Introduction
Phagocytosis, first described by Metchnikoff >100
years ago, is the engulfing and internalizing of particles with a
size of ≥0.5 µm by cells. Unlike micropinocytosis, phagocytosis is
initiated by the recognition and binding of cell surface receptors
to target particles (1,2). Professional phagocytes, including
macrophages, neutrophils and dendritic cells, participate in the
first line of defense against infection by clearing pathogens from
the sites of infection (3).
Nonprofessional phagocytes, such as epithelial and endothelial
cells, and fibroblasts, do not internalize pathogens, but clear
senescent cells by engulfing apoptotic bodies (4,5).
Therefore, nonprofessional phagocytes avoid the release of
inflammatory contents of apoptotic cells in this way, and thus
serve an important role in maintaining tissue homeostasis.
Recently, Juncadella et al (6) reported that the function of airway
epithelial cells to phagocytose apoptotic cells was attenuated in
asthmatic mice, thereby forming interleukin (IL)-33-dependent
allergic airway inflammation. This indicates that the engulfment of
apoptotic cells by airway epithelial cells is an important
mechanism for the regression of airway inflammation.
Insulin-like growth factor 1 (IGF-1) is a small
7.6-kDa peptide that exhibits 50% homology with insulin. It is a
hormone with local activity following autocrine or paracrine
secretion, and regulates cell survival, metabolism, proliferation
and differentiation (7,8). IGF-1 is predominantly produced in the
liver, while it is also produced in certain other organs, such as
the lungs, where it is elevated under conditions of acute lung
injury, pulmonary fibrosis and asthma (9). IGF-1 promotes the proliferation and
differentiation of alveolar epithelial cells during the repair of
hypoxia-induced lung injury (10).
Although IGF-1 has been demonstrated to promote the phagocytic
activity of mouse macrophages (11), its effects on nonprofessional
phagocytes, such as alveolar epithelial cells, have not been
described to date. Recently, it has been reported that the
concentration of IGF-1 was elevated in the lung tissues of
asthmatic mice (12).
As the engulfment of apoptotic cells by the airway
epithelium affects the progression of airway inflammation in
asthmatic mice, the effect of IGF-1 on the phagocytic function of
alveolar epithelial cells was investigated in the present study.
The study revealed that IGF-1 inhibited the phagocytosis of
fluorescent microspheres and apoptotic cells by MLE-12 alveolar
epithelial cells and by mouse alveolar epithelial cells from
primary cultures. IGF-1 was elevated in the lung tissue and
bronchoalveolar lavage fluid (BALF) of asthmatic mice, whereas
IGF-1 blockade promoted phagocytosis by alveolar epithelial cells
and reduced airway inflammation.
Materials and methods
Cell culture
Primary alveolar epithelial cells and MLE-12
alveolar epithelial cells (Shanghai Jining Shine Biotechnology
Corporation) were cultured in Dulbecco's modified Eagle's medium
with 10% FBS (HyClone; GE Healthcare Life Sciences) containing 1%
penicillin and streptomycin (Beyotime Institute of Biotechnology)
in a 5% CO2 incubator at 37°C. When the cultures reached
80–90% confluency, the cells were harvested with 0.25% pancreatin
for subculturing MLE-12 cells. The cells were passaged every 1–2
days.
Animal model and treatment
protocol
A total of 30 female BALB/c mice were purchased from
the Experimental Animal Center of Bengbu Medical College. The mice
(4-weeks-old) were housed in pathogen-free grade conditions with
60–70% humidity under a 12 h light/dark cycle at 25°C with free to
access food and water. The process for establishing an asthma model
is illustrated in Fig. 1. Briefly,
mice weighing 15–30 g were intraperitoneally injected with 200 µl
of a sensitizing solution containing 50 mg ovalbumin
(Sigma-Aldrich; Merck KGaA) and 2 mg aluminum hydroxide (Thermo
Fisher Scientific, Inc.) on days 0, 7 and 14. Beginning on day 21,
the mice were placed in an inhalation chamber and treated with
aerosolized 5% ovalbumin for 30 min once a day for 1 week. Control
mice received a mock challenge with phosphate-buffered saline
(PBS). To deplete alveolar macrophages, asthma model mice received
2-chloroadenosine (2-CA; Sigma-Aldrich; Merck KGaA) administered in
four doses of 1 µg/20 µl once every 3 days, beginning on day 20.
For IGF-1 blockade, asthma model mice were administered anti-IGF-1
antibody (1:10; cat. no. ab9572; Abcam) by intranasal drip in a
dose of 1 µg/20 µl once every 3 days, beginning on day 20. Animal
experiments in the present study were approved by the Ethics
Committee of the Bengbu Medical College (Bengbu, China).
Collection of BALF and alveolar
macrophages
Mice were anesthetized with 1% chloral hydrate prior
to tracheal intubation, and bronchoalveolar lavage with 0.6 ml PBS
was performed for six consecutive times. The BALF samples obtained
from each lavage were centrifuged at 233 × g for 5 min at 4°C. The
supernatant was collected for cytokine assays. Next, the red blood
cells were lysed, and the pellet was resuspended in RPMI-1640
medium (HyClone; GE Healthcare Life Sciences) and then seeded onto
6-well plates at a final density of 5×105 cells/well.
The cells were allowed to adhere for at least 2 h before washing
away the nonadherent cells to expose the adherent alveolar
macrophages. The purity of the isolated macrophages was
>95%.
Isolation of primary alveolar
epithelial cells
Mice were anesthetized with chloral hydrate prior to
tracheal intubation and injection of 10–15 ml of air directly to
the lungs. The pulmonary artery was lavaged with PBS, and
bronchoalveolar lavage was performed through the trachea until the
lavage fluid was clear and transparent. Intrapulmonary digestion
was performed by two successive injections of 15 ml 0.5% trypsin
into the lungs for 10 min each. Digestion was stopped with
RPMI-1640 medium containing 20% fetal calf serum (FCS). The trachea
and bronchi were removed, following which the lung tissue was
dissected and placed in RPMI-1640 medium. The lung tissue was cut
into sections of <1 mm3 in size, ground between
frosted glass sheets, and passed through 200 mesh and 400 mesh
screens to obtain a suspension of primary alveolar epithelial
cells.
Induction and assay of apoptotic
cells
MLE-12 and primary alveolar epithelial cells
(4×105 cells/well) were stimulated with 0.25 µM curcumin
(Sigma-Aldrich; Merck KGaA) for 72 h. Subsequently, the cells were
stained for 20 min with Annexin V-fluorescein isothiocyanate (FITC)
and propidium iodide using an assay kit (Beyotime Institute of
Biotechnology), following the manufacturer's protocol. Apoptosis
was assayed by flow cytometry (FACSCalibur; BD Biosciences,
Franklin Lakes, NJ, USA) within 1 h of staining.
Phagocytosis assay
MLE-12 and freshly isolated alveolar epithelial
cells (5×104 cells/well) were stimulated for 48 h with
100, 200, 300, 500 and 1,000 ng/ml IGF-1 (Abcam). The cells were
then incubated with 1 µl of a suspension of 4.55×107
fluorescent microspheres (1 µm in diameter) for 2 h before washing
twice in PBS with 5% FCS and fixing in 4% paraformaldehyde (PFA).
Cells that had induced phagocytosis of fluorescent microspheres
were detected with a FACSCalibur flow cytometer.
Furthermore, to assay the phagocytosis of apoptotic
cells, activated MLE-12 or primary alveolar epithelial cells
(5×104 cells/well) were seeded into 6-well plates, and
cocultured for 4 h with FITC-stained apoptotic MLE-12 cells. The
cells were then washed twice with PBS containing 5% FCS and fixed
with 4% PFA. The phagocytosis of FITC-labeled apoptotic cells was
detected by flow cytometry.
Western blot assay
Isolated mouse lung tissue was washed with PBS, and
protein extracts were prepared with NP-40 cell lysis buffer
(Beyotime Institute of Biotechnology). The protein concentration of
the cell lysates was measured using a bicinchoninic acid protein
assay kit (Beyotime Institute of Biotechnology). Following
separation by 12% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis, the protein samples were transferred to
polyvinylidene difluoride (PVDF) membranes. Next, the PVDF
membranes were blocked in Tris-buffered saline and Tween 20 (TBST)
with 5% skim milk powder at 25°C for 2 h. The membranes were washed
with TBST solution and incubated overnight at 4°C with IGF-1
(1:1,000; cat. no. ab9572; Abcam) and β-actin (1:1,000; cat. no.
AF0003; Beyotime Institute of Biotechnology) primary antibodies.
Membranes were washed and then incubated with horseradish
peroxidase-conjugated goat anti-mouse secondary antibody (1:1,000;
cat. no. A0208; Beyotime Institute of Biotechnology) at 25°C for 2
h. Subsequently, the protein bands were read by enhanced
chemiluminescence using a BeyoECL Plus kit, and strip density
analysis was performed with ImageJ software 6.0 (https://imagej.nih.gov/ij/).
Enzyme-linked immunosorbent assay
(ELISA)
The content of IGF-1 and IL-33 in the BALF
supernatants, as well as IGF-1 in lung tissues were assayed using
commercially available ELISA kits (cat. nos. K02016571 and
M27131086; Cusabio Biotech Co., Ltd.), following the manufacturer's
protocol.
Lung histology
The mouse lungs were surgically obtained from mice
24 h after final antigen or mock challenge. Lung tissue was fixed
at 25°C for 1 week in 4% PFA, dehydrated in a graded alcohol
series, embedded in paraffin and sectioned at 5 µm. Following
dewaxing and rehydration, the sections were stained with
hematoxylin-eosin, and observed and photographed under a light
microscope.
Statistical analysis
The results are expressed as the mean ± standard
deviation. Similar results were obtained from three independent
experiments. Statistical analysis was performed with SPSS software,
version 16.0 (SSPS, Inc., Chicago, IL, USA). One-way analysis of
variance was conducted to evaluate multiple group comparisons,
while Student's t-tests were used to evaluate comparisons between
two groups. P-values of <0.05 were considered to denote
statistically significant differences.
Results
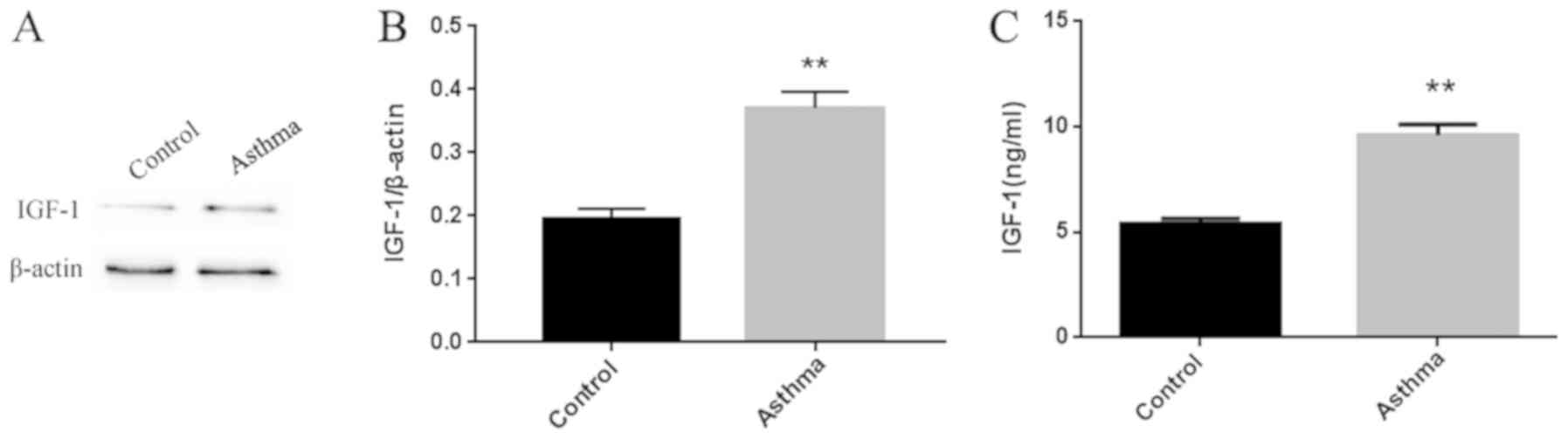
Increased IGF-1 protein expression in
the lungs of asthmatic mice
As shown in Fig.
2A, IGF-1 protein expression was significantly higher in the
lung tissue of asthmatic mice as compared with that in normal
control mice. IGF-1 protein expression was also markedly higher in
the BALF of asthmatic mice compared with that in normal mice
(Fig. 2B).
Elevated IGF-1 expression in the lungs
of asthmatic mice primarily results from synthesis in alveolar
macrophages
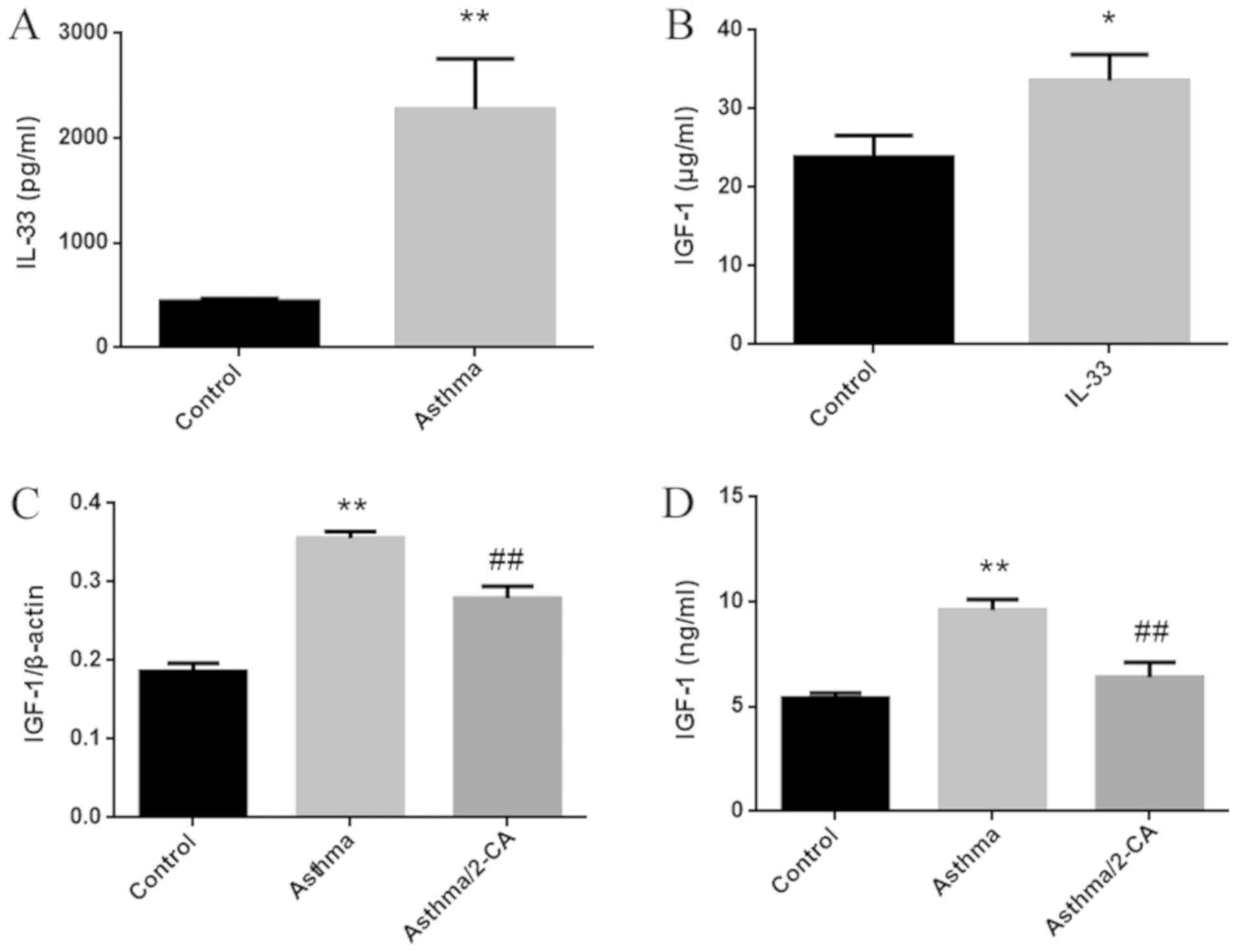
IL-33, a cytokine that promotes airway inflammation,
is elevated in the serum of asthma patients, and is associated with
disease severity (13). In the
current study, IL-33 was significantly elevated in the BALF of
asthma model mice (Fig. 3A). IGF-1
expression was also significantly increased in primary alveolar
macrophages stimulated by IL-33 in vitro (Fig. 3B). Following the depletion of
alveolar macrophages by 2-CA, the elevation of IGF-1 expression in
the lung tissues of asthmatic mice decreased by 50%, while the
elevation of IGF-1 in the BALF almost completely disappeared
(Fig. 3C and D). The results of
Fig. 3C and D indicated that the
increase in IGF-1 in the lung tissues of asthma model mice
primarily resulted from synthesis in alveolar macrophages.
IGF-1 inhibits the phagocytic activity
of alveolar epithelial cells
The phagocytosis of apoptotic cells by alveolar
epithelial cells has been demonstrated to reduce airway
inflammation, and IGF-1 significantly inhibited the phagocytosis of
MLE-12 and primary alveolar epithelial cells. The results of the
treatment of MLE-12 cells with 100–1,000 ng/ml IGF-1 on the
phagocytosis of fluorescent microspheres are displayed in Fig. 4A and B. After 12 h of treatment,
the phagocytic activity of MLE-12 cells significantly decreased at
doses of 200, 300 and 500 ng/ml, with the inhibitory effect
reaching a peak at 300 ng/ml IGF-1. Following stimulation of MLE-12
cells with 100 ng/ml curcumin for 24 h, the percentage of cells in
late apoptosis was 97.8% compared with that of 2.95% for
unstimulated MLE-12 cells (Fig.
4C). Upon treatment with 300 ng/ml IGF-1 for 4 h, the
phagocytosis decreased to 32.6% (Fig.
4D and E). However, when treatment with 300 ng/ml IGF-1 was
performed for 12 h, it resulted in a significant reduction in the
phagocytosis of fluorescent microspheres and apoptotic cells by
primary alveolar epithelial cells (Fig. 4F-I).
 | Figure 4.IGF-1 inhibits phagocytosis of MLE-12
and primary alveolar epithelial cells. (A) Percentage of
phagocytosis of fluorescent microspheres, and (B) the corresponding
flow cytometry results. MLE-12 cells were stimulated for 48 h with
100, 200, 300, 500 or 1000 ng/ml IGF-1, incubated with fluorescent
microspheres (4.55×107 particles) for 12 h and then
examined by flow cytometry to assess the phagocytosis. (C) MLE-12
cells were stimulated with curcumin for 72 h, stained with Annexin
V-FITC and propidium iodide, and then apoptosis was assayed by flow
cytometry. (D) Phagocytosis of apoptotic cells, and (E) the
corresponding flow cytometry results of MLE-12 cells stimulated for
6 h with 300 ng/ml IGF-1. Following treatment, the cells were
cocultured with FITC-stained apoptotic MLE-12 cells for 4 h, and
then apoptosis was assayed by flow cytometry. (F) Phagocytosis of
fluorescent microspheres and (G) corresponding flow cytometry
results, as well as the (H) phagocytosis of apoptotic cells and (I)
corresponding flow cytometry results, of primary alveolar
epithelial cells treated with 300 ng/ml IGF-1 for 12 h. Following
treatment, the cells were cocultured with fluorescent microspheres
or apoptotic MLE-12 cells for 4 h, and then assessed by flow
cytometry. Data are expressed as the mean ± standard deviation of
three independent experiments. *P<0.05 and **P<0.01 vs.
unstimulated control group. IGF-1, insulin-like growth factor
1. |
Antibody blocking of IGF-1 antibody
promotes the phagocytic activity of primary alveolar epithelial
cells and reduces lung inflammation in asthma model mice
Anti-IGF-1 blocking antibody was administered by
nasal drops during the development of the asthma model.
Subsequently, primary alveolar epithelial cells were isolated from
mouse lung tissue, and their phagocytic activity was assayed with
fluorescent microspheres and apoptotic cells. Blocking IGF-1 in the
asthma model mice significantly enhanced the phagocytosis of
fluorescent microspheres and apoptotic cells by primary alveolar
epithelial cells (Fig. 5A and B).
Compared with the normal mice, the asthmatic mice exhibited
extensive lung inflammation with distinct perivascular and
peribronchial cuffing. Intranasal instillation of IGF-1 blocking
antibody significantly decreased the infiltration of inflammatory
cells around the airways and blood vessels, and decreased the
thickness of the bronchial mucosa and airway secretions (Fig. 5C). Hence, IGF-1 blockade also
significantly reduced lung airway inflammation in asthma model
mice.
Discussion
In the mouse model established in the present study,
IGF-1 levels were found to be significantly elevated in the lung
tissues and BALF of asthmatic mice compared with the normal
controls. IGF-1 is a peptide of approximately 70 amino acids
containing four domains; it mediates cell growth, metabolism,
proliferation and differentiation following binding to the IGF-1
receptor (IGF-1R) (8,14). It is active in lung development and
various disease states, such as inflammation, fibrosis and tumors
(9). Numerous studies have
reported increased IGF-1 mRNA expression in intrabronchial biopsy
tissue obtained from asthmatic patients and patients with tracheal
epithelial fibrosis (15). In
addition, Yao et al (12)
reported increased IGF-1 protein expression, assayed by
immunohistochemistry, in lung tissues from asthma model mice. The
results of the present study are consistent with previous reports,
all of which support further study of IGF-1 as a therapeutic target
in asthma.
IL-33 was found to be elevated in the BALF of asthma
model mice in the current study (Fig.
3A). It is known that IL-33 promotes systemic T helper 2 (Th2)
cell responses and is constitutively expressed in a variety of
tissues, including the airways of asthmatic patients, particularly
those with severe disease (16,17).
IL-33 activity is mediated by binding to its ST2 receptor, which is
present on macrophages (18).
IL-33 binding to alveolar macrophages can increase the expression
of the mannose receptor IL-4Ra, as well as the production of C-C
motif chemokine ligand 24 (CCL24) and CCL17, thus contributing to
allergic inflammation (19,20).
In the asthma model mice investigated in the present study
(Fig. 3B), IL-33 promoted IGF-1
expression by alveolar macrophages. IGF-1 production has been
reported in endothelial cells, epithelial cells, fibroblasts and
macrophages, among others (21,22).
In addition, Wang et al (23) reported that epidermal T cells can
produce IGF-1. In the present study, the increase in IGF-1 that was
detected in the asthma model animals was mainly produced in
alveolar macrophages. This result is in line with the findings of
Fritz et al (24), who
demonstrated that alveolar macrophage-derived IGF-1 induced the
proliferation of lung epithelial cells.
In the asthma model established in the current
study, epithelial cells were nonprofessional phagocytes that helped
to maintain tissue homeostasis by clearing apoptotic bodies.
Allergens can stimulate the apoptosis of airway epithelial cells,
whereas the surrounding intact epithelial cells can phagocytose the
apoptotic cells and secrete anti-inflammatory cytokines. Airway
epithelial cells, thus, regulate inflammation by phagocytosis
(6). In the established model,
IGF-1 prevented the phagocytosis of apoptotic cells by alveolar
epithelial cells, thus increasing the release of inflammatory
contents from apoptotic cells, thereby aggravating airway
inflammation. Therefore, blocking IGF-1 improved airway
inflammation and lung histopathology in asthmatic mice.
IGF-1R-deficient mice are less susceptible to skin inflammation in
comparison with normal mice, indicating that IGF-1 signaling
promotes inflammation (25). A
previous study has reported reduced airway hyperresponsiveness,
mucus secretion and eosinophil infiltration of airway tissues in
IGF-1R-deficient asthmatic mice (26). These observations are consistent
with the involvement of IGF-1 in the development of asthma.
Aerobic training is recommended as an adjuvant
therapy for asthma patients, since it reduces the expression of
proinflammatory signals, including IGF-1 and peribronchial
leukocyte activation, leading to reduced airway inflammation and
Th2 responses (27,28). The results of the mouse model
described in the current study add to the evidence obtained from
other asthma models that IGF-1 is a proinflammatory signal. Such
evidence suggested that creatine supplementation promotes goblet
cell proliferation, and upregulates the expression levels of IL-5,
inducible nitric oxide synthase and proinflammatory mediators, such
as IGF-1, in epithelial cells (29,30).
To date, few studies have investigated the
association of IGF-1 signaling with phagocytosis. A study by Xiao
et al (31) did not
identify an effect of IGF-1 on the phagocytosis of peritoneal
macrophages. Furthermore, Dos Santos Reis et al (11) reported that IGF-1 promoted
phagocytosis by J774 macrophage cells. By contrast, in the current
study model, IGF-1 inhibited phagocytosis by MLE-12 alveolar
epithelial cells and primary alveolar epithelial cells. The
differences in the findings of these studies may be the result of
using different target cells.
In conclusion, the effects of IGF-1 on the
phagocytosis of alveolar epithelial cells may represent a novel
regulatory mechanism of airway inflammation, which is illustrated
in Fig. 6. The results suggested
that IL-33 was elevated in the airway of asthmatic mice and induced
IGF-1 production by alveolar macrophages. IGF-1 then prevented the
phagocytosis of apoptotic cells by alveolar epithelial cells and
increased the release of inflammatory contents of apoptotic cells,
thus leading to increased airway inflammation. Recurrent airway
inflammation leads to airway hyperresponsiveness and airway
remodeling, which exacerbates asthma. Nevertheless, in order to
further explain the role of IGF-1 in the pathogenesis of asthma, it
is necessary to construct an asthma model using IGF-1-deficient
mice, and then observe airway inflammation and airway remodeling,
which will be the focus of our future work. These efforts will
support IGF-1 as a potential therapeutic target for the treatment
of asthma.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
National Science Foundation of China (grant nos. 81273273 and
81801573), and the Anhui Provincial Natural Science Foundation
(grant nos. 1708085MH218 and 1808085QH253).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MM, FW, JH and XT performed the experiments. HM, SG
and CS conducted data interpretation and analysis. CS wrote the
manuscript. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Bengbu Medical College (Bengbu, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that there is no conflict of
interest.
References
|
1
|
Flannagan RS, Jaumouillé V and Grinstein
S: The cell biology of phagocytosis. Annu Rev Pathol. 7:61–98.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rosales C and Uribe-Querol E:
Phagocytosis: A fundamental process in immunity. Biomed Res Int.
2017:90428512017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gordon S: Phagocytosis: An immunobiologic
process. Immunity. 44:463–475. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shklover J, Levy-Adam F and Kurant E:
Apoptotic cell clearance in development. Curr Top Dev Biol.
114:297–334. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Alva-Murillo N, López-Meza JE and
Ochoa-Zarzosa A: Nonprofessional phagocytic cell receptors involved
in Staphylococcus aureus internalization. Biomed Res Int.
2014:5385462014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Juncadella IJ, Kadl A, Sharma AK,
Hochreiter-Hufford A, Borish L and Ravichandran KS: Apoptotic cell
clearance by bronchial epithelial cells critically influences
airway inflammation. Nature. 493:547–551. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yakar S and Adamo ML: Insulin-like growth
factor 1 physiology: Lessons from mouse models. Endocrinol Metab
Clin North Am. 41:231–247. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Troncoso R, Ibarra C, Vicencio JM,
Jaimovich E and Lavandero S: New insights into IGF-1 signaling in
the heart. Trends Endocrinol Metab. 25:128–137. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang Z, Li W, Guo Q, Wang Y, Ma L and
Zhang X: Insulin-like growth factor-1 signaling in lung development
and inflammatory lung diseases. Biomed Res Int.
2018:60575892018.PubMed/NCBI
|
|
10
|
Narasaraju TA, Chen H, Weng T, Bhaskaran
M, Jin N, Chen J, Chen Z, Chinoy MR and Liu L: Expression profile
of IGF system during lung injury and recovery in rats exposed to
hyperoxia: a possible role of IGF-1 in alveolar epithelial cell
proliferation and differentiation. J Cell Biochem. 97:984–998.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dos Santos Reis MD, Dos Santos YMO, de
Menezes CA, Borbely KSC and Smaniotto S: Resident murine macrophage
migration and phagocytosis are modulated by growth hormone. Cell
Biol Int. 42:615–623. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yao X, Wang W, Li Y, Huang P, Zhang Q,
Wang J, Wang W, Lv Z, An Y, Qin J, et al: IL-25 induces airways
angiogenesis and expression of multiple angiogenic factors in a
murine asthma model. Respir Res. 16:392015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
BahramiMahneh S, Movahedi M, Aryan Z,
Bahar MA, Rezaei A, Sadr M and Rezaei N; Universal Scientific
Education and Research Network (USERN), : Serum IL-33 is elevated
in children with asthma and is associated with disease severity.
Int Arch Allergy Immunol. 168:193–196. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jung HJ and Suh Y: Regulation of IGF-1
signaling by microRNAs. Front Genet. 5:4722015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hoshino M, Nakamura Y, Sim JJ, Yamashiro
Y, Uchida K, Hosaka K and Isogai S: Inhaled corticosteroid reduced
lamina reticularis of the basement membrane by modulation of
insulin-like growth factor (IGF)-I expression in bronchial asthma.
Clin Exp Allergy. 28:568–577. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Préfontaine D, Nadigel J, Chouiali F,
Audusseau S, Semlali A, Chakir J, Martin JG and Hamid Q: Increased
IL-33 expression by epithelial cells in bronchial asthma. J Allergy
Clin Immunol. 125:752–754. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Préfontaine D, Lajoie-Kadoch S, Foley S,
Audusseau S, Olivenstein R, Halayko AJ, Lemière C, Martin JG and
Hamid Q: Increased expression of IL-33 in severe asthma: Evidence
of expression by airway smoothmuscle cells. J Immunol.
183:5094–5103. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kurowska-Stolarska M, Stolarski B, Kewin P
Murphy G, Corrigan CJ, Ying S, Pitman N, Mirchandani A, Rana B, van
Rooijen N, et al: IL-33 amplifies the polarization of alternatively
activated macrophages that contribute to airway inflammation. J
Immunol. 183:6469–6477. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lloyd CM: IL-33 family members and
asthma-bridging innate and adaptive immune responses. Curr Opin
Immunol. 22:800–806. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nile CJ, Barksby E, Jitprasertwong P,
Preshaw PM and Taylor JJ: Expression and regulation of
interleukin-33 in human monocytes. Immunology. 130:172–180. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ahluwalia A, Jones MK, Hoa N and Tarnawski
AS: NGF protects endothelial cells from indomethacin-induced injury
through activation of mitochondria and upregulation of IGF-1. Cell
Signal. 40:22–29. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Szczęsny E, Slusarczyk J, Głombik K,
Budziszewska B, Kubera M, Lasoń W and Basta-Kaim A: Possible
contribution of IGF-1 to depressive disorder. Pharmacol Rep.
65:1622–1631. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang Y, Bai Y, Li Y, Liang G, Jiang Y, Liu
Z, Liu M, Hao J, Zhang X, Hu X, et al: IL-15 enhances activation
and IGF-1 production of dendritic epidermal T cells to promote
wound healing in diabetic mice. Front Immunol. 8:15572017.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fritz JM, Dwyer-Nield L and Malkinson AM:
Stimulation of neoplastic mouse lung cell proliferation by alveolar
macrophage-derived, insulin-like growth factor-1 can be blocked by
inhibiting MEK and PI3K activation. Mol Cancer. 10:762011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Knuever J, Willenborg S, Ding X Akyüz MD,
Partridge L, Niessen CM, Brüning JC and Eming SA: Myeloid
cell-restricted Insulin/IGF-1 receptor deficiency protects against
skin inflammation. J Immunol. 195:5296–5308. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Piñeiro-Hermida S, Gregory JA, López IP,
Torrens R, Ruíz-Martínez C, Adner M and Pichel JG: Attenuated
airway hyperresponsiveness and mucus secretion in HDM-exposed
Igf1r-deficientmice. Allergy. 72:1317–1326. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Vieira RP, Silva RA, Oliveira-Junior MC,
Greiffo FR, Ligeiro-Oliveira AP, Martins MA and Carvalho CR:
Exercise deactivates leukocytes in asthma. Int J Sports Med.
35:629–635. 2014.PubMed/NCBI
|
|
28
|
Vieira RP, de Andrade VF, Duarte AC, Dos
Santos AB, Mauad T, Martins MA, Dolhnikoff M and Carvalho CR:
Aerobic conditioning and allergic pulmonary inflammation in mice.
II. Effects on lung vascular and parenchymal inflammation and
remodeling. Am J Physiol Lung Cell Mol Physiol. 295:L670–L679.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Vieira RP, Duarte AC, Claudino RC, Perini
A, Santos AB, Moriya HT, Arantes-Costa FM, Martins MA, Carvalho CR
and Dolhnikoff M: Creatine supplementation exacerbates allergic
lung inflammation and airway remodeling in mice. Am J Respir Cell
Mol Biol. 37:660–667. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ferreira SC, Toledo AC, Hage M, Santos AB,
Medeiros MC, Martins MA, Carvalho CR, Dolhnikoff M and Vieira RP:
Creatine activates airway epithelium in asthma. Int J Sports Med.
31:906–912. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Xiao W, Chen P, Wang R and Dong J:
Overload training inhibits phagocytosis and ROS generation of
peritoneal macrophages: Role of IGF-1 and MGF. Eur J Appl Physiol.
113:117–125. 2013. View Article : Google Scholar : PubMed/NCBI
|