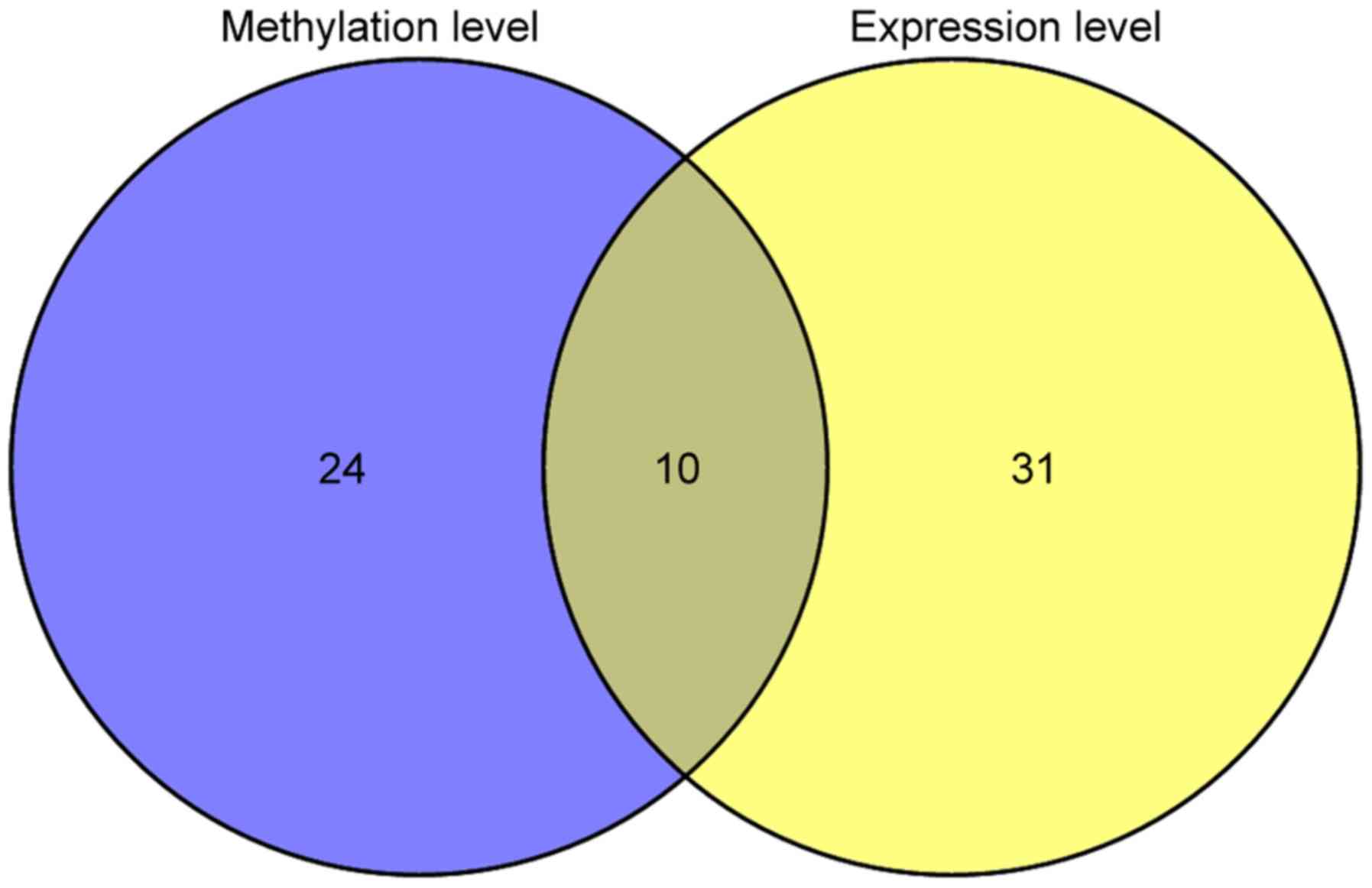
methylation and gene expression of individual genes for 165 genes.
Univariate Cox regression analysis revealed that 34 genes were
significantly related to prognosis in methylation levels, 41 genes
had a significant association with prognosis in gene expression
levels, and 10 were overlapped (Fig.
4).
A lasso Cox-PH model was fit using the overlapped 10
genes to identify the optimal panel of methylation genes for
prognosis prediction. The parameter λ value was tuned to 0.1845 by
conducting 1,000 simulations of cross-validation to obtain a
maximal cross-validation likelihood (cvl) value of −720.354. A
panel of four genes was obtained under this condition, including
zinc finger protein (ZNF)10, transmembrane serine protease
(TMPRSS)12, endoplasmic reticulum-Golgi intermediate
compartment protein (ERGIC)2, and ring finger protein
(RNF)215.
It was investigated whether the methylation level of
the four genes was associated with prognosis. As revealed in
Fig. 5A, all patients from the
training set were divided into hypermethylated and hypomethylated
groups based on the median methylation level of each gene. A
Kaplan-Meier plot revealed that the patients with hypomethylated
ZNF10 had significantly better survival outcome than those
with hypermethylated ZNF10 (P=0.0434). The same result was
observed for TMPRSS12 (P=0.0386) and ERGIC2
(P=3.093×10−4). Conversely, a significantly worse
prognosis was observed in the patients with hypomethylated
RNF215 as compared to those with hypermethylated
RNF215 (P=0.0129).
The relationship between gene expression of each of
the four genes and survival was explored. Based on the median gene
expression level, all samples in the training set were classified
into high expression and low expression groups. ZNF10,
TMPRSS12 and ERGIC2 were associated with significantly
longer OS in the high expression group than in the low expression
group (P=1.125×10−4, P=0.0471, P=1.196×10−3;
Fig. 5B). An opposite result was
observed for RNF215; patients with high RNF215
expression had significantly shorter OS than those with low
expression (P=0.0391; Fig.
5B).
A risk prediction model was constructed that
included risk score based on the Cox-PH prognostic correlation of
the optimal four-gene panel (Table
V):
| Table V.Risk score model based on a four-gene
methylation signature. |
Table V.
Risk score model based on a four-gene
methylation signature.
| ID | Gene | Chr. | Position | Location | Coef | Hazard ratio (95%
CI) | P-value |
|---|
| cg25577680 | ZNF10 | chr12 | 132217652 | Promoter | 0.964 | 6.259
(1.274–10.74) | 0.0230 |
| cg27261219 | TMPRSS12 | chr12 | 49522905 | TSS | 1.035 | 3.179
(1.131–8.935) | 0.0277 |
| cg25338581 | ERGIC2 | chr12 | 29425828 | TSS | 7.166 | 7.576
(4.142–10.506) | 0.0023 |
| cg25964984 | RNF215 | chr22 | 29113371 | TSS | −4.896 | 0.437
(0.0405–0.719) | 0.0223 |
Risk score=(0.9639) ×
Methylationcg25577680 + (1.035) ×
Methylationcg27261219 + (7.1664) ×
Methylationcg25338581 + (−4.8961) ×
Methylationcg25964984
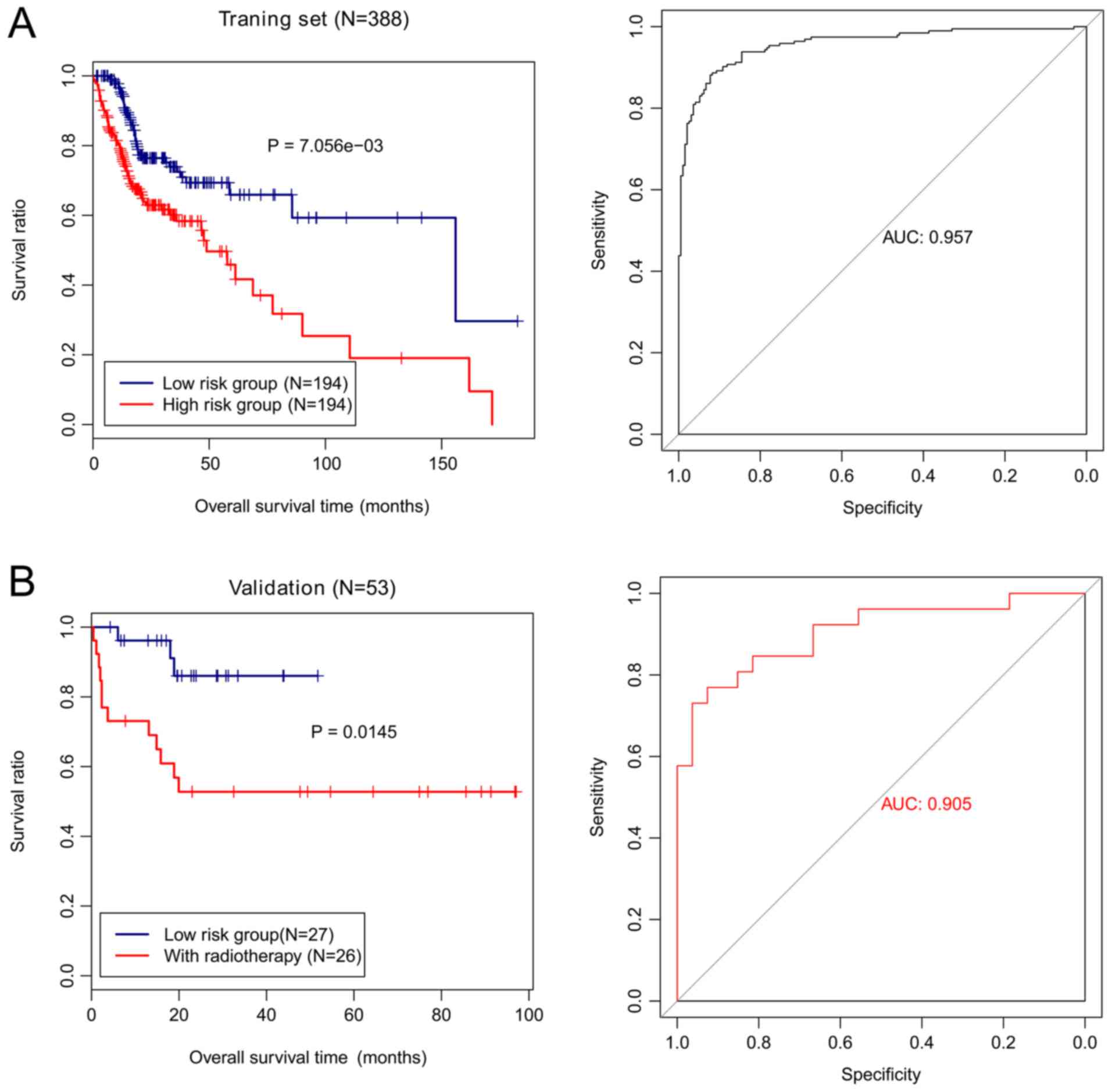
The risk score was calculated for each patient. The
training cohort was grouped based on the risk score into high-risk
and low-risk groups. Samples in the low-risk group exhibited a
significantly longer OS than those from the high-risk group
(P=7.056×10−3; Fig.
6A). The AUC value of the methylation risk score was 0.957,
suggestive of the high accuracy of the four-gene methylation
signature in predicting the survival of patients with HNSCC
(Fig. 6A). The risk stratification
ability of the four-gene methylation signature in the validation
cohort was assessed. The validation cohort was classified based on
the methylation risk score into high-risk and low-risk groups. The
samples in the low-risk group also had markedly longer OS than
those from the high-risk group (P=0.0145; Fig. 6B) with an AUC value of 0.905.
Independence of the four-gene
prognostic methylation signature from radiotherapy
To investigate whether the prognostic performance of
this four-gene methylation signature was independent of
radiotherapy, the methylation risk score was applied to patients
with or without radiotherapy. In particular, the patients without
radiotherapy were classified based on the methylation risk score
into high-risk and low-risk groups, and the OS of patients in the
low-risk group was longer than that of patients from the high-risk
group (P=3.632×10−4; Fig.
7A). The patients receiving radiotherapy were also separated as
per the methylation risk score into two risk groups, and those from
the low-risk group had a significantly longer OS than the patients
from the high-risk group (P=0.0281; Fig. 7B). These results demonstrated that
the prognostic value of this four-gene methylation signature was
independent of radiotherapy.
Discussion
Despite advancements in the diagnosis and treatment
of HNSCC, the prognosis remains poor (24). Radiation therapy is used as a
standard adjuvant treatment for HNSCC (25). The present study focused on
radiotherapy-related aberrant methylation of genes in HNSCC to
identify a prognostic methylation signature. A total of 976 DMGs
were revealed between the patients with survival of 12 months or
shorter without radiotherapy and those surviving for 48 months or
longer and receiving radiotherapy. Moreover, four co-methylation
modules that were markedly enriched with DMGs were retrieved by
WGCNA analysis. The DMGs in the four modules were functionally
associated with protein-DNA complex assembly, nucleosome
organization, chromatin assembly, and the cell cycle process. These
results may improve our understanding of the mechanisms underlying
these DNA methylation alterations in HNSCC.
Following correlation analysis and multivariate Cox
regression analysis, a LASSO-penalized Cox-PH model was used to
identify the most informative genes for the prediction of survival.
LASSO is a popular algorithm with a property of simultaneous
variable selection and shrinkage, leading to the identification of
prognostic signatures (26). It
has been used in Cox-PH model for the survival analysis of patients
with breast cancer (27). A
four-gene panel predictive of prognosis with the lasso Cox-PH model
was established. The risk score derived from the four-gene
methylation signature stratified the patients into two risk groups
with significantly different OS in both the training and validation
sets. ROC curves demonstrated high specificity and sensitivity of
the four-gene methylation signature in predicting OS of patients
with HNSCC.
The four methylation genes of prognostic value were
ZNF10, TMPRSS12, ERGIC2 and RNF215. Zinc finger
protein 10 encoded by the ZNF10 gene acts as a transcription
repressor and is a member of the zinc finger proteins, the largest
transcription factor family involved in development,
differentiation, and metabolism. This protein plays versatile roles
in cancer progression (28). Chung
et al provided evidence that the glioma-associated oncogene
family zinc finger 1 is a biomarker in HNSCC (29). However, to the best of our
knowledge, the role of ZNF10 in HNSCC is yet unknown. Based
on our results, the hypermethylated CpG sites in ZNF10 were
associated with poor prognosis of patients with HNSCC, suggesting
that the hypermethylation of ZNF10 may serve as an indicator
of poor HNSCC prognosis. The TMPRSS12 gene encodes
transmembrane protease, serine 12, a member of the serine protease
family participating in diverse functions such as immune response
and blood coagulation and production (30). It has been recognized as an
important gene associated with human infertility by genome-wide
analyses (31). TMPRSS12 is
used as one of the CG signatures in the identification of
cancer-associated aberrant DNA methylation that influences gene
expression (32). In our study, it
had methylated CpG sites and the hypermethylation was related to
poor OS outcome. This observation may suggest that the
hypermethylated CpGs in TMPRSS12 may be the causative factor
for poor prognosis of HNSCC. ERGIC2, namely PTX1, is
a gene determined by subtractive hybridization. The ERGIC2 protein
is an endoplasmic reticulum (ER) resident protein implicated in
protein trafficking between ER and Golgi bodies (33,34).
PTX1 is an alias for the gene PITX1, which has been
identified as a predictive indicator of the response to
chemotherapy in HNSCC (35). In
the present study, as reported for the aforementioned two genes,
ERGIC2 also carried methylated CpG sites, and its
hypermethylation was associated with poor survival outcome. Thus,
methylated ERGIC2 may be a predictive factor for HNSCC
prognosis. The RNF125 gene encodes a novel E3
ubiquitin-protein ligase that may be involved in the T-cell
receptor signaling pathway (36).
Ring finger ubiquitin protein ligases play a role in tumorigenesis
and metastasis (37). Yang et
al suggested that RNF125 strengthens p53 degradation and
suppresses p53 function (38). The
proto-oncoprotein S-phase kinase-associated protein2 (Skp2) has
been revealed to be overexpressed in many malignancies such as
HNSCC. Skp2 plays an important role in the degradation of p27 and
p21 via the ubiquitin-proteasome pathway (39). In the present study, the
hypomethylation and high expression of RNF125 were
associated with poor OS, indicative of its function as an oncogene
in HNSCC. Further studies are warranted to determine if it
functions through the ubiquitin-related pathway.
In the present study, it was revealed that the
four-gene methylation signature related to radiotherapy can
successfully discriminate between the patients with high risk and
low risk. The prognostic ability of this methylation signature was
independent of radiotherapy. These findings may improve the risk
stratification of patients with HNSCC and is of a particular
clinical relevance. Despite these valuable findings, the present
study has a few limitations. First, the number of patient samples
with available clinical information was small. In addition, these
predictive results need to be further validated through substantial
experiments.
In conclusion, a radiotherapy-related four-gene
methylation signature was identified for predicting HNSCC survival.
This platform may serve as a guide for the development of
individualized therapy for patients with HNSCC. Translation of our
findings into future clinical trials requires validation using
large populations of patients.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JM performed data analyses and wrote the manuscript.
RL contributed significantly to data analyses and manuscript
revision. JW conceived and designed the study. All authors read and
approved the manuscript, and agree to be accountable for all
aspects of the research in ensuring that the accuracy or integrity
of any part of the work are appropriately investigated and
resolved.
Ethics approval and consent to
participate
In the original article of the datasets, the trials
were approved by the local institutional review boards of all
participating centers, and informed consent was obtained from all
patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
HNSCC
|
head and neck squamous cell
carcinoma
|
|
TCGA
|
The Cancer Genome Atlas
|
|
DMG
|
differentially methylated CpG site
|
|
WGCNA
|
Weighted Gene Co-expression Network
Analysis
|
|
HPV
|
human papilloma virus
|
|
OS
|
overall survival
|
|
FDR
|
false discovery rate
|
|
FC
|
fold change
|
References
|
1
|
GBD 2015 Disease, Injury Incidence and
Prevalence Collaborators: Global, regional, and national incidence,
prevalence, and years lived with disability for 310 diseases and
injuries, 1990–2015: A systematic analysis for the Global Burden of
Disease Study 2015. Lancet. 388:1545–1602. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Leemans CR, Snijders PJF and Brakenhoff
RH: The molecular landscape of head and neck cancer. Nat Rev
Cancer. 18:269–282. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
López-Verdín S, Lavalle-Carrasco J,
Carreón-Burciaga RG, Serafín-Higuera N, Molina-Frechero N,
González-González R and Bologna-Molina R: Molecular markers of
anticancer drug resistance in head and neck squamous cell
carcinoma: A literature review. Cancers (Basel). 10:E3762018.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Beyzadeoglu M, Selek U and Ozyigit G:
Radiation Therapy for Head and Neck Cancers: A Case-Based Review.
Springer; New York, NY: pp. pp2442014
|
|
5
|
Delpu Y, Cordelier P, Cho WC and Torrisani
J: DNA methylation and cancer diagnosis. Int J Mol Sci.
14:15029–15058. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Klutstein M, Nejman D, Greenfield R and
Cedar H: DNA methylation in cancer and aging. Cancer Res.
76:3446–3450. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lechner M, Fenton T, West J, Wilson G,
Feber A, Henderson S, Thirlwell C, Dibra HK, Jay A, Butcher L, et
al: Identification and functional validation of HPV-mediated
hypermethylation in head and neck squamous cell carcinoma. Genome
Med. 5:152013. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Steinmann K, Sandner A, Schagdarsurengin U
and Dammann RH: Frequent promoter hypermethylation of tumor-related
genes in head and neck squamous cell carcinoma. Oncol Rep.
22:1519–1526. 2009.PubMed/NCBI
|
|
9
|
Kostareli E, Hielscher T, Zucknick M,
Baboci L, Wichmann G, Holzinger D, Mücke O, Pawlita M, Del Mistro
A, Boscolo-Rizzo P, et al: Gene promoter methylation signature
predicts survival of head and neck squamous cell carcinoma
patients. Epigenetics. 11:61–73. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kostareli E, Holzinger D, Bogatyrova O,
Hielscher T, Wichmann G, Keck M, Lahrmann B, Grabe N,
Flechtenmacher C, Schmidt CR, et al: HPV-related methylation
signature predicts survival in oropharyngeal squamous cell
carcinomas. J Clin Invest. 123:2488–2501. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Esposti DD, Sklias A, Lima SC, Beghelli-de
la Forest Divonne S, Cahais V, Fernandez-Jimenez N, Cros MP, Ecsedi
S, Cuenin C, Bouaoun L, et al: Unique DNA methylation signature in
HPV-positive head and neck squamous cell carcinomas. Genome Med.
9:332017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shuryak I, Hall EJ and Brenner DJ: Dose
dependence of accelerated repopulation in head and neck cancer:
Supporting evidence and clinical implications. Radiother Oncol.
127:20–26. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhu X, Wang Y, Tan L and Fu X: The pivotal
role of DNA methylation in the radio-sensitivity of tumor
radiotherapy. Cancer Med. 7:3812–3819. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Huang KH, Huang SF, Chen IH, Liao CT, Wang
HM and Hsieh LL: Methylation of RASSF1A, RASSF2A, and HIN-1 is
associated with poor outcome after radiotherapy, but not surgery,
in oral squamous cell carcinoma. Clin Cancer Res. 15:4174–4180.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Krishnan NM, Dhas K, Nair J, Palve V,
Bagwan J, Siddappa G, Suresh A, Kekatpure VD, Kuriakose MA and
Panda B: A minimal DNA methylation signature in oral tongue
squamous cell carcinoma links altered methylation with tumor
attributes. Mol Cancer Res. 14:805–819. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: Limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Langfelder P and Horvath S: WGCNA: An R
package for weighted correlation network analysis. BMC
Bioinformatics. 9:5592008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Massart R, Dymov S, Millecamps M, Suderman
M, Gregoire S, Koenigs K, Alvarado S, Tajerian M, Stone LS and Szyf
M: Overlapping signatures of chronic pain in the DNA methylation
landscape of prefrontal cortex and peripheral T cells. Sci Rep.
6:196152016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cao J and Zhang S: A bayesian extension of
the hypergeometric test for functional enrichment analysis.
Biometrics. 70:84–94. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gene Ontology Consortium: Gene ontology
consortium: Going forward. Nucleic Acids Res. 43:D1049–D1056. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Huang DW, Sherman BT, Tan Q, Kir J, Liu D,
Bryant D, Guo Y, Stephens R, Baseler MW, Lane HC and Lempicki RA:
DAVID Bioinformatics Resources: Expanded annotation database and
novel algorithms to better extract biology from large gene lists.
Nucleic Acids Res. 35:W169–W175. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tibshirani R: The lasso method for
variable selection in the Cox model. Stat Med. 16:385–395. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Goel MK, Khanna P and Kishore J:
Understanding survival analysis: Kaplan-Meier estimate. Int J
Ayurveda Res. 1:274–278. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Marur S and Forastiere AA: Head and neck
squamous cell carcinoma: Update on epidemiology, diagnosis, and
treatment. Mayo Clin Proc. 91:386–396. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Marur S and Forastiere AA: Head and neck
cancer: Changing epidemiology, diagnosis, and treatment. Mayo Clin
Proc. 83:489–501. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bøvelstad HM, Nygård S, Størvold HL,
Aldrin M, Borgan Ø, Frigessi A and Lingjaerde OC: Predicting
survival from microarray data-a comparative study. Bioinformatics.
23:2080–2087. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Goeman JJ: L1 penalized estimation in the
Cox proportional hazards model. Biom J. 52:70–84. 2010.PubMed/NCBI
|
|
28
|
Jen J and Wang YC: Zinc finger proteins in
cancer progression. J Biom Sci. 23:532016. View Article : Google Scholar
|
|
29
|
Chung CH, Dignam JJ, Hammond ME, Klimowicz
AC, Petrillo SK, Magliocco A, Jordan R, Trotti A, Spencer S, Cooper
JS, et al: Glioma-associated oncogene family zinc finger 1
expression and metastasis in patients with head and neck squamous
cell carcinoma treated with radiation therapy (RTOG 9003). J Clin
Oncol. 29:1326–1334. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hedstrom L: Serine protease mechanism and
specificity. Chem Rev. 34:4501–4524. 2003.
|
|
31
|
Liu M, Hu Z, Qi L, Wang J, Zhou T, Guo Y,
Zeng Y, Zheng B, Wu Y, Zhang P, et al: Scanning of novel
cancer/testis proteins by human testis proteomic analysis.
Proteomics. 13:1200–1210. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Saghafinia S, Mina M, Riggi N, Hanahan D
and Ciriello G: Pan-cancer landscape of aberrant DNA methylation
across human tumors. Cell Rep. 25:1066–1080.e8. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kwok SC, Liu X, Mangel P and Daskal I:
PTX1(ERGIC2)-VP22 fusion protein upregulates interferon-beta in
prostate cancer cell line PC-3. DNA Cell Biol. 25:523–529. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kwok SC, Kumar S and Dai G:
Characterization of a variant of ERGIC2 transcript. DNA Cell Biol.
33:73–78. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Takenobu M, Osaki M, Fujiwara K, Fukuhara
T, Kitano H, Kugoh H and Okada F: PITX1 is a novel predictor of the
response to chemotherapy in head and neck squamous cell carcinoma.
Mol Clin Oncol. 5:89–94. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chu P, Pardo J, Zhao H, Li CC, Pali E,
Shen MM, Qu K, Yu SX, Huang BC, Yu P, et al: Systematic
identification of regulatory proteins critical for T-cell
activation. J Biol. 2:212003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Fang S, Lorick KL, Jensen JP and Weissman
AM: RING finger ubiquitin protein ligases: Implications for
tumorigenesis, metastasis and for molecular targets in cancer.
Semin Cancer Biol. 13:5–14. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yang L, Zhou B, Li X, Lu Z, Li W, Huo X
and Miao Z: RNF125 is a ubiquitin-protein ligase that promotes p53
degradation. Cell Physiol Biochem. 35:237–245. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Khan AQ, Siveen KS, Prabhu KS,
Kuttikrishnan S, Akhtar S, Shaar A, Raza A, Mraiche F, Dermime S
and Uddin S: Curcumin-mediated degradation of s-phase kinase
protein 2 induces cytotoxic effects in human
papillomavirus-positive and negative squamous carcinoma cells.
Front Onco. 8:3992018. View Article : Google Scholar
|