Introduction
Portal hypertension (PHT) is one of the most severe
clinical consequences of patients with liver cirrhosis, which
results in life-threatening complications including variceal
bleeding and hepatic encephalopathy (1). Previous studies have indicated that
increased intrahepatic vascular resistance (IHVR) is the initial
and determinant factor of PHT (1–3).
Various structural and functional factors are responsible for the
IHVR increase (2,4–6). The
formation of fibrous septa and regenerative nodules causes the
distortion and compression of the venous system, which increases
the resistance to portal venous blood flow (1,2,6). The
progressive process of cirrhosis is characterized by the excessive
deposition of extracellular matrix (ECM) proteins including type I
collagen (Col I), Col III and fibronectin (FN). Hepatic stellate
cells (HSCs) are the primary cells responsible for liver cirrhosis
(7). Upon liver injury, quiescent
HSCs acquire an activated phenotype, migrate to the damaged region,
proliferate and produce ECM proteins (8–10).
Transforming growth factor β1 (TGFβ1), which is the most potent
profibrogenic cytokine in the liver, promotes the synthesis of ECM
proteins in HSCs (11,12). Multiple signaling pathways,
including TGFβ1/SMAD, PI3K/AKT and ERK, are involved in TGFβ1
signal transduction.
Carvedilol, a novel non-selective β-blocker (NSBB),
is an antagonist of non-selective β- and selective
α1-adrenoreceptors that effectively reduces portal
pressure (13). Studies of the
role of carvedilol in the reduction of portal pressure mainly focus
on hemodynamics. Previous studies have reported that carvedilol
improves myocardial and renal fibrosis (14,15).
A small number of studies have examined the antifibrogenic effect
of carvedilol on liver cirrhosis, and the underlying mechanisms are
not well described (16). In
chronic liver disease, portal pressure is mainly determined by the
severity of the destruction of hepatic architecture (17). Therefore, it was hypothesized that
the antifibrogenic effect of carvedilol may be involved in reducing
the portal pressure. The present study aimed to investigate the
antifibrogenic effect of carvedilol in vivo and in
vitro. The rat model of liver cirrhosis induced by carbon
tetrachloride was used to investigate the antifibrogenic effect of
carvedilol in vivo. In vitro, the human line LX-2 was
used to explore the mechanisms underlying carvedilol function.
Materials and methods
Materials
Carbon tetrachloride (CCl4) was purchased
from Sinopharm Chemical Reagent Co., Ltd. Carvedilol used in animal
experiments was obtained from Qilu Pharmaceutical Co., Ltd.
Carvedilol used in the cell-based experiments was purchased from
Sigma-Aldrich (Merck KGaA). Recombinant human TGFβ1 was obtained
from PeproTech, Inc. Specific inhibitor of SMAD3 (SIS3) was
purchased from Medchem Express.
Animals
A total of 40 male Wistar rats (weight, 180–200 g;
age, 8 weeks) were purchased from the Central Animal Care Facility
of Shandong University (Jinan, China). The rats were housed in the
animal care facility under temperature- and humidity-controlled
conditions (temperature, 22–24°C; humidity, 50±5) with a 12-h
light-dark cycle and were provided free access to food and water.
The mortality rate was 20%, and 13 rats were sacrificed for
histological evaluation of liver cirrhosis and measurement of
portal pressure. All rats were sacrificed under anesthesia induced
by intraperitoneal injection of pentobarbital (30 mg/kg). All
animal experiments were carried out in accordance with the Guide
for the Care and Use of Laboratory Animals. All procedures were
approved by the Animal Care and Use Committee of Shandong
Provincial Hospital affiliated to Shandong University (approval no.
2018-005).
Induction of liver cirrhosis by
CCl4 and administration of carvedilol
Cirrhosis was induced by the intraperitoneal (i.p.)
injection of CCl4, as previously described (18). Rats were randomly divided into
three groups: i) Control, which received an i.p. injection of olive
oil (0.5 ml/kg body weight) twice weekly for 9 weeks; ii)
CCl4-intoxicated, which received an i.p. injection of
CCl4 (1 ml/kg; CCl4 to olive oil v/v ratio,
1:1) twice weekly for 9 weeks; and iii) CCl4 +
carvedilol-treated, which received an i.p. injection of
CCl4 (1 ml/kg; CCl4 to olive oil v/v ratio,
1:1) twice weekly for 9 weeks as well as concurrent treatment with
carvedilol (10 mg/kg) via gavage daily for 9 weeks. Rats in the
control and CCl4-intoxicated groups received the vehicle
(2 ml saline) by gavage daily for 9 weeks.
Serum assays
At the end of the experiment, rats were weighed and
anesthetized. Laparotomy was performed to expose the inferior vena
cava. In total, 3 ml venous blood was collected from each rat into
procoagulant vacuum tubes from the inferior vena cava and
centrifuged at 3,000 × g for 10 min at 4°C. Supernatant was
collected and stored at −80°C until biochemical assays were
performed. Serum aspartate aminotransferase (AST), alanine
aminotransferase (ALT) and albumin (ALB) levels were measured using
an AU1000 fully automatic biochemical analyzer (Olympus
Corporation).
Histological examination
Following blood sample collection, rats were
sacrificed and livers were harvested. Liver specimens were fixed in
4% paraformaldehyde for 24 h at room temperature, embedded in
paraffin and cut into 4-µm-thick sections. Sections were stained
with hematoxylin and eosin (H&E) for 5 min and for 30 sec,
respectively, at room temperature for histopathological
examination, and photographed under an Olympus BX63F light
microscope (Olympus Corporation; magnification, ×100). Sections
were stained with sirius red (S-R) dye for 1 h at room temperature
and with hematoxylin for 3 min at room temperature in order to
visualize collagen deposition; the sections were photographed under
a light microscope and under a Nikon Eclipse Ci-E polarized light
microscope (Nikon Corporation; magnification, ×200). The
picrosirius-polarization method was used to evaluate the
distribution of Col I (thick, strongly birefringent, yellow or red
fibers) and Col III (thin, weakly birefringent, green fibers), as
previously described (19). The
collagen-positive area to total area ratio was quantified using
Image-Pro Plus 6.0 (Media Cybernetics, Inc.).
Immunohistochemical analysis
Immunohistochemistry was performed using a Polink-2
Plus Polymer-Horseradish Peroxidase (HRP) Anti-Rabbit
Immunoglobulin G (IgG) Detection System (Beijing Zhongshan Golden
Bridge Biotechnology Co., Ltd.), according to the manufacturer's
protocol. Liver sections were deparaffinized and rehydrated in
descending series of ethanol. After heat-mediated antigen retrieval
with citrate buffer at 120°C for 3 min, the sections were incubated
with 3% hydrogen peroxide for 10 min at room temperature to
suppress endogenous peroxidase activity. The sections were
incubated with polyclonal rabbit anti-rat α-smooth muscle actin
(α-SMA; 1:300; cat. no. A03744; Boster Biological Technology) at
4°C overnight, and were subsequently warmed at 37°C for 30 min.
Following incubation with the goat anti-rabbit IgG HRP-conjugated
secondary antibody included in the kit at 37°C for 30 min, the
sections were stained with diaminobenzidine solution,
counterstained with hematoxylin and dehydrated through an
increasing gradient of ethanol, according to the manufacturer's
protocol. Images were captured under an Olympus BX63F light
microscope. Sections incubated with PBS instead of the primary
antibody were used as negative controls.
Western blotting
Liver tissue samples were stored in liquid nitrogen.
Proteins were extracted from cells and liver tissues using the
Tissue or Cell Total Protein Extraction kit (Sangon Biotech Co.,
Ltd.) according to the manufacturer's protocol. Protein
concentrations were measured using a Bicinchoninic Acid Protein
assay kit (Beyotime Institute of Biotechnology). Equal amounts (50
µg/well) of proteins were separated by 8% SDS-PAGE and transferred
to PVDF membranes (EMD Millipore). Following blocking in 5% skimmed
milk for 1 h at room temperature, the membranes were incubated with
the primary antibodies at 4°C overnight, followed by incubation
with the HRP-conjugated secondary antibodies goat anti-rabbit
(Beijing Zhongshan Golden Bridge Biotechnology Co., Ltd.; cat. no.
ZB-5301; 1:5,000) or rabbit anti-goat (Beijing Zhongshan Golden
Bridge Biotechnology Co., Ltd.; cat. no. ZB-2306; 1:5,000) for 2 h
at room temperature. The signals of the target proteins were
detected by enhanced chemiluminescence using Amersham Imager 600
(GE Healthcare). ImageJ software (version 1.46r; National
Institutes of Health, Bethesda, MD, USA) was used to perform
densitometric analysis. Band intensities were normalized to GAPDH.
The primary antibodies used were as follows: Monoclonal rabbit
anti-total AKT (cat. no. 4691; 1:1,000), monoclonal rabbit
anti-phosphorylated (p)-AKT (cat. no. 4060; 1:1,000), polyclonal
rabbit anti-total-p44/42 MAPK (t-ERK1/2; cat. no. 9102; 1:1,000),
polyclonal rabbit anti-phospho-p44/42 MAPK (p-ERK1/2; cat. no.
9101; 1:1,000), monoclonal rabbit anti-total SMAD3 (cat. no. 9523;
1:1,000), monoclonal rabbit anti-p-SMAD2 (cat. no. 3108; 1:1,000)
and monoclonal rabbit anti-GAPDH (cat. no. 5174; 1:1,000) purchased
from Cell Signaling Technology, Inc.; polyclonal rabbit anti-FN
(cat. no. ab2413; 1:1,000), monoclonal rabbit anti-α-SMA (cat. no.
ab32575; 1:1,000), monoclonal rabbit anti-total SMAD2 (cat. no.
ab40855; 1:2,000) and monoclonal rabbit anti-p-SMAD3 (cat. no.
ab52903; 1:2,000) purchased from Abcam; and monoclonal goat
anti-Col I (cat. no. 1310-01; 1:1,000) purchased from
SouthernBiotech (Birmingham, AL, USA).
Cell culture
LX-2 is an activated human HSC cell line that is
widely used as a model for hepatic fibrosis (9). LX-2 cells were a gift from Professor
Weifen Xie (Shanghai Changzheng Hospital, The Second Military
Medical University, Shanghai, China). Cells were cultured in DMEM
(Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 2% fetal bovine serum (FBS; Gibco; Thermo Fisher
Scientific, Inc.), 100 U/ml of penicillin and 100 µg/ml of
streptomycin (Gibco; Thermo Fisher Scientific, Inc.) at 37°C in a
humidified incubator with 5% CO2.
Cell Counting Kit-8 (CCK-8)
LX-2 cells were plated in 96-well culture plates
(5×103 cells/well) in triplicate and incubated at 37°C
overnight. Subsequently, the cells were incubated with 0, 1, 2, 5
or 10 µM carvedilol in a humidified incubator with 5%
CO2 at 37°C for 24 h. Following the treatment, 10 µl
CCK-8 solution (Dojindo Molecular Technologies, Inc.) was added to
each well. The plates were incubated in a humidified incubator with
5% CO2 at 37°C for 2 h. Cell viability was calculated
according to the manufacturer's protocol (Dojindo Molecular
Technologies, Inc.). Optical density was measured at 450 nm using a
spectrophotometer (Thermo Fisher Scientific, Inc.). The experiment
was repeated three times.
Transwell invasion assay
Serum-starved LX-2 cells (1×106/ml)
treated with 0, 1, 2, 5 or 10 µM carvedilol in 100 µl serum-free
culture medium were seeded into the upper chamber of a 24-well
Transwell plate, and the membranes of the upper chamber were coated
with Matrigel (BD Biosciences). Culture medium with 10% FBS was
added to the lower chamber. Serum-free culture medium was used in
the lower chamber as a non-induced control. Cells were incubated in
a humidified incubator with 5% CO2 at 37°C for 24 h.
Subsequently, the cell medium was discarded, and cells in the upper
chamber were removed by a cotton swab. The cells on the lower
surface of the chamber were fixed with 4% paraformaldehyde for 20
min at room temperature and stained with hematoxylin for 10 min at
room temperature. The migrated cells were assessed in six randomly
selected fields under an Olympus BX63F light microscope.
Statistical analysis
All data are presented as the mean ± SD. SPSS
Statistics 20.0 (IBM Corp.) was used for statistical analyses.
Comparisons were performed using one-way ANOVA followed by
Dunnett's test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Effects of carvedilol on body weight
and biochemical parameters of CCl4-intoxicated rats
The body weight of the CCl4-intoxicated
group was significantly lower compared with the control group
(P<0.01; Table I). The body
weight of the CCl4 + carvedilol-treated group was lower
compared with the CCl4-intoxicated group (P<0.05;
Table I). Compared with the
control group, CCl4 treatment significantly increased
the serum levels of ALT and AST (P<0.01), whereas co-treatment
with carvedilol decreased the elevated levels of ALT and AST
(P<0.05 and P<0.01, respectively; Table I). Compared with the control group,
CCl4 significantly decreased the serum level of ALB
(P<0.01; Table I); however, not
significant difference was identified in the levels of ALB between
the CCl4-intoxicated and CCl4 +
carvedilol-treated groups.
 | Table I.Effects of CARV on body weight and
biochemical parameters in CCl4-intoxicated cirrhotic
rats. |
Table I.
Effects of CARV on body weight and
biochemical parameters in CCl4-intoxicated cirrhotic
rats.
| Parameter | Control (n=7) |
CCl4-intoxicated (n=6) | CCl4 +
CARV (n=6) |
|---|
| Bodyweight (g) | 434.3±21.9 |
380.0±25.8a |
341.8±40.5b |
| ALT (U/l) | 50.98±12.76 |
99.04±14.04a |
79.76±11.68b |
| AST (U/l) | 90.45±12.87 |
170.45±42.50a |
118.65±21.70c |
| ALB (g/l) | 27.63±2.89 |
22.53±1.20a | 21.20±1.84 |
Carvedilol improves
CCl4-induced structural distortion and fibrosis in the
liver
Histological differences among the groups were
demonstrated by H&E staining of liver sections. Liver sections
of the control group exhibited normal lobular architecture, whereas
liver sections of the CCl4-intoxicated group exhibited
typical architectural distortions with regenerative nodules
surrounded by proliferative connective tissue; co-treatment with
carvedilol notably improved the architectural destruction induced
by CCl4 (Fig. 1A). S-R
staining revealed that, compared with control group rats,
CCl4 treatment resulted in excessive collagen deposition
in cirrhotic livers, which was notably reduced by co-treatment with
carvedilol (Fig. 1B). Collagen
deposition was quantified by the picrosirius-polarization method.
The collagen-positive area to total area ratio in the
CCl4-intoxicated group was significantly higher compared
with the control group (P<0.01; Fig. 1C), whereas co-treatment with
carvedilol significantly lowered the CCl4-induced
collagen accumulation (P<0.05).
Carvedilol inhibits HSC activation in
vivo
α-SMA is a marker of HSC activation (8). Immunohistochemical assay results
demonstrated that the expression of α-SMA was notably higher in
liver tissues of the CCl4-intoxicated group compared
with the control group, whereas co-treatment with carvedilol
suppressed the CCl4-induced increase of α-SMA (Fig. 2A). Compared with the control group,
the protein expression of α-SMA was significantly upregulated by
CCl4 (P<0.01; Fig.
2B), determined by western blotting, and co-treatment with
carvedilol decreased the upregulated α-SMA protein expression
levels (P<0.01). These results demonstrated that carvedilol may
have an inhibitory effect on HSC activation in cirrhotic livers of
rats.
Carvedilol inhibits HSC activation in
vitro
LX-2 cells exhibit an activated HSC phenotype, which
was assessed as previously described (9). LX-2 cells were treated with
carvedilol at concentrations of 0, 1, 2, 5 and 10 µM for 24 h.
α-SMA protein expression in LX-2 cells was significantly reduced by
carvedilol at 5 and 10 µM compared with untreated control cells
(P<0.01; Fig. 2C). This result
further demonstrated that carvedilol may have an inhibitory effect
on HSCs activation in in vitro.
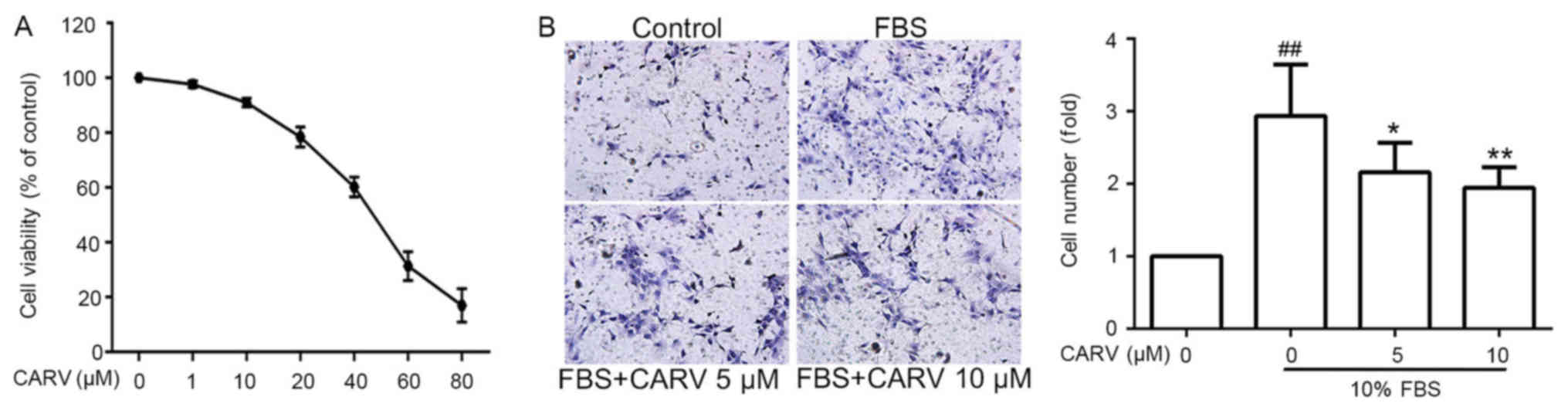
Carvedilol inhibits HSC
proliferation
LX-2 cells were treated with carvedilol at
concentrations ranging from 1 to 80 µM for 24 h. Carvedilol
inhibited the proliferation of LX-2 cells in a dose-dependent
manner (Fig. 3A). Carvedilol
concentrations of ≤10 µM were selected for subsequent cell
experiments as carvedilol did not significantly reduce cell
viability at this concentration.
Carvedilol decreases the invasive
ability of HSCs
The number of migrated FBS-induced LX-2 cells was
significantly higher compared with the non-induced control group
(P<001; Fig. 3B), whereas
FBS-induced migration was significantly inhibited by carvedilol at
concentrations of 5 and 10 µM (P<0.05 and P<0.01,
respectively). This result demonstrated that carvedilol may inhibit
the invasive ability of HSCs.
Carvedilol inhibits TGFβ1-induced
collagen synthesis of HSCs
LX-2 cells were seeded in 6-well plates and
incubated overnight. The cells were stimulated with TGFβ1 at
concentrations of 10, 20, 30 and 50 ng/ml for 24 h. Compared with
the control group, stimulation with TGFβ1 significantly increased
the protein expression levels of Col I and FN (P<0.05 and
P<0.01, respectively; Fig. 4A).
TGFβ1 (20 ng/ml) was used to stimulate LX-2 cells for 24 h with or
without co-treatment with carvedilol. The TGFβ1-induced
upregulation of Col I and FN was downregulated by carvedilol at 5
and 10 µM (P<0.05 and P<0.01, respectively; Fig. 4B).
Carvedilol inhibits TGFβ1-induced HSC
collagen synthesis via the TGFβ1/SMAD pathway
Signaling molecules downstream of TGFβ1 were
screened in LX-2 cells treated with TGFβ1 (20 ng/ml) for 0.5, 1 and
2 h (Fig. 5). AKT and ERK
phosphorylation levels did not change significantly (Fig. 5B and C, respectively), whereas the
expression levels of p-SMAD2 and p-SMAD3 were upregulated in LX-2
cells stimulated with TGFβ1 compared with the untreated control
(P<0.0; Fig. 5D and E,
respectively).
TGFβ1 was used to stimulate LX-2 cells for 0.5 h
with or without co-treatment with carvedilol. The phosphorylation
of SMAD2 and SMAD3 induced by TGFβ1 was suppressed by pretreatment
with carvedilol at concentrations of 5 and 10 µM (P<0.05 and
P<0.01, respectively; Fig. 6A and
B). SIS3 is a potent and selective inhibitor of SMAD3 (20). TGFβ1 was used to stimulate LX-2
cells for 24 h with or without co-treatment with 10 µM SIS3.
Pretreatment with SIS3 significantly decreased the TGFβ1-induced
upregulation of Col I and FN (P<0.01; Fig. 6C).
Discussion
Recent advances in our understanding of the
pathophysiology of PHT and liver cirrhosis has resulted in improved
management for patients with cirrhosis (2). As a novel NSBB, carvedilol reduces
portal pressure more effectively than traditional NSBBs, such as
propranolol and nadolol (21). For
patients with compensated cirrhosis, the goal of treatment is to
delay the development of liver cirrhosis and PHT. Therefore, the
effects of carvedilol on liver cirrhosis were explored in
vivo and in vitro to discover the potential role of
carvedilol in treating early-stage liver cirrhosis.
Previous studies have reported that carvedilol has
antioxidant, anti-proliferative, anti-inflammatory, anti-angiogenic
and antifibrogenic effects (14,15,22–24).
Hamdy et al demonstrated that carvedilol had potent
antifibrotic effects in chronic CCl4-induced liver
damage, but the underlying mechanisms were not described (25). In the present study, carvedilol not
only improved the hepatotoxicity indicators, but also improved
hepatic architectural distortion and liver fibrosis in cirrhotic
rats. Structural changes are important factors in the development
of IHVR, which is the determinant factor of PHT (6). Previous studies have demonstrated
that improvement of liver fibrosis can reduce portal pressure
(17,26). Therefore, the antifibrogenic effect
of carvedilol may be involved in reducing portal pressure. HSCs are
the target of the antifibrogenic therapy for hepatic fibrosis
(27,28). A number of agents targeting
activated HSCs have demonstrated their antifibrogenic effect in
animal models (29–32). Preventing HSC activation,
proliferation, migration and collagen synthesis are major
objectives in the treatment of liver fibrosis (27,28,33).
The present study demonstrated that carvedilol may exert
antifibrogenic effects in cirrhotic rats by inhibiting the
proliferation, migration, activation and collagen synthesis in
HSCs.
HSCs are the resident perisinusoidal cells in the
space of Disse, and are the central effector in hepatic fibrosis
(7). In response to liver injury,
HSCs are activated and undergo phenotypic transformation to a
myofibroblastic phenotype characterized by proliferation, migration
to sites of injury, increased production of profibrogenic
cytokines, and elevated accumulation of ECM components including
Col I and FN (10,34). Among the profibrogenic cytokines,
TGFβ1 is the most potent profibrogenic cytokine; TGFβ1 promotes the
accumulation of ECM proteins in the progression of liver fibrosis
(12). TGFβ1 was used in the
present study to stimulate LX-2 cells to explore the mechanisms
underlying the antifibrogenic effect of carvedilol. The results
demonstrated that TGFβ1 upregulated the collagen synthesis in LX-2
cells, which was consistent with previous studies (12,35).
The present study results demonstrated that
pretreatment with carvedilol decreased TGFβ1-induced collagen
synthesis in LX-2 cells. TGFβ1 activates SMAD-dependent and
SMAD-independent pathways, including PI3K/AKT and MAPK pathways
such as ERK (36–38). The TGFβ/SMAD pathway is a major
signaling pathway in the liver in both normal and pathological
conditions (36,37). In the SMAD-dependent pathway,
members of the SMAD family transmit signals from the cell surface
into the nucleus. Following stimulation by TGFβ1, SMAD 2 and SMAD3
are phosphorylated and form a heterotrimeric complex with SMAD4
(36,37). This complex translocates into the
nucleus and regulates the expression of target genes (36,37).
The results of the present study revealed that TGFβ1 activated the
SMAD-dependent pathway in LX-2 cells, and pretreatment with
carvedilol decreased the TGFβ1-induced phosphorylation of SMAD2 and
SMAD3. As a potent and selective inhibitor of SMAD3, SIS3 blocked
the upregulation of collagen synthesis in TGFβ1-stimulated LX-2
cells. These results demonstrated that carvedilol may reduce the
TGFβ1-induced increase of collagen synthesis in LX-2 cells by
inhibiting the TGFβ1/SMAD pathway.
In conclusion, the present study demonstrated that
carvedilol may improve liver cirrhosis in rats by inhibiting HSCs
proliferation, invasion, activation and collagen synthesis.
Furthermore, carvedilol may inhibit collagen synthesis in HSCs by
suppressing the TGFβ1/SMAD pathway. Therefore, the application of
carvedilol in chronic liver diseases may be extended beyond the
pharmacological treatment of PHT in patients with decompensated
cirrhosis, and carvedilol may be applied in the treatment of
early-stage liver cirrhosis.
Acknowledgements
Not applicable.
Funding
The present study was supported by The National
Nature Science Foundation of China (grant no. 81370590).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
LL performed experiments and wrote the manuscript.
GL and GW performed the experiments. DM and ZL maintained the
animals and established the liver cirrhosis animal model. CZ
designed the study. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
All experimental procedures and protocols were
approved by the Animal Medical Ethics Committee of Shandong
Provincial Hospital affiliated to Shandong University (Jinan,
China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Fernandez M: Molecular pathophysiology of
portal hypertension. Hepatology. 61:1406–1415. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bosch J, Groszmann RJ and Shah VH:
Evolution in the understanding of the pathophysiological basis of
portal hypertension: How changes in paradigm are leading to
successful new treatments. J Hepatol. 62:S121–S130. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Reiberger T, Ferlitsch A, Payer BA, Pinter
M, Homoncik M and Peck-Radosavljevic M; Vienna Hepatic Hemodynamic
Lab, : Non-selective β-blockers improve the correlation of liver
stiffness and portal pressure in advanced cirrhosis. J
Gastroenterol. 47:561–568. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bosch J, Abraldes JG, Fernández M and
García-Pagán JC: Hepatic endothelial dysfunction and abnormal
angiogenesis: New targets in the treatment of portal hypertension.
J Hepatol. 53:558–567. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Iwakiri Y and Groszmann RJ: Vascular
endothelial dysfunction in cirrhosis. J Hepatol. 46:927–934. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sanyal AJ, Bosch J, Blei A and Arroyo V:
Portal hypertension and its complications. Gastroenterology.
134:1715–1728. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lee UE and Friedman SL: Mechanisms of
hepatic fibrogenesis. Best Pract Res Clin Gastroenterol.
25:195–206. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Senoo H, Yoshikawa K, Morii M, Miura M,
Imai K and Mezaki Y: Hepatic stellate cell (vitamin A-storing cell)
and its relative-past, present and future. Cell Biol Int.
34:1247–1272. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xu L, Hui AY, Albanis E, Arthur MJ,
O'Byrne SM, Blaner WS, Mukherjee P, Friedman SL and Eng FJ: Human
hepatic stellate cell lines, LX-1 and LX-2: New tools for analysis
of hepatic fibrosis. Gut. 54:142–151. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Reynaert H, Thompson MG, Thomas T and
Geerts A: Hepatic stellate cells: Role in microcirculation and
pathophysiology of portal hypertension. Gut. 50:571–581. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Friedman SL: Molecular regulation of
hepatic fibrosis, an integrated cellular response to tissue injury.
J Biol Chem. 275:2247–2250. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bissell DM, Roulot D and George J:
Transforming growth factor beta and the liver. Hepatology.
34:859–867. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
de Franchis R; Baveno VI Faculty, :
Expanding consensus in portal hypertension: Report of the Baveno VI
Consensus Workshop: Stratifying risk and individualizing care for
portal hypertension. J Hepatol. 63:743–752. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Arozal W, Sari FR, Watanabe K, Arumugam S,
Veeraveedu PT, Ma M, Thandavarayan RA, Sukumaran V, Lakshmanan AP,
Kobayashi Y, et al: Carvedilol-afforded protection against
daunorubicin-induced cardiomyopathic rats in vivo: Effects on
cardiac fibrosis and hypertrophy. ISRN Pharmacol. 2011:4305492011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wong VY, Laping NJ, Nelson AH, Contino LC,
Olson BA, Gygielko E, Campbell WG Jr, Barone F and Brooks DP:
Renoprotective effects of carvedilol in hypertensive-stroke prone
rats may involve inhibition of TGF beta expression. Br J Pharmacol.
134:977–984. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
El-Demerdash E, Abdel-Sattar SA, El-Bakly
WM and Mohamed EA: Antifibrotic effects of carvedilol and impact of
liver fibrosis on carvedilol pharmacokinetics in a rat model. Eur J
Drug Metab Pharmacokinet. 42:767–779. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Krogsgaard K, Gluud C, Henriksen JH and
Christoffersen P: Correlation between liver morphology and portal
pressure in alcoholic liver disease. Hepatology. 4:699–703. 1984.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Starkel P and Leclercq IA: Animal models
for the study of hepatic fibrosis. Best Pract Res Clin
Gastroenterol. 25:319–333. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Andrade GB, Montes GS, Conceição GM and
Saldiva PH: Use of the Picrosirius-polarization method to age
fibrotic lesions in the hepatic granulomas produced in experimental
murine schistosomiasis. Ann Trop Med Parasitol. 93:265–272. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jinnin M, Ihn H and Tamaki K:
Characterization of SIS3, a novel specific inhibitor of Smad3, and
its effect on transforming growth factor-beta1-induced
extracellular matrix expression. Mol Pharmacol. 69:597–607. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tripathi D and Hayes PC: Beta-blockers in
portal hypertension: New developments and controversies. Liver Int.
34:655–667. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Carlson W and Oberg K: Clinical
pharmacology of carvedilol. J Cardiovasc Pharmacol Ther. 4:205–218.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ding Q, Tian XG, Li Y, Wang QZ and Zhang
CQ: Carvedilol may attenuate liver cirrhosis by inhibiting
angiogenesis through the VEGF-Src-ERK signaling pathway. World J
Gastroenterol. 21:9566–9576. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Barone FC, Campbell WG Jr, Nelson AH and
Feuerstein GZ: Carvedilol prevents severe hypertensive
cardiomyopathy and remodeling. J Hypertens. 16:871–884. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hamdy N and El-Demerdash E: New
therapeutic aspect for carvedilol: Antifibrotic effects of
carvedilol in chronic carbon tetrachloride-induced liver damage.
Toxicol Appl Pharmacol. 261:292–299. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Di Pascoli M, Divi M, Rodriguez-Vilarrupla
A, Rosado E, Gracia-Sancho J, Vilaseca M, Bosch J and García-Pagán
JC: Resveratrol improves intrahepatic endothelial dysfunction and
reduces hepatic fibrosis and portal pressure in cirrhotic rats. J
Hepatol. 58:904–910. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wu J and Zern MA: Hepatic stellate cells:
A target for the treatment of liver fibrosis. J Gastroenterol.
35:665–672. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Schuppan D and Popov Y: Hepatic fibrosis:
From bench to bedside. J Gastroenterol Hepatol. 17 (Suppl
3):S300–S305. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kaji K, Yoshiji H, Ikenaka Y, Noguchi R,
Aihara Y, Douhara A, Moriya K, Kawaratani H, Shirai Y, Yoshii J, et
al: Dipeptidyl peptidase-4 inhibitor attenuates hepatic fibrosis
via suppression of activated hepatic stellate cell in rats. J
Gastroenterol. 49:481–491. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li J, Li X, Xu W, Wang S, Hu Z, Zhang Q,
Deng X, Wang J, Zhang J and Guo C: Antifibrotic effects of luteolin
on hepatic stellate cells and liver fibrosis by targeting
AKT/mTOR/p70S6K and TGFβ/Smad signalling pathways. Liver Int.
35:1222–1233. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhu J, Wu J, Frizell E, Liu SL, Bashey R,
Rubin R, Norton P and Zern MA: Rapamycin inhibits hepatic stellate
cell proliferation in vitro and limits fibrogenesis in an in vivo
model of liver fibrosis. Gastroenterology. 117:1198–1204. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Son G, Hines IN, Lindquist J, Schrum LW
and Rippe RA: Inhibition of phosphatidylinositol 3-kinase signaling
in hepatic stellate cells blocks the progression of hepatic
fibrosis. Hepatology. 50:1512–1523. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Trautwein C, Friedman SL, Schuppan D and
Pinzani M: Hepatic fibrosis: Concept to treatment. J Hepatol.
62:S15–S24. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Greuter T and Shah VH: Hepatic sinusoids
in liver injury, inflammation, and fibrosis: New pathophysiological
insights. J Gastroenterol. 51:511–519. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kajdaniuk D, Marek B, Borgiel-Marek H and
Kos-Kudła B: Transforming growth factor β1 (TGFβ1) in physiology
and pathology. Endokrynol Pol. 64:384–396. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Inagaki Y and Okazaki I: Emerging insights
into transforming growth factor beta Smad signal in hepatic
fibrogenesis. Gut. 56:284–292. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Derynck R and Zhang YE: Smad-dependent and
Smad-independent pathways in TGF-beta family signalling. Nature.
425:577–584. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bakin AV, Tomlinson AK, Bhowmick NA, Moses
HL and Arteaga CL: Phosphatidylinositol 3-kinase function is
required for transforming growth factor beta-mediated epithelial to
mesenchymal transition and cell migration. J Biol Chem.
275:36803–36810. 2000. View Article : Google Scholar : PubMed/NCBI
|