Introduction
Inadequate production of insulin and insulin
resistance are the main causes of type 2 diabetes (1). β-cells, the insulin producing cells
of the pancreas, maintain glucose homeostasis (2). Both a decrease in the number of
β-cells and their functional impairment play key roles in the
pathogenesis of diabetes (2).
Similar to other cells in the human body, β-cells are tightly
regulated by cell cycle progression, a process that is not well
understood (2). Improving
pancreatic islet function and increasing the number of β-cells are
potential strategies in the treatment of diabetes mellitus
(2).
A number of studies have shown that cell cycle
regulating factors are pivotal in β-cell function, proliferation
and apoptosis (3,4). Nyblom et al (3) found that several pancreatic islet
proteins controlling islet cell regeneration and proliferation
changed significantly in patients with type 2 diabetes, including
activation of polo-like kinase 1 (PLK1). In another study, Misfeldt
et al (4) discovered an
increase in PLK1 expression and islet cell hyperplasia after a 60%
resection of rat pancreatic islet cells.
PLKs are a family of serine/threonine protein
kinases that orchestrate a plethora of cellular processes in
eukaryotic organisms (5,6). PLKs have an N-terminal catalytic
kinase domain and one or more C-terminal polo boxes that are well
conserved from budding yeast to Drosophila, Xenopus and
mammals. The most well studied and universally expressed mammalian
subfamily member, PLK1, plays a critical role in mitotic
spatiotemporal regulation (7,8).
PLK1 knockdown by RNA interference or inhibition with small
molecules results in a failure to establish a bipolar spindle or to
properly attach kinetochores to microtubules (9,10).
Defective development is observed in PLK1-null mice due to massive
mitotic arrest (11). PLK1 is also
widely recognized as an oncogene, as high PLK1 expression in cancer
correlates with a poor prognosis (12,13).
In eukaryotic cells, cell division cycle protein 14
(Cdc14) is essential for faithful cell cycle progression. As a
member of the dual-specificity phosphatase family, Cdc14 has a
conserved phosphatase domain located at the N-terminal. The
function of human Cdc14A has not yet been fully elucidated;
however, Cdc14A has many critical functions, including in DNA
damage checkpoint control, DNA repair, centrosome maturation and
separation, and the regulation of cytokinesis (14–16).
Cdc14A also plays a role in cell migration and adhesion, and may
regulate tumor metastasis (17).
In a previous study, the interrelationship between PLK1 and Cdc14A
was identified and characterized (18). The C-terminal domain of Cdc14A
auto-inhibits the phosphatase activity when bound to the
N-terminal, which illustrates a self-inhibitory association
(18). When phosphorylated by
PLK1, the inhibitory self-association of Cdc14A, as judged by its
phosphatase activity, was found to be disrupted in both in
vitro and in vivo studies (18). The co-localization of PLK1 and
Cdc14A in the centrosome were also revealed (19). In addition, faithful chromosome
segregation during mitosis relies on the spatiotemporal interaction
between PLK1 and Cdc14A (18).
Therefore, it is of great interest to elucidate the physiological
effects of the interrelationship between these two important
proteins.
In this study, the roles of PLK1 in cell
proliferation, apoptosis and insulin secretion regulation were
investigated in the β-TC3 cell line under both low and high glucose
conditions. Additionally, whether the phosphorylation of Cdc14A by
PLK1 is involved in these processes was investigated. The data
presented here reveal that the phospho-regulation of Cdc14A by PLK1
is involved in β-cell function and cell cycle regulation under high
glucose conditions.
Materials and methods
cDNA construction
The cDNA of Cdc14A (NM_033312) was kindly donated by
Jiri Lukas. To generate the plasmid encoding GFP-fused Cdc14A, the
respective open reading frame was amplified by PCR using Taq Plus
DNA polymerase (cat. no. ET105; Tiangen Biotech Co., Ltd.), and
inserted into pEGFP-C1 vector (Clontech Laboratories, Inc.) by
ligating EcoRI-SalI sites in Cdc14A cDNA. The
following PCR cycling conditions were used: 94°C for 5 min; 35
cycles consisting of 94°C for 30 sec, 55°C for 30 sec and 72°C for
2 min; 72°C for 5 min; and finally, cooling to 16°C. The Cdc14A
primers were 5′-CATGAATTCATGGCAGCGGAGTCAGGGGA-3′ (forward) and
5′-CGGGTCGACTCAGAAGGCTTCCTTGGCAC-3′ (reverse). A green fluorescent
protein-tagged non-phosphorylatable Cdc14AS351A/363A
(Cdc14AAA) mutant was constructed using a QuikChange
Site-directed Mutagenesis kit (cat. no. 210518; Stratagene; Agilent
Technologies Inc.).
Cell lines and culture conditions
The mouse β-TC3 cell line was purchased from CHI
Scientific Inc. The cells were cultured in DMEM (HyClone; GE
Healthcare Life Sciences) supplemented with 10% heat-inactivated
FBS (cat. no. 16000-044; Gibco; Thermo Fisher Scientific Inc.) and
5% CO2 at 37°C. In addition, 1% L-glutamine and 1%
antibiotics were also added to the culture medium. The culture
medium was refreshed every 3 days.
Cell transfections
Cdc14A plasmids (2 µg/ml) and PLK1 siRNA (50 nM/l)
were transfected into β-TC3 cells at 80% confluency using
Lipofectamine® 2000 reagent (Invitrogen; Thermo Fisher
Scientific, Inc.), according to the manufacturer's instructions.
PLK1 siRNA (siRNA329, siRNA1566 and siRNA1568) and a control siRNA
were designed and synthesized by Invitrogen (Thermo Fisher
Scientific, Inc.). The sense strand of PLK1 siRNA329 was
5′-GAUUGUGCCUAAGUCUCUGTT-3′, and its antisense strand was
5′-CAGAGACUUAGGCACAAUCTT-3′. The sense strand of PLK1 siRNA1566 was
5′-UGAAGAUCUGGAGGUGAAATT-3′, and its antisense strand was
5′-UUUCACCUCCAGAUCUUCATT-3′. The sense strand of PLK1 siRNA1568 was
5′-AUUGUGCUUGGCUGCCAGUTT-3′, and its antisense strand was
5′-ACUGGCAGCCAAGCACAAUTT-3′. The sense strand of the control siRNA
was 5′-UUCUCCGAACGUGUCACGUTT-3′, and its antisense strand was
5′-ACGUGACACGUUCGGAGAATTT-3′. After transfection of PLK1 siRNA for
72 h, the cells were either collected or transfected with Cdc14A
wild type or mutant plasmids for a further 48 h. The efficiency of
PLK1 siRNA transfection was determined by reverse transcription
quantitative (RT-q)PCR.
Isolation of RNA and RT-qPCR
Total RNA was extracted from β-TC3 cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.), according to the manufacturer's instructions, and was
reverse-transcribed using a PrimeScript RT-PCR kit (Thermo Fisher
Scientific, Inc.). RT was performed at 37°C for 15 min, followed by
85°C for 5 sec. The following PCR cycling conditions were used:
95°C for 30 sec, followed by 40 cycles of 95°C for 5 sec, 60°C for
40 sec and 95°C for 15 sec. The PLK1 primers were
5′-GAGTGCCACCTTAGTGACTTGCT-3′ and 5′-CTTGTCCGAATAGTCCACCCAC-3′. The
GAPDH primers were 5′-AGAGGGAAATCGTGCGTGAC-3′ and
5′-CCAAGAAGGAAGGCTGGAAAA-3′. qPCR was performed using an
SYBR® Premix Ex Taq™ II kit [(Tli RNaseH Plus),
ROX plus; cat. no. RR82LR; Takara Biotechnology Co., Ltd.) and a
7300 Real-Time PCR system (Thermo Fisher Scientific, Inc.). The
data were analyzed by the relative standard curve method and
normalized to GAPDH expression. Relative RNA expression was
calculated using the 2−ΔΔCq method (20). All experiments were performed in
triplicate.
Cell cycle analysis
β-TC3 cells were fixed in ice-cold 70% ethanol for 1
h. Following this, 1 mg/ml RNase (Beyotime Institute of
Biotechnology) was added to the culture medium and the cells were
incubated at 37°C for 30 min. Cells were analyzed using FACScan
cytofluorometry equipment (Becton-Dickinson and Company) and FlowJo
version 7.6.5 software (FlowJo LLC) after staining with propidium
iodide (0.5 mg/ml) at 4°C for 30 min.
Apoptosis analysis
Annexin V/PI double staining was used to determine
the number of dead cells. Transfected β-TC3 cells were harvested as
described earlier and washed with ice-cold PBS. The cells were
stained with FITC-conjugated Annexin V (BD Pharmingen; BD
Biosciences) in binding buffer (10 mM HEPES, 140 mM NaCl, 2.5 mM
CaCl2, pH 7.4), followed by FACScan cytofluorometry (BD
Biosciences) and CellQuest Pro version 5.1 software (BD
Biosciences) analysis. Each experiment was conducted in
triplicate.
Cell proliferation assay
Cell proliferation was analyzed using the Cell
Counting Kit-8 (CCK-8; Dojindo Molecular Technologies, Inc.) assay.
β-TC3 cells (3×103) were transferred into each well of a
96-well plate in DMEM. Each group of cells was plated into five
wells and the test was repeated three times. A total of 10 µl of
CCK-8 reagent was added to each well at each time-point followed by
incubation for 4 h. The optical density was measured using a
microplate reader (Bio-Rad Laboratories, Inc.) at a wavelength of
450 nm. The cell proliferation rate was plotted as a curve for each
group of cells.
Western blot analysis
Total cell extracts were denatured in RIPA lysis
buffer (Beyotime Institute of Biotechnology). The protein
concentration was quantified using a BCA assay. Protein (30
µg/lane) was loaded and separated on 15% polyacrylamide gels. The
proteins were then transferred to nitrocellulose filter membranes,
blocked in blocking buffer [1% BSA (Sigma-Aldrich; Merck KGaA), 10
mM Tris pH 7.5, 100 mM NaCl, 0.1% Tween-20] for 1 h at 37°C, and
incubated with primary antibodies overnight at 4°C. The primary
antibodies for western blotting included anti-Cdc14A (1:500; cat.
no. 13660-1-AP; ProteinTech Group, Inc.) and anti-GAPDH (1:10,000;
cat. no. MAB374; EMD Millipore). The membrane was incubated with
horseradish peroxidase-conjugated secondary antibody (1:5,000; cat.
no. BA1050; Wuhan Boster Biological Technology, Ltd.) for 1 h at
room temperature. Antigen-antibody complexes were visualized using
ECL reagent (Pierce; Thermo Fisher Scientific, Inc.). Western blot
bands were quantified by ImageJ Version 1.50 software (National
Institutes of Health).
Insulin secretion assay
First, β-TC3 cells were incubated in DMEM at a low
glucose concentration (5.5 mmol/l) for 24 h. Then, cells were
cultivated for 30 min under high glucose conditions (25 mmol/l)
before being transfected with PLK1 siRNA and/or Cdc14A plasmids. At
3–5 days following transfection, aliquots of the incubation media
were collected for the insulin assays. Insulin levels were
determined using a mouse insulin radioimmunoassay (RIA) kit (cat.
no. K4271; BioVision, Inc.) according to the manufacturer's
instructions (CIS Bio International).
Statistical analysis
Each experiment was performed in triplicate, and the
results are expressed as the mean ± SD. One-way ANOVA and the least
significant difference post hoc test was applied when comparing
normally distributed data with homogeneity of variance among three
or more groups. Kruskal-Wallis and Dunn's pairwise post hoc tests
were used when making multiple comparisons among non-normally
distributed measurement data. χ2 test followed by
Bonferroni tests were applied when comparing cell cycle
distributions among different groups. P<0.05 was considered to
indicate a statistically significant difference. Statistical
analyses were performed using SPSS version 23 (SPSS, Inc.).
Results
PLK1 promotes cell proliferation while
inhibiting cellular apoptosis in β-TC3 cell lines under high
glucose conditions
At 72 h post transfection with siRNA targeting PLK1
(siRNA329, siRNA1566 and siRNA1568), whole cell lysates and culture
media were collected. RT-qPCR analysis was performed in triplicate
to detect the effect of the PLK1 siRNA. As shown in Fig. 1A, PLK1 levels were the lowest when
its expression was depleted with siRNA329. Therefore, siRNA329 was
utilized in the following experiments.
To determine the function of PLK1 in β-TC3 cells,
the effects of PLK1 siRNA on the cell cycle, cell proliferation and
cellular apoptosis under high glucose conditions were first
examined. Compared with low glucose, high glucose significantly
promoted cell proliferation (P<0.05). PLK1 depletion
significantly inhibited cell proliferation compared with the
control siRNA under both low and high glucose conditions
(P<0.05). The results of the CCK-8 assay indicate that PLK1
promotes β-TC3 cell proliferation under both low and high glucose
conditions, with the maximum effect observed on day 4(Fig. 1B and C).
Cell cycle distribution was also determined on day
4; the results are shown in Fig. 2
and Table I. The results showed
that PLK1 siRNA increased the population of cells in the
G1phase and decreased the population of cells in the
G2 and S phases under low glucose conditions
(P<0.05). Under high glucose conditions, PLK1 siRNA also
decreased the number of cells in the G2 phase, but had
no significant effect on the number of cells in the G1
and S phases. High glucose conditions significantly decreased the
population of cells in the G1 phase and increased the
number of cells in the G2 and S phases (P<0.05).
These results indicated that PLK1 controls the cell cycle
progression of β-TC3 cells.
 | Table I.Cell cycle distributions of different
groups. |
Table I.
Cell cycle distributions of different
groups.
|
| Cell cycle
phase |
|---|
|
|
|
|---|
| Group | G1
(%) | S (%) | G2
(%) |
|---|
| Control
siRNA+low-glucose | 82.79 | 9.64 | 7.55 |
| PLK1
siRNA+low-glucose | 89.52 | 5.86 | 4.59 |
| Control
siRNA+high-glucose | 79.28 | 9.87 | 10.82 |
| PLK1
siRNA+high-glucose | 82.00 | 9.07 | 8.90 |
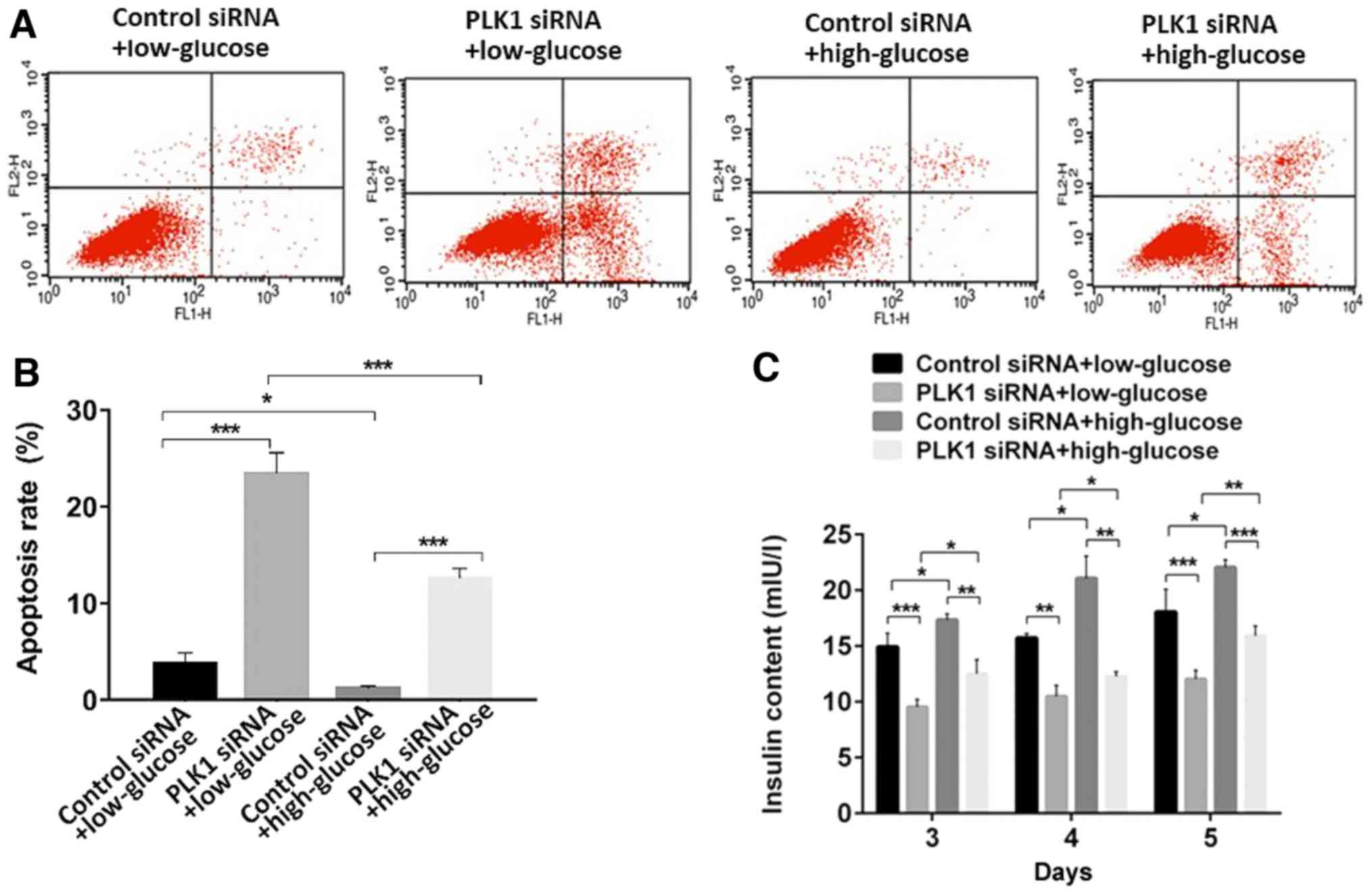
Additionally, flow cytometry analysis revealed that,
when transfected with control siRNA, high glucose conditions
inhibited β-TC3 cell apoptosis compared with the rate of apoptosis
in the low glucose conditions (P<0.05). PLK1 siRNA promoted
cellular apoptosis compared with control siRNA under both low and
high glucose conditions (P<0.05). These results indicated that
PLK1 inhibits β-TC3 cell apoptosis under both low or high glucose
conditions (Fig. 3A and B).
PLK1 promotes insulin secretion in
β-TC3 cell lines under high glucose conditions
To determine insulin release in response to glucose
stimulation and PLK1 depletion, aliquots of the culture media of
different groups of cells 3–5 days after transfection were taken to
be used to assess insulin levels using a mouse insulin RIA kit.
Exposure of β-TC3 cells to a high concentration of glucose promoted
insulin secretion compared to exposure to a low concentration of
glucose when transfected with control siRNA (P<0.05). Depletion
of PLK1 inhibited insulin secretion under both low and high glucose
conditions (P<0.05). These results indicated that PLK1 promotes
insulin section in β-TC3 cells under both low and high glucose
conditions (Fig. 3C).
Phospho-regulation of Cdc14A by PLK1
is involved in β-TC3 cell cycle regulation and insulin secretion
under high glucose conditions
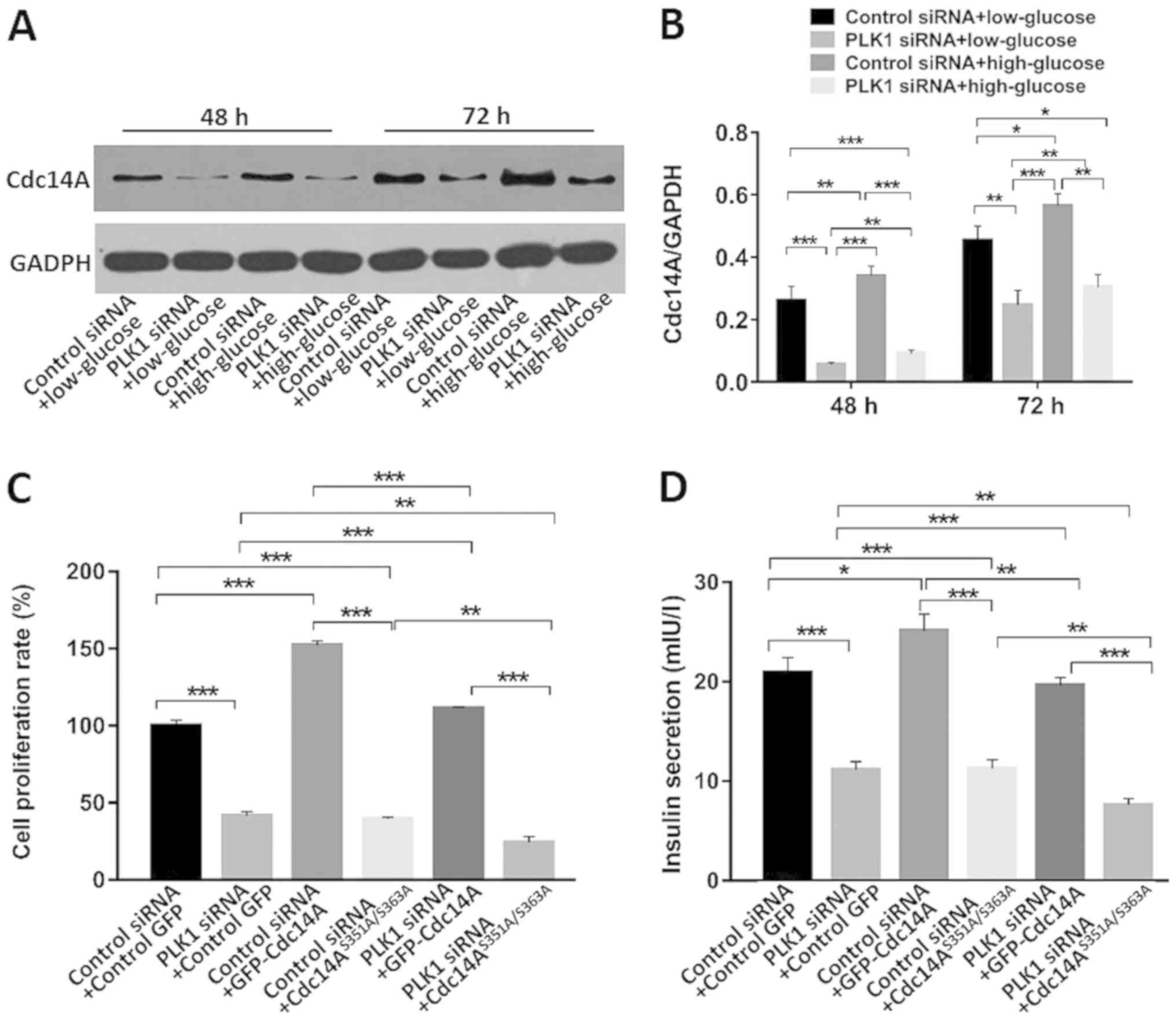
Cdc14A is a substrate of PLK1 (18). PLK1 phosphorylates Cdc14A,
stimulating its phosphatase activity (18). In this study the interactions of
these two proteins in β-TC3 cells were investigated. As shown in
Fig. 4A and B, PLK1 silencing
decreased Cdc14A expression in both low and high glucose conditions
compared with control siRNA at 48 and 72 h after transfection
(P<0.05). High glucose conditions promoted Cdc14A expression
compared with low glucose conditions when transfected with control
siRNA (P<0.05).
Serine351 and Serine 363 of Cdc14A are the
phosphorylatable sites for PLK1 (18). Both serine residues were mutated to
alanine in order to generate a GFP-tagged non-phosphorylatable
Cdc14A, Cdc14AAA, which exhibited significantly
attenuated phosphatase activity (18). β-TC3 cell proliferation and insulin
secretion was monitored under high glucose conditions on day 4
after transfection with the indicated siRNAs and plasmids. As shown
in Fig. 4C, compared with the
control siRNA and empty GFP-vector transfected groups,
co-transfection with control siRNA and Cdc14A under high glucose
conditions promoted β-TC3 cell proliferation, while transfection
with Cdc14AAA inhibited cell proliferation (P<0.05).
When co-transfected with PLK1 siRNA, similar results were observed
(P<0.05). Moreover, PLK1 siRNA partially reversed the
proliferation-promoting effects of Cdc14A compared with those
associated with the control siRNA + Cdc14A group or with the PLK1
siRNA + Cdc14A group (P<0.05). PLK1 silencing further
intensified the proliferation inhibition of
Cdc14AAA-transfected cells compared with the control
siRNA + Cdc14AAA group or with the PLK1 siRNA +
Cdc14AAA group (P<0.05). These results indicated that
the phospho-regulation of Cdc14A is involved in β-TC3 cell
proliferation and that it is regulated by PLK1 under high glucose
conditions.
As shown in Fig.
4D, PLK1 depletion or Cdc14AAA overexpression
inhibited β-TC3 cell insulin secretion compared with that in the
control siRNA + GFP-vector group (P<0.05). Overexpression of
wild type Cdc14A promoted insulin secretion compared to that in the
control group under high glucose conditions and with that in the
control siRNA + GFP-vector group (P<0.05). Cdc14A partially
reversed the insulin inhibiting effect of PLK1 siRNA compared to
that of the control siRNA + Cdc14A group and with that of the PLK1
siRNA + Cdc14A group (P<0.05). Cdc14AAA showed a
synergistic effect with PLK1 siRNA compared to that of the control
siRNA + Cdc14AAA group and with that of the PLK1 siRNA +
Cdc14AAA group (P<0.05). These results indicated that
the phospho-regulation of Cdc14A by PLK1 is also involved in the
regulation of β-TC3 cell insulin secretion under high glucose
conditions.
Discussion
Type 2 diabetes has become a common disease due to
lifestyle factors and poses a serious threat to human health. A
lack of functional β-cells ultimately results in both type 1 and
type 2 diabetes. Scientists are searching for more sources of islet
cells or β-cells to treat diabetes; however, cadaveric islet cell
transplantation remains promising (19). Although sources of newly formed
islet cells remain elusive, it is clear that β-cells have the
capacity to replicate themselves (21,22).
Thus, it may be possible for diabetic patients to benefit from the
manipulation of β-cell proliferation.
However, it is difficult to increase β-cell mass
directly. The accumulation of double-stranded DNA damage may be
observed when a substantial number of β-cells enter mitosis,
resulting in apoptosis and not in functional β-cell expansion
(23). More work is needed to
control β-cell proliferation. New pathways and compounds have been
identified by high-throughput screening methods and must be studied
further. The G1/S and G2/M transitions are
key cell cycle checkpoints that can be regulated. However, there is
still an outstanding question as to whether these strategies affect
other cell types within the body. Many pathways have been
implicated, including the adenylyl cyclase/protein kinase A
pathway, the mitogen-activated protein kinase pathway, the JAK-STAT
pathway, the PI3-kinase-PKB/Akt pathway and the insulin receptor
substrate-2 pathway. The basic cell cycle machinery of β-cells has
been outlined in a previous review (24).
It is well known that acute high glucose stimulation
promotes insulin secretion by β-cells. The compensatory increase in
β-cell mass that is, in part, accounted for by β-cell replication
corrects the transient hyperglycemia caused by sub-partial or
partial pancreatectomy in rodents (25–27).
Glucotoxicity is a phenomenon in which chronic hyperglycemia causes
β-cell dysfunction and death (28,29).
The precise molecular mechanisms underlying glucotoxicity are not
completely understood.
Although PLK1 and Cdc14A are both essential for the
cell cycle and are conserved in invertebrates and mammals, little
is known about their crosstalk and mutual regulation in β-TC3
cells, especially under high glucose conditions.
The objective of this study was to provide a basis
for understanding the mechanism underlying diabetes. The effects of
PLK1 siRNA on cell proliferation, the cell cycle, apoptosis and
insulin secretion in β-TC3 cells was investigated as was whether
the phosphorylation of Cdc14A by PLK1 is involved in these
processes. β-TC3 cells were treated in culture media with low or
high concentrations of glucose. It was found that exposure of β-TC3
cells to high glucose for 3–5 days promoted cell proliferation,
apoptosis and insulin release compared with exposure to a lower
glucose concentration. PLK1 siRNA inhibited β-TC3 cell
proliferation, apoptosis and insulin release and decreased Cdc14A
expression under both low and high glucose conditions. Cdc14A
overexpression enhanced β-TC3 cell proliferation and insulin
secretion under high glucose conditions. PLK1 silencing intensified
the inhibition of proliferation and insulin secretion of
Cdc14AAA.
In this study, the effects of PLK1 siRNA on
proliferation, apoptosis and insulin secretion were observed under
both low and high glucose conditions. Therefore, the effects of
PLK1 siRNA may only partially result from high glucose stimulation.
Considering that high glucose alone stimulates β-TC3 cell
proliferation and insulin secretion, the effect of PLK1 siRNA on
insulin secretion may simply reflect effects on cell proliferation.
More studies should be designed to test whether PLK1 is involved in
glucose stimulation and insulin secretion. The results presented
here are consistent with those of another study, in which PLK1 was
significantly activated during β cell replication in individuals
with type 2 diabetes and partial pancreatectomy-treated rats
(4).
It was also found that PLK1 siRNA decreased the
numbers of β-TC3 cells in the G2- and S phases of the
cell cycle while significantly increasing the G1 population under
low glucose conditions. High glucose had the opposite effects, but
only in G2 cells. This finding may be because high
glucose conditions have the opposite effect on β-TC3 cells. These
results suggested that cell cycle regulation may be associated with
the interaction of PLK1 siRNA and high glucose, which is a
relationship that requires further study.
In this study, PLK1 silencing decreased Cdc14A
expression. Cdc14A can be phosphorylated by PLK1, and this
phosphorylation partially releases the self-inhibition of Cdc14A,
thereby enhancing its phosphatase activity (18). Therefore, PLK1 silencing would
change not only the expression but also the phosphorylation status
of Cdc14A. More research is needed to investigate the crosstalk
between Cdc14A and PLK1. It is possible that the phosphorylation of
Cdc14A is involved in the stability or degradation of Cdc14A;
alternatively, PLK1 may regulate the promoter activity of Cdc14A.
The molecular mechanisms of the reduced expression of Cdc14A should
be further explored and discussed.
The present study was limited due to the cell line
type and techniques used in the experiments. Therefore, the
isolation, purification and primary culture of human β-cells is
required. Animal studies are also needed to test the crosstalk
between Cdc14A and PLK1.
In conclusion, PLK1 and Cdc14A may play important
roles in β-TC3 cell cycle regulation. PLK1 and Cdc14A promoted
β-TC3 cell proliferation under high glucose conditions. Decreased
insulin secretion was also observed when PLK1 was silenced. The
phosphorylation of Cdc14A by PLK1 is involved in these processes.
These findings may lead to new therapeutic strategies to understand
how the number of β-cells is regulated and how this can be
manipulated.
Acknowledgements
The authors would like to thank Dr Jiri Lucas (Novo
Nordisk Foundation Center for Protein Research, University of
Copenhagen, Denmark) for Cdc14A cDNA.
Funding
This work was supported by grants from the Natural
Science Foundation for Young Scientists of China (grant no.
81700705); the Natural Science Foundation for Zhejiang Province of
China (grant no. LY17H070001); and the Projects of Medical and
Health Science and Technology for Zhejiang Province of China (grant
no. 2015KYA107).
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
HH and JC designed the experiments. HH, LW, FH, XH,
YL, XX, SZ and PZ performed the experiments. HH, DS, JL and JC
analyzed the data. HH wrote the manuscript. All authors have read
and approved the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Deng S, Vatamaniuk M, Huang X, Doliba N,
Lian MM, Frank A, Velidedeoglu E, Desai NM, Koeberlein B, Wolf B,
et al: Structural and functional abnormalities in the islets
isolated from type 2 diabetic subjects. Diabetes. 53:624–632. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Prentki M and Nolan CJ: Islet beta cell
failure in type 2 diabetes. J Clin Invest. 116:1802–1812. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nyblom HK, Bugliani M, Fung E, Boggi U,
Zubarev R, Marchetti P and Bergsten P: Apoptotic, regenerative, and
immune-related signaling in human islets from type 2 diabetes
individuals. J Proteome Res. 8:5650–5656. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Misfeldt A, Costa RH and Gannon M:
Beta-cell proliferation, but not neogenesis, following 60% partial
pancreatectomy is impaired in the absence of FoxM1. Diabetes.
57:3069–3077. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Barr FA, Silljé HH and Nigg EA: Polo-like
kinases and the orchestration of cell division. Nat Rev Mol Cell
Biol. 5:429–440. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zitouni S, Nabais C, Jana SC, Guerrero A
and Bettencourt-Dias M: Bettencourt-Dias M. Polo-like kinases:
Structural variations lead to multiple functions. Nat Rev Mol Cell
Biol. 15:433–452. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bruinsma W, Raaijmakers JA and Medema RH:
Switching Polo-like kinase-1 on and off in time and space. Trends
Biochem Sci. 37:534–542. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Petronczki M, Lénárt P and Peters JM: Polo
on the rise from mitotic entry to cytokinesis with Plk1. Dev Cell.
14:646–659. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu X and Erikson RL: Activation of
Cdc2/cyclin B and inhibition of centrosome amplification in cells
depleted of Plk1 by siRNA. Proc Natl Acad Sci USA. 99:8672–8676.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lénárt P, Petronczki M, Steegmaier M, Di
Fiore B, Lipp JJ, Hoffmann M, Rettig WJ, Kraut N and Peters JM: The
small-molecule inhibitor BI 2536 reveals novel insights into
mitotic roles of polo-like kinase 1. Curr Biol. 17:304–315. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
de Cárcer G, Manning G and Malumbres M:
From Plk1 to Plk5: Functional evolution of polo-like kinases. Cell
Cycle. 10:2255–2262. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Strebhardt K and Ullrich A: Targeting
polo-like kinase 1 for cancer therapy. Nat Rev Cancer. 6:321–330.
2006. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Eckerdt F, Yuan J and Strebhardt K:
Polo-like kinases and oncogenesis. Oncogene. 24:267–276. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bassermann F, Frescas D, Guardavaccaro D,
Busino L, Peschiaroli A and Pagano M: The Cdc14B-Cdh1-Plk1 axis
controls the G2 DNA-damage-response checkpoint. Cell. 134:256–267.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mocciaro A, Berdougo E, Zeng K, Black E,
Vagnarelli P, Earnshaw W, Gillespie D, Jallepalli P and Schiebel E:
Vertebrate cells genetically deficient for Cdc14A or Cdc14B retain
DNA damage checkpoint proficiency but are impaired in DNA repair. J
Cell Biol. 189:631–639. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mocciaro A and Schiebel E: Cdc14: A highly
conserved family of phosphatases with non-conserved functions? J
Cell Sci. 123:2867–2876. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen NP, Uddin B, Voit R and Schiebel E:
Human phosphatase CDC14A is recruited to the cell leading edge to
regulate cell migration and adhesion. Proc Natl Acad Sci USA.
113:990–995. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yuan K, Hu H, Guo Z, Fu G, Shaw AP, Hu R
and Yao X: Phospho-regulation of HsCdc14A By Polo-like kinase 1 is
essential for mitotic progression. J Biol Chem. 282:27414–27423.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shapiro AM, Ricordi C, Hering BJ,
Auchincloss H, Lindblad R, Robertson RP, Secchi A, Brendel MD,
Berney T, Brennan DC, et al: International trial of the Edmonton
protocol for islet transplantation. N Engl J Med. 355:1318–1330.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hanley NA, Hanley KP, Miettinen PJ and
Otonkoski T: Weighing up beta-cell mass in mice and humans:
Self-renewal, progenitors or stem cells? Mol Cell Endocrinol.
288:79–85. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xu X, D'Hoker J, Stangé G, Bonné S, De Leu
N, Xiao X, Van de Casteele M, Mellitzer G, Ling Z, Pipeleers D, et
al: Beta cells can be generated from endogenous progenitors in
injured adult mouse pancreas. Cell. 132:197–207. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rieck S, Zhang J, Li Z, Liu C, Naji A,
Takane KK, Fiaschi-Taesch NM, Stewart AF, Kushner JA and Kaestner
KH: Overexpression of hepatocyte nuclear factor-4α initiates cell
cycle entry, but is not sufficient to promote β-cell expansion in
human islets. Mol Endocrinol. 26:1590–1602. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sharma A, Yerra VG and Kumar A: Emerging
role of Hippo signalling in pancreatic biology: YAP re-expression
and plausible link to islet cell apoptosis and replication.
Biochimie. 133:56–65. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
García-Ocaña A, Vasavada RC, Takane KK,
Cebrian A, Lopez-Talavera JC and Stewart AF: Using beta-cell growth
factors to enhance human pancreatic Islet transplantation. J Clin
Endocrinol Metab. 86:984–988. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Montaña E, Bonner-Weir S and Weir GC:
Transplanted beta cell response to increased metabolic demand.
Changes in beta cell replication and mass. J Clin Invest.
93:1577–1582. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Vasavada RC, Gonzalez-Pertusa J, Fujinaka
Y, Fiaschi-Taesch N, Cozar-Castellano I and Garcia-Ocaña A: Growth
factors and beta cell replication. Int J Biochem Cell Biol.
38:931–950. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lowell BB and Shulman GI: Mitochondrial
dysfunction and type 2 diabetes. Science. 307:384–387. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Maedler K, Sergeev P, Ris F, Oberholzer J,
Joller-Jemelka HI, Spinas GA, Kaiser N, Halban PA and Donath MY:
Glucose-induced beta cell production of IL-1beta contributes to
glucotoxicity in human pancreatic islets. J Clin Invest.
110:851–860. 2002. View Article : Google Scholar : PubMed/NCBI
|