Introduction
Volatile anesthetics are known to reduce myocardial
ischemia-reperfusion injury (1–3).
Mitochondria exert significant cardioprotective roles (4); the cardioprotective effects caused by
pretreatment of volatile anesthetics (anesthetic preconditioning,
APC) may be triggered by the induction of reactive oxygen species
(ROS) and reactive nitrogen species formation, and by the release
of nitric oxide (NO) (1,5). NO exhibits several beneficial roles
in myocardial ischemia-reperfusion injury (1,6,7). In
addition, isoflurane increases myocardial protection following
ischemic post-conditioning via a NO-dependent mechanism (2,3).
Mitochondria contain nitric oxide synthase (NOS), an enzyme which
produces large amounts of NO and regulates respiratory function
(8). NO inhibits mitochondrial
respiration by binding to the respiratory chain enzyme and
regulating the function of mitochondrial ATP-dependent
K+ (mKATP) channels (9). The interaction of NO with the
mitochondrial electron transport chain may serve an important role
in APC-induced myocardial protection.
Reversible changes in mitochondrial function caused
by volatile anesthetics are associated with APC-induced
cardioprotective mechanisms (10–12).
Volatile anesthetics are known to affect mitochondrial respiration
and electron transport chain (ETC) activity (12–14).
Riess et al (12) reported
that the administration of sevoflurane in isolated cardiac
mitochondria during state 2 respiration resulted in an attenuated
state 3 respiration. This attenuation was proposed to be mediated
by ROS and was not associated with mKATP channel opening
(12). Although it is known that
ROS induces the protective effects of APC on ischemia-reperfusion
injury (1), the mechanism of its
action remains unclear. Volatile anesthetics increase the signals
generated by ROS, which is a mandatory step in triggering APC
(15,16). The use of ROS scavengers blocks
APC-induced cardioprotection (15–17).
Collectively, these studies suggested that volatile anesthetics may
directly affect mitochondrial complexes and ROS production, thereby
regulating mitochondrial respiration and bioenergetics. Recently,
we reported that isoflurane induced the attenuation of state 3
respiration (18). This occurred
in the presence of the complex I substrates glutamate and malate,
and was independent of endogenous mitochondrial NO production
(18); however, the effects of
volatile anesthetics on mitochondrial respiration and ROS
production in the presence of complex II substrates require further
investigation. Furthermore, the roles of NO production and
mKATP channel function in this process have not been
elucidated. In the present study, we aimed to determine the effects
of isoflurane on mitochondrial respiration, membrane potential
(ΔΨm) and ROS production in mitochondria isolated from
cardiac cells. The cells were analyzed in the presence of the
complex II-associated substrates succinate (Suc) with or without
the complex I inhibitor rotenone (Rote). Furthermore, the roles of
mKATP channel opening in the effects of isoflurane were
also examined.
Materials and methods
Experimental animals
This experimental study was approved by the Animal
Care and Use Committee of the Suzhou Science and Technology Town
Hospital. Male Sprague-Dawley rats (weight: 250±50 g; n=112), aged
8–10 weeks, were purchased from the Animal Center of Suzhou
University and were housed at 25°C with 60% humidity under a 12 h
light-dark cycle. Two rats were housed in each cage and were
allowed access to food and water ad libitum. The rats used
in the present study underwent humane care in accordance with the
National Institute Health Guide for the Care and Use of Laboratory
Animals (https://grants.nih.gov/grants/olaw/Guide-for-the-Care-and-use-of-laboratory-animals.pdf)
(2).
Drugs and chemicals
Isoflurane was purchased from Abbot Laboratories.
The potent NOS inhibitor, L-N5-(1-Iminoethyl)-ornithine
(L-NIO) was obtained from Calbiochem. The superoxide dismutase
mimetic manganese (III) tetrakis (4-benzoic acid) porphyrin
chloride (TBAP) was purchased from OxisResearch. The
negatively-charged spin trap N-tert-Butyl-a-(2osulfophenyl)nitrone
sodium (SPBN), adenosine-diphosphate (ADP), the selective inhibitor
of NO-sensitive guanylyl cyclase
1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ), the
mitochondrial KATP channel blocker 5-hydroxydecanoic
acid (5-HD), the nonspecific KATP channel blocker
glibenclamide (GLB), the complex II substrate Suc, the complex II
inhibitor malonate and the complex I blocker Rote were obtained
from Sigma-Aldrich (Merck KGaA). Rhodamine 123 and Amplex red were
purchased from Molecular Probes (Thermo Fisher Scientific, Inc.). A
total of 10 µl of isoflurane was originally dispersed by sonication
in 1 ml respiration buffer (200 mM mannitol, 50 mM sucrose, 5 mM
KH2PO4, 1 mM EGTA, 5 mM
3-(n-morpholino)propane sulfonic acid, and 0.1% bovine serum
albumin; pH 7.15, adjusted with potassium hydroxide) and 5 µl of
this solution was added to the chamber with 500 µl of mitochondrial
suspension. The concentration of isoflurane was determined by gas
chromatography (0.25±0.02 mM; Schimadzu).
Isolation of mitochondria
The animals were anesthetized with an
intraperitoneal injection of pentobarbital sodium (50 mg/kg) prior
to decapitation. The heart was quickly removed and placed in an
ice-cold isolation buffer containing mannitol (200 mM), sucrose (50
mM), KH2PO4 (5 mM) EGTA (1 mM),
3-(N-Morpholino)propane sulfonic acid (MOPS; 5 mM) and 0.1% bovine
serum albumin (Sigma-Aldrich; Merck KGaA), pH 7.3, adjusted by
potassium hydroxide. The discarded large blood vessels and auricles
were removed of any remaining blood with isolation buffer. The
ventricle was cut into small pieces of 1 mm3 in volume.
Mitochondrial isolation was carried out at 4°C as described
previously (18). The total
protein concentration was determined by a bicinchoninic protein
assay using bovine serum albumin as a standard.
Mitochondrial oxygen (O2)
consumption
Mitochondrial respiration was measured via
polarography at 28°C with a computer-controlled Clark-type
O2 electrode (Hansatech Instruments Ltd.) (14) in 0.5 ml of respiratory buffer
containing KCl (130 mM), K2PH4 (5 mM), MOPS
20 (mM), EGTA (2.5 mM), Na4Pl2O7
(0.001 mM), EDTA (1 mM) and 0.1% of bovine serum albumin (pH 7.2,
adjusted with potassium hydroxide). The solution was mixed with 0.5
mg of mitochondrial protein per milliliter and with Suc (5 mM) as
the complex II substrate (19).
State 3 respiration was initiated by addition of ADP (0.25 mM).
State 4 respiration was monitored following depletion of ADP
(Fig. 1, raw recording data). The
representative traces of the respiration indicated a high
functional quality and structural integrity of the mitochondria
after the isolation procedure, as demonstrated by the respiratory
control ratio (RCR, state 3/state 4). The RCR was estimated at
9.83±1.51 in the presence of Suc. The value of each state was based
on the mean of each slope. The RCR was calculated as follows: State
3/state 4. The data were stored online using the manufacture's
software.
Experimental protocols of
mitochondrial O2 consumption
To elucidate the mechanism of action of isoflurane
on mitochondrial respiration, immediately before each experiment,
an aliquot of the concentrated mitochondria was added to
respiration buffer at 27°C. The compound was mixed with Suc in the
presence or absence of the complex I inhibitor Rote (10 µM)
according to the protocols presented in Fig. 2 (n=8 each protocol). The following
compounds were also added: NOS inhibitor L-NIO (10 µM), cyclic
guanosine monophosphate (cGMP) inhibitor ODQ (10 µm), the ROS
scavengers TBAP (10 µM) and SPBN (10 µM), and the KATP
channel inhibitors 5-HD (300 µM) and GLB (2 µM). These experiments
used the conventional definition state 2 as the closely analogous
steady state in the presence of respiratory substrates and in the
absence of additional ADP. The definition state 3 represented the
state of maximal phosphorylation after the addition of ADP, while
state 4 referred to the state of minimal phosphorylation following
depletion of ADP. RCR indicated the degree of the coupling
respiration and of the phosphorylation process.
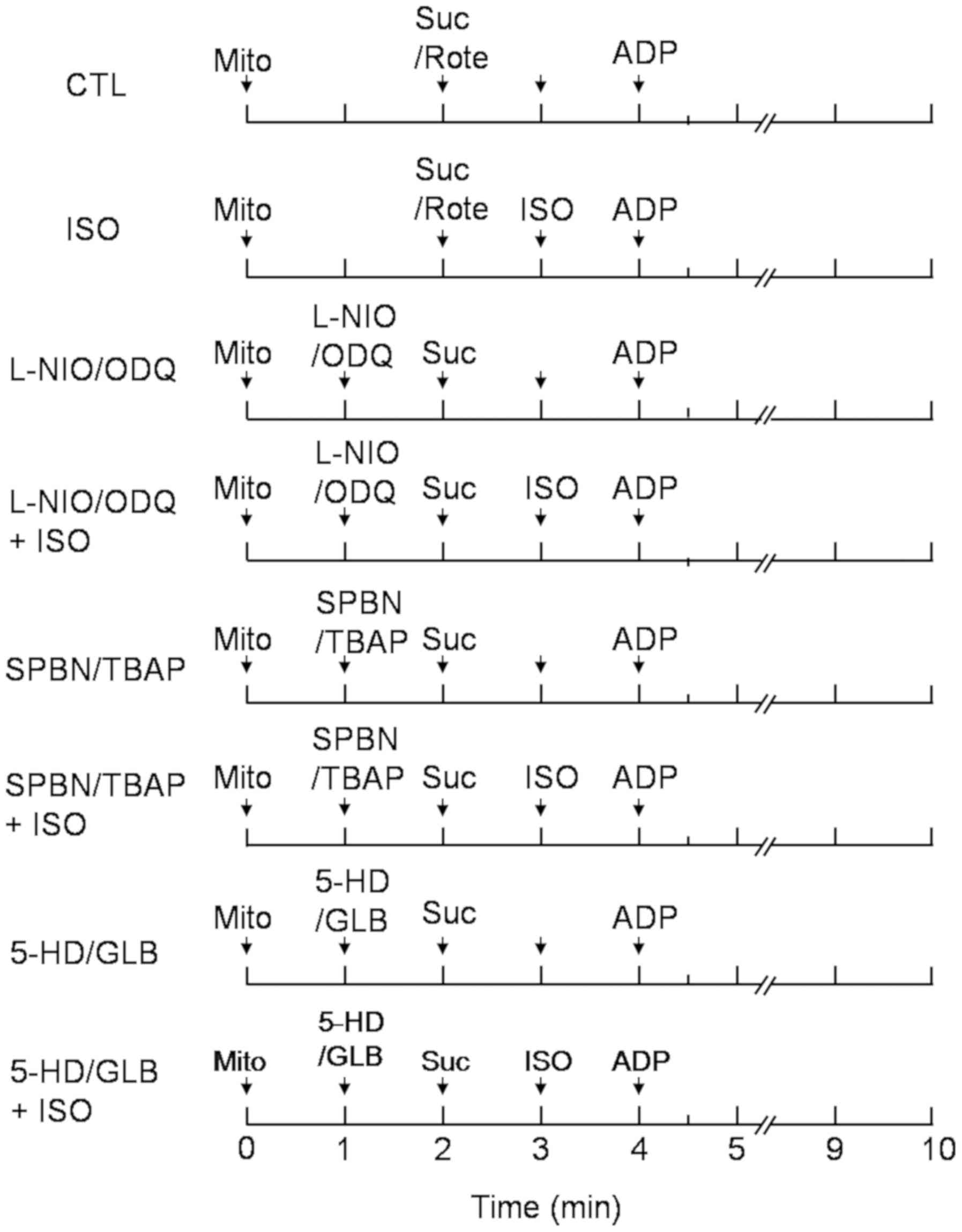 | Figure 2.Experimental protocol used for
measurement of the mitochondrial respiration state. ISO (0.25 mM)
was added at the 2 min period (state 2). The NOS inhibitor L-NIO
(10 µM), the guanylyl cyclase inhibitor ODQ (10 µm), the reactive
oxygen species scavengers TBAP (10 µM) and SPBN (10 µM), and the
KATP channel inhibitors 5-HD (300 µM) and GLB (2 µM)
were added at 1 min (state 1) with or without isoflurane. State 2
respiration was initiated by addition of 5 mM Suc or Suc with the
complex I inhibitor rotenone (Rote, 10 µM). State 3 respiration was
initiated by addition of 250 µM ADP at 4 min. N=8 in each group.
5-HD, 5-hydroxydecanoic acid; CTL, control; GLB, glibenclamide;
ISO, isoflurane; Mito, mitochondria; ODQ,
1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one; Suc, succinate; SPBN,
N-tert-Butyl-a-(2osulfophenyl)nitrone sodium; TBAP, tetrakis
(4-benzoic acid) porphyrin chloride. |
The control group included the substrate Suc alone,
or in the presence the complex I (nicotinamide adenine dinucleotide
dehydrogenase) inhibitor Rote. These compounds were added at 2 min,
while ADP was added at 4 min. Isoflurane was added during state 2
respiration in order to monitor the effect of isoflurane on state 3
and 4 respiration. All other drugs were added prior to isoflurane
treatment at 1 min in order to observe the effects of NO, ROS and
KATP channel opening on isoflurane-mediated
mitochondrial respiration.
Mitochondrial membrane potential
(ΔΨm)
Mitochondria (0.5 mg/ml) were suspended in
respiratory buffer. During state 2–4 respiration, ΔΨm was monitored
in the presence of the substrate Suc alone or in the presence of
Suc and Rote in a cuvette-based spectrophotometer (Model QM-8,
Photon Technology International) operating at excitation and
emission wavelengths of 503 and 527 nm, respectively. The detection
was conducted in the presence of the fluorescent dye rhodamine 123
(50 nM) (20). ΔΨm was expressed
as the percentage of rhodamine 123 fluorescence. The relative
mitochondrial fluorescence was subtracted from the value obtained
following the addition of the mitochondrial uncoupler, carbonyl
cyanide-m-chlorophenylhydrazenone (CCCP, 4 µM).
Mitochondrial
H2O2 release
During state 2 and 3 respiration, the rates of
mitochondrial H2O2 release in the presence of
Suc or Suc and Rote were measured spectrophotometrically (Model
QM-8, Photon Technology International) using the fluorescent dye
Amplex red (12.5 µM) in the presence of 0.1 U/ml of horseradish
peroxidase at 30°C. Mitochondria (0.5 mg/ml) were suspended in the
respiration buffer, and the reaction was started by the addition of
5 mM succinate (19,21). The excitation and emission
wavelengths were set to 530 and 583 nm, respectively. The
H2O2 release rate was expressed as the
percentage of baseline H2O2 release
(following addition of the substrate). The baseline
H2O2 levels were calibrated via a standard
curve. Specifically three standard curves of photon counts were
plotted that were in the range of 10–200 nm of
H2O2 (Amplex red and horseradish peroxidase
were added to the reactants). Each regression was linear and the
mean of the standard curves was used (R>0.99). The control group
(dye vehicle) contained 0.1% dimethyl sulfoxide.
Statistical analysis
All data are expressed as mean ± standard deviation.
Two-way analysis of variance was used to evaluate the differences
between the groups. A Student-Newman-Keuls multiple comparison
post-hoc test was used to assess the differences within the groups.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Mitochondrial respiration
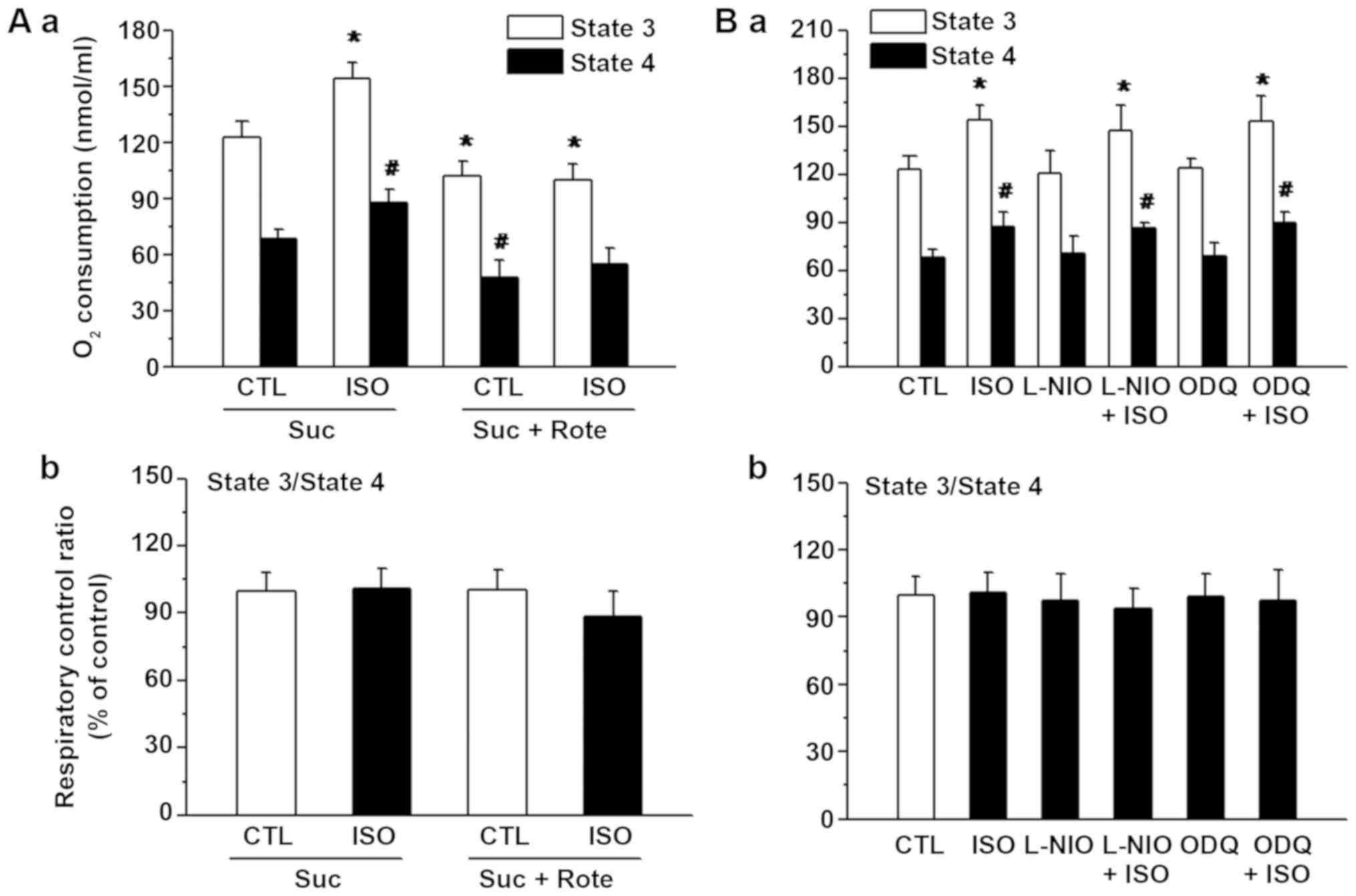
When Suc was used as a substrate, isoflurane
significantly increased the state 3 and 4 respiration rates
compared with those of the control group (Fig. 3A). Compared with Suc treatment
alone, the rate of state 3 respiration was significantly decreased
in the control and isoflurane groups in the presence of Suc and
Rote. The RCR was markedly reduced in the isoflurane and Rote
group; however, this effect was not significantly different
compared with that of the control group.
The NOS inhibitor L-NIO or the cyclic cGMP inhibitor
ODQ was added prior to the administration of Suc (Fig. 3B). The administration of L-NIO or
ODQ did not significantly alter the rates of state 3 and 4
respiration in the L-NIO+ISO group or ODQ+ISO group compared with
isoflurane alone group.
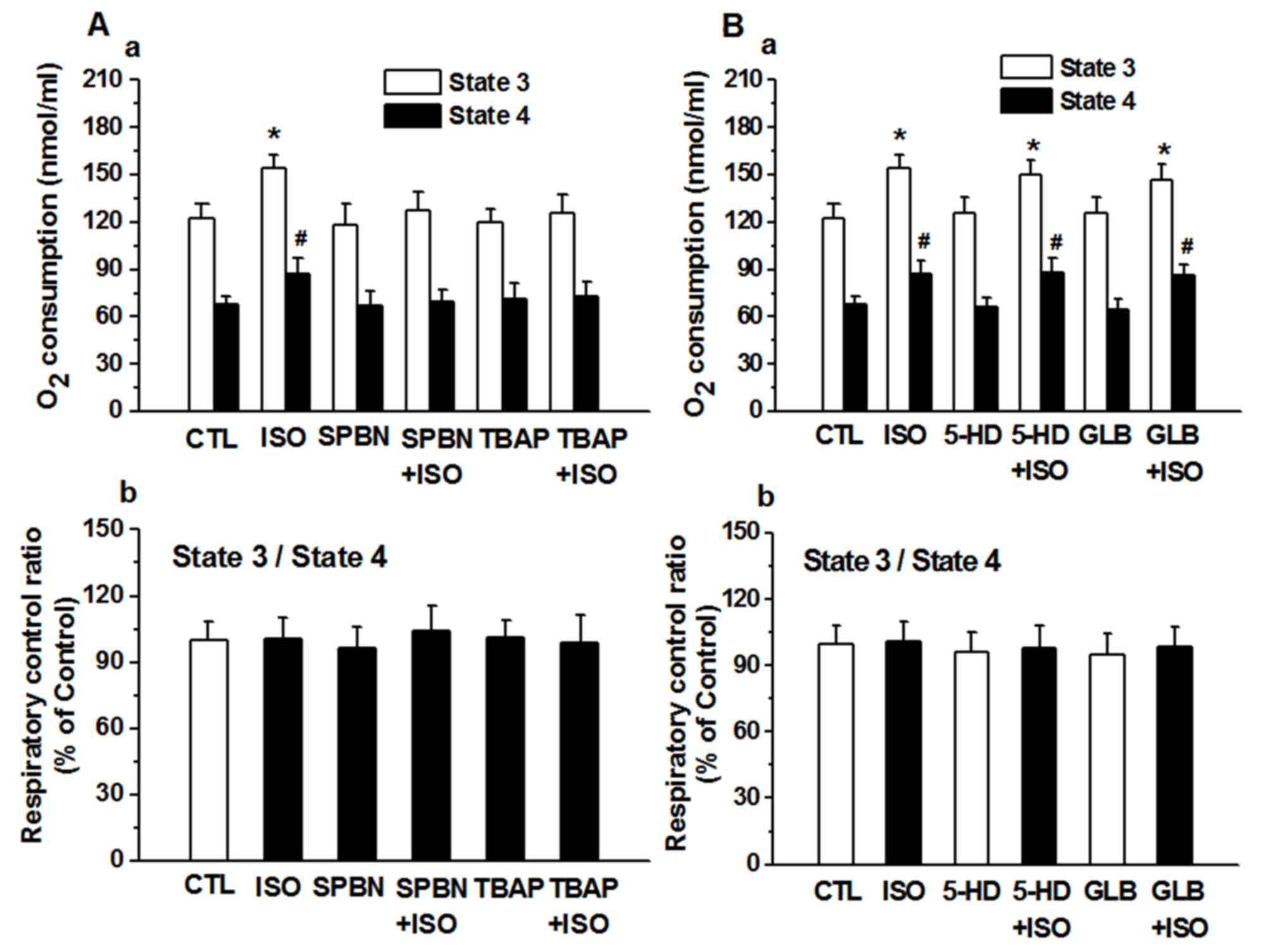
The ROS scavengers SPBN or TBAP did not
significantly alter the states of mitochondrial respiration
compared with that of the control group. Isoflurane significantly
increased state 3 and 4 respiration rates; however, SPBN and TBAP
inhibited the increase in the isoflurane-induced state 3 and 4
respiration rates in the presence of the substrate Suc (Fig. 4Aa). There was no significance
difference between ISO group and SPBN+ISO group or TBAP+ISO group.
The putative KATP channel blockers 5-HD and GLB did not
inhibit the increase in the isoflurane-induced state 3 and 4
respiration rates in the presence of the substrate Suc (Fig. 4B).
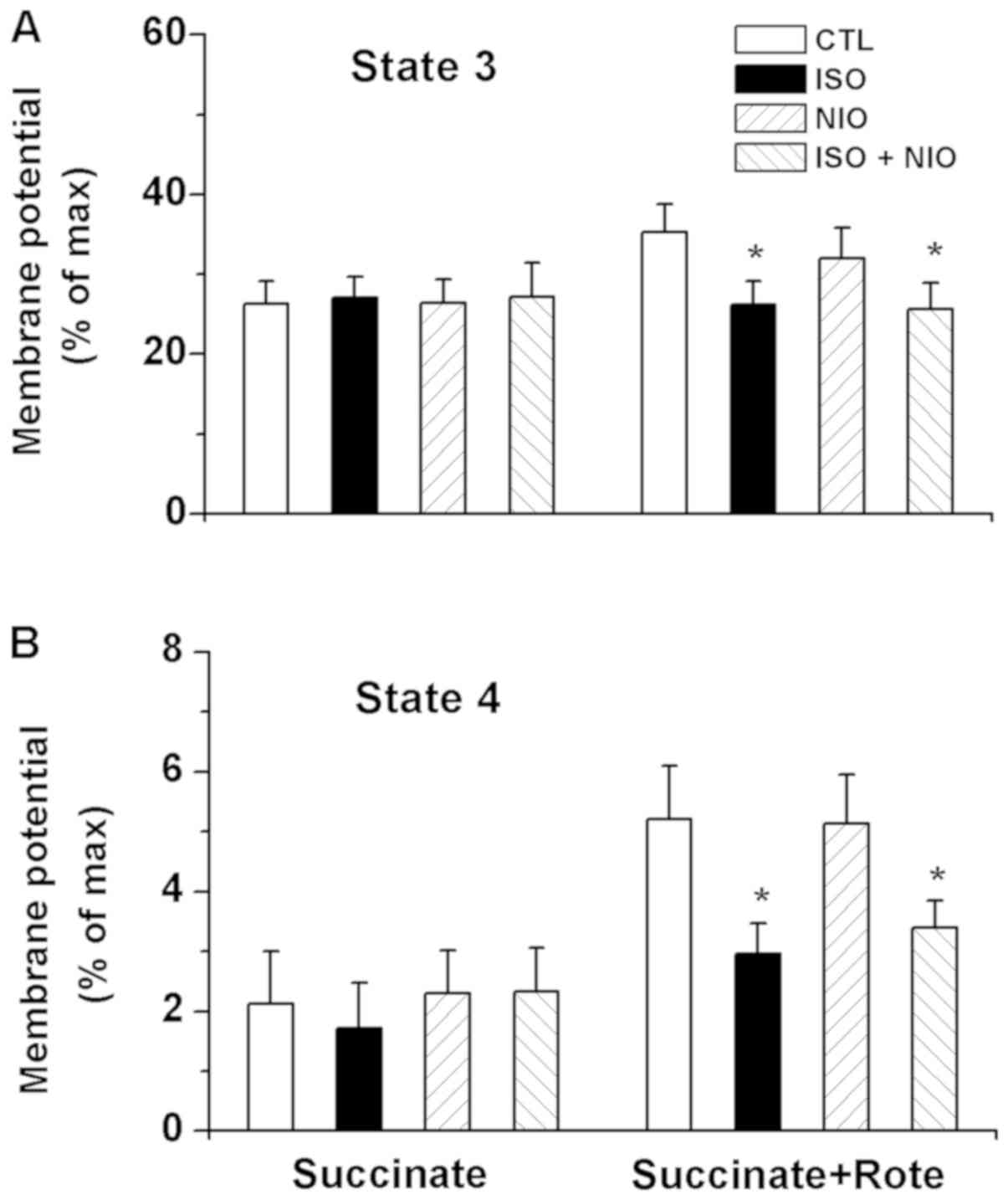
ΔΨm. The fluorescence of the membrane
potential-sensitive dye rhodamine 123 was measured in order to
examine the effects of isoflurane on ΔΨm. The data are presented as
the percentage of maximal depolarization by carbonyl
cyanide-m-chlorophenylhydrazenone (Fig. 5). Administration of isoflurane at
state 2 respiration caused no effects on the ΔΨm noted at state 3
and 4 respiration in the presence of Suc. However, depolarization
was significantly promoted by isoflurane at state 3 respiration in
the presence of Suc and Rote compared with the control; L-NIO did
not significantly alter the effects of isoflurane (Fig. 5A). Of note, significant
depolarization of ΔΨm at state 4 respiration was detected in the
Suc and Rote group compared with the control; L-NIO did not notably
inhibit depolarization induced by isoflurane (Fig. 5B).
Mitochondrial ROS generation
The baseline levels of mitochondrial
H2O2 release at state 2 respiration were
significantly higher in the Suc group compared with the Suc+Rote
group. Isoflurane induced a significant decrease in the
concentration of H2O2 compared with the
control, while L-NIO markedly inhibited the isoflurane-induced
decrease in H2O2 concentration levels
(Fig. 6A). A significant increase
in H2O2 release was observed compared with
the control in response to isoflurane with Suc and Rote; this
effect was not inhibited by L-NIO (Fig. 6B). Isoflurane and L-NIO did not
significantly alter H2O2 release rates during
ADP-initiated state 3 respiration in the Suc or Suc+Rote groups
(Fig. 6B).
Discussion
We recently reported that isoflurane exerts specific
effects on different states of mitochondrial respiration during the
oxidation of complex I substrates, which may not depend on
endogenous mitochondrial NO (18).
The results of the current study supported previous findings in
which: i) Isoflurane increased mitochondrial state 3 respiration in
isolated intact mitochondria in the presence of the complex II
substrate Suc; ii) this effect of isoflurane was inhibited by Rote;
iii) The NOS inhibitor L-NIO or the NO-sensitive guanylyl cyclase
ODQ did not inhibit the increase in the respiration rate (state 3)
caused by isoflurane in the presence of Suc; iv) the ROS scavengers
SPBN and TBAP inhibited the increase in the respiration rate
(states 3 and 4) caused by isoflurane in the presence of Suc. This
effect was not noted by the putative KATP channel
blockers 5-HD and GLB; v) isoflurane did not alter state 3
ΔΨm depolarization in the presence of Suc, whereas this
peak depolarization was decreased in the presence of Suc and Rote;
and vi) isoflurane reduced the concentration of
H2O2 during ADP-initiated state 3
respiration; this effect was not inhibited by L-NIO.
Volatile anesthetics have been reported to protect
the myocardium from infrared radiation damage by regulating
mitochondrial respiratory function (18,19,20,22).
Isoflurane attenuated state 3 respiration and reduced the RCR
during the oxidation of the complex I-chain substrate
glutamate/malic acid (18). In the
present study, isoflurane significantly enhanced state 3
respiration in the presence of Suc alone. This result is consistent
with the findings of Hirata et al (19). The analysis of mitochondrial
respiration in the presence of Suc alone indicated a higher rate of
ROS production, mainly due to the reverse transport of electrons
into complex I (23). Agarwal
et al (20) demonstrated
that isoflurane inhibited the O2 consumption of complex
I-chain substrates, but not of complex-II linked substrates
(20). In the current study, the
results revealed that Suc combined with Rote exhibited no
difference in the state 3 respiration rate or the RCR between the
control and the isoflurane-treated groups, consistent with the
aforementioned study (20). When
Suc was used without Rote, isoflurane increased O2
consumption. These results suggested that complex I was the target
of isoflurane. The Krebs cycle causes the conversion of Suc to
fumarate and malate, and subsequently to oxaloacetate (24). This occurs in the absence of Rote;
oxaloacetate causes a decrease in Suc oxidation by inhibiting
complex II (19). When complex I
activity is inhibited, NADH is oxidized, thereby lowering the
levels of NAD+, which in turn disrupts the oxidation of
malate to oxaloacetate (25).
Therefore, isoflurane may inhibit complex I by attenuating the
formation of oxaloacetate, thereby increasing the complex
II-associated respiration.
In the presence of the complex I substrates pyruvate
and malate, the ROS scavengers TBAP (simulated superoxide
dismutase) and SPBN (free radical spin trap) reduce the effects of
sevoflurane on state 3 respiration. The application of Suc in the
presence of Rote eliminates the effects of the two ROS scavengers
on the sevoflurane-induced attenuation of state 3 respiration
(18). In the present study
isoflurane induced an increase in the state 3 respiration rate with
the complex II substrate Suc (in the absence of Rote), whereas TBAP
and SPBN (ROS scavengers) inhibited the effects of isoflurane. This
suggested that complex I function was promoted by superoxide
formation in the absence of Rote during isoflurane exposure,
whereas the effects of isoflurane were inhibited in the presence of
the scavengers TBAP and SPBN. The results are in agreement with the
study by Riess et al (12)
in which a positive feedback mechanism of ROS formation was noted
in complex III. This feedback effect attenuated the function of
complex I, thereby producing additional ROS.
The opening of the mKATP channel
increases the production of ROS in the mitochondria isolated from
heart and liver, while ROS is produced in the electron transport
chain and notably BY complex I (26). However, Riess et al
(12) reported that complex
I-associated attenuation of mitochondrial respiration by the
volatile anesthetic sevoflurane was mediated by ROS and was
independent of mKATP channel opening. Volatile
anesthetics increase ROS formation via complex I and/or complex III
by affecting the reversible attenuation of mitochondrial electron
transport (12). ROS can affect
mitochondrial respiration by acting on downstream effectors, such
as the mKATP channel, which is normally closed at
physiological ATP levels (27). In
agreement with the study by Riess et al (12), the results of the present study
indicated that the putative KATP channel blockers 5-HD
and GLB did not inhibit isoflurane-induced state 3 and 4
respiration increase in the presence of Suc. Therefore, the
involvement of KATP channel or other potassium channel
opening with regards to the isoflurane-induced mitochondrial
respiratory state can be ruled out.
NO is produced by mitochondrial NOS, and regulates
mitochondrial O2 consumption and transmembrane potential
(28). The reaction of NO with
superoxide anion produces peroxynitrite, which induces oxidation
and/or nitrosation by irreversibly altering susceptible targets in
the mitochondria (28). The
activation of mitochondrial NOS by the volatile anesthetic
isoflurane produces NO, which affects respiration. Our previous
study demonstrated that the potent NOS inhibitor L-NIO did not
alter the effects of isoflurane on mitochondrial respiration and
did not affect normal mitochondrial respiration in the presence of
complex I substrates (18).
Guanylate cyclase (GC) converts GTP to cGMP and catalyzes the
synthesis of cGMP. NO activates soluble GC (sGC) and promotes cGMP
synthesis; sGC is inhibited by ODQ (29). In the current study, the NOS
inhibitor L-NIO or the sGC inhibitor ODQ were added prior to the
administration of isoflurane in the presence of complex II
substrates. These compounds demonstrated the same results as those
noted in the presence of the complex I substrates. This finding
further confirmed that the production of endogenous mitochondrial
NO did not influence the effects of isoflurane on mitochondrial
respiration.
The high production of ROS relies on high internal
ΔΨm, which is sensitive to complex I inhibition. Therefore, Rote
could prevent ROS production under reverse electron flow conditions
(30,31). In the presence of high proton
motive force, electrons are passed to NAD until the electron pool
is completely depleted and NAD is reduced to NADH (31). Subsequently, all upstream redox
centers are fully reduced and semiquinone transfers its unpaired
electrons to O2 (31).
It is known that additional ROS are produced under the
FADH2-ligating substrates during mitochondrial respiration
(30,32). ROS production in the presence of
Suc occurs due to the reverse electron flow into the complex I of
ETC, which is mainly dependent on the high ΔΨm (33,34).
The results of the present study supported the aforementioned
observations. A mitochondrial uncoupler was used, namely CCCP,
which allowed protons to re-enter to the inner mitochondrial
membrane and inhibit the production of H2O2,
which led to the collapse of ΔΨm. Any decrease in ΔΨm would abolish
ROS generation at this site. In addition, reverse electron
flow-induced ROS production can be highly attenuated by the complex
I blocker Rote and ADP. This rationale is partially in agreement
with the present findings, as the data indicated that the
depolarization of ΔΨm was reduced by isoflurane and L-NIO in the
presence of Suc and Rote. The decrease in mitochondrial damage was
caused by the block of the electron flow from complex I during
myocardial ischemia-reperfusion, which in turn occurred due to the
reduction in ROS production (35).
The production of ROS in the mitochondria is
initiated in the form of superoxide, by transferring electrons
directly to O2 (electron leakage) and converting them to
H2O2 via superoxide dismutase (28). A previous study has shown that
isoflurane reduced ROS production in intact mitochondria in the
presence of Suc, whereas in the presence of Rote, opposing effects
were noted (ROS increase) (19).
The results of the present study further indicated that isoflurane
significantly decreased the baseline levels of
H2O2 release in the Suc group during state 2
respiration, whereas it increased the baseline levels of
H2O2 release during state 3 respiration in
the Suc and Rote group. This effect was possibly mediated via
reverse electron flow. Endogenous mitochondrial NO production was
not involved in the decreased H2O2
concentration levels induced by isoflurane, since the NOS inhibitor
L-NIO did not affect the levels of H2O2
release. Due to the significant increase in the concentration of
Suc during hypoxia, the effect of isoflurane may be particularly
relevant during myocardial ischemia (36). Therefore, isoflurane could reduce
ROS production and may contribute to APC-induced cardioprotective
mechanisms. It has been reported in isolated cardiac mitochondria
that ROS serve different roles in sevoflurane-induced ETC
attenuation (12).
The present study indicated that isoflurane
increased mitochondrial state 3 respiration in isolated cardiac
mitochondria in the presence of the complex II substrate and
further confirmed that the effect of isoflurane on the
mitochondrial respiration was not associated with endogenous
mitochondrial NO production. Isoflurane decreased the
depolarization of ΔΨm, thus reducing ROS production caused by
reverse electron flow; however, this effect was determined to be
independent of mKATP channel opening. Altered
mitochondrial function is induced by volatile anesthetics and
attenuates ischemia/reperfusion injury (20). This provides an important component
in deciphering the complexity of cardiac protection (12,20).
The findings reported in the present study suggest a pivotal role
for isoflurane in regulating mitochondrial respiration and ROS
production, and provide useful information of the possible
synergistic action of volatile anesthetics at multiple sites of the
mitochondria. These synergistic effects may be the basis of the
cardioprotective effects induced by such compounds. Furthermore,
the effects of volatile anesthetics on mitochondrial function under
pathological conditions, and those involved in induced cardiac
protection require further investigation.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants (grant no.
SS201756 to Dr An and grant no. SS201613 to Dr Wang) from the
Suzhou Science and Technology Development Plan. In addition, grants
from the National Science and Technology Development Plan (grant
no. NSFC 81703501 to Dr Qiao), and from the Suzhou New District
Science and Technology Project (grant no. 2017Z004 to Dr An and
grant no. 2017Q003 to Lei Hong).
Authors' contributions
JW, JS, TC, CW and JA participated in conceiving and
designing the experiments; JW, JS, HL and SQ conducted the
experiments; JW, JS, TC, CW and JA were involved in the
interpretation of the studies, the analysis of the data and
reviewed of the manuscript; JW, JS and JA prepared the manuscript
draft. All authors read and approved the final version of the
manuscript.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Ethics approval and consent to
participate
The present study was approved by the Animal Care
and Use Committee of the Suzhou Science and Technology Town
Hospital.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Swyers T, Redford D and Larson DF:
Volatile anesthetic-induced preconditioning. Perfusion. 29:10–15.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ge ZD, Pravdic D, Bienengraeber M, Pratt
PF Jr, Auchampach JA, Gross GJ, Kersten JR and Warltier DC:
Isoflurane postconditioning protects against reperfusion injury by
preventing mitochondrial permeability transition by an endothelial
nitric oxide synthase-dependent mechanism. Anesthesiology.
112:73–85. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cao J, Xie H, Sun Y, Zhu J, Ying M, Qiao
S, Shao Q, Wu H and Wang C: Sevoflurane post-conditioning reduces
rat myocardial ischemia reperfusion injury through an increase in
NOS and a decrease in phopshorylated NHE1 levels. Int J Mol Med.
36:1529–1537. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Camara AK, Bienengraeber M and Stowe DF:
Mitochondrial approaches to protect against cardiac ischemia and
reperfusion injury. Front Physiol. 2:132011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li H and Lang XE: Protein kinase C
signaling pathway involvement in cardioprotection during isoflurane
pretreatment. Mol Med Rep. 11:2683–2688. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bolli R: Cardioprotective function of
inducible nitric oxide synthase and role of nitric oxide in
myocardial ischemia and preconditioning: An overview of a decade of
research. J Mol Cell Cardiol. 33:1897–1918. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chiari PC, Bienengraeber MW, Weihrauch D,
Krolikowski JG, Kersten JR, Warltier DC and Pagel PS: Role of
endothelial nitric oxide synthase as a trigger and mediator of
isoflurane-induced delayed preconditioning in rabbit myocardium.
Anesthesiology. 103:74–83. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Brown GC: Nitric oxide and mitochondrial
respiration. Biochim Biophys Acta. 1411:351–369. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shiva S, Oh JY, Landar AL, Ulasova E,
Venkatraman A, Bailey SM and Darley-Usmar VM: Nitroxia: The
pathological consequence of dysfunction in the nitric
oxide-cytochrome c oxidase signaling pathway. Free Radic Biol Med.
38:297–306. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
An J, Camara AK, Riess ML, Rhodes SS,
Varadarajan SG and Stowe DF: Improved mitochondrial bioenergetics
by anesthetic preconditioning during and after 2 hours of 27
degrees C ischemia in isolated hearts. J Cardiovasc Pharmacol.
46:280–287. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
An J, Camara AK, Rhodes SS, Riess ML and
Stowe DF: Warm ischemic preconditioning improves mitochondrial
redox balance during and after mild hypothermic ischemia in guinea
pig isolated hearts. Am J Physiol Heart Circ Physiol.
288:H2620–H2627. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Riess ML, Eells JT, Kevin LG, Camara AK,
Henry MM and Stowe DF: Attenuation of mitochondrial respiration by
sevoflurane in isolated cardiac mitochondria is mediated in part by
reactive oxygen species. Anesthesiology. 100:498–505. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hanley PJ, Ray J, Brandt U and Daut J:
Halothane, isoflurane and sevoflurane inhibit NADH: Ubiquinone
oxidoreductase (complex I) of cardiac mitochondria. J Physiol.
544:687–693. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ljubkovic M, Mio Y, Marinovic J, Stadnicka
A, Warltier DC, Bosnjak ZJ and Bienengraeber M: Isoflurane
preconditioning uncouples mitochondria and protects against
hypoxia-reoxygenation. Am J Physiol Cell Physiol. 292:C1583–C1590.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tanaka K, Weihrauch D, Kehl F, Ludwig LM,
LaDisa JF Jr, Kersten JR, Pagel PS and Warltier DC: Mechanism of
preconditioning by isoflurane in rabbits: A direct role for
reactive oxygen species. Anesthesiology. 97:1485–1490. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kevin LG, Novalija E, Riess ML, Camara AK,
Rhodes SS and Stowe DF: Sevoflurane exposure generates superoxide
but leads to decreased superoxide during ischemia and reperfusion
in isolated hearts. Anesth Analg. 96:949–955. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Novalija E, Kevin LG, Eells JT, Henry MM
and Stowe DF: Anesthetic preconditioning improves adenosine
triphosphate synthesis and reduces reactive oxygen species
formation in mitochondria after ischemia by a redox dependent
mechanism. Anesthesiology. 98:1155–1163. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xu F, Qiao S, Li H, Deng Y, Wang C and An
J: The effect of mitochondrial complex I-linked respiration by
isoflurane is independent of mitochondrial nitric oxide production.
Cardiorenal Med. 28:113–122. 2018. View Article : Google Scholar
|
|
19
|
Hirata N, Shim YH, Pravdic D, Lohr NL,
Pratt PF Jr, Weihrauch D, Kersten JR, Warltier DC, Bosnjak ZJ and
Bienengraeber M: Isoflurane differentially modulates mitochondrial
reactive oxygen species production via forward versus reverse
electron transport flow: Implications for preconditioning.
Anesthesiology. 115:531–540. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Agarwal B, Dash RK, Stowe DF, Bosnjak ZJ
and Camara AK: Isoflurane modulates cardiac mitochondrial
bioenergetics by selectively attenuating respiratory complexes.
Biochim Biophys Acta. 1837:354–365. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Heinen A, Camara AK, Aldakkak M, Rhodes
SS, Riess ML and Stowe DF: Mitochondrial Ca2+-induced K+ influx
increases respiration and enhances ROS production while maintaining
membrane potential. Am J Physiol Cell Physiol. 292:C148–C156. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Harisseh R, Chiari P, Villedieu C, Sueur
P, Abrial M, Fellahi JL, Ovize M and Gharib A: Cyclophilin D
modulates the cardiac mitochondrial target of isoflurane,
sevoflurane, and desflurane. J Cardiovasc Pharmacol. 69:326–334.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Saborido A, Soblechero L and Megias A:
Isolated respiring heart mitochondria release reactive oxygen
species in states 4 and 3. Free Radic Res. 39:921–931. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ackrell BA, Kearney EB and Mayr M: Role 3f
oxalacetate in the regulation of mammalian succinate dehydrogenase.
J boil Chem. 249:2021–2027. 1974.
|
|
25
|
Esteitie N, Hinttala R, Wibom R, Nilsson
H, Hance N, Naess K, Tear-Fahnehjelm K, von Döbeln U, Majamaa K and
Larsson NG: Secondary metabolic effects in complex I deficiency.
Ann Neurol. 58:544–552. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Andrukhiv A, Costa AD, West IC and Garlid
KD: Opening mitoKATP increases superoxide generation from complex I
of the electron transport chain. Am J Physiol Heart Circ Physiol.
291:H2067–H2074. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zaugg M, Lucchinetti E, Spahn DR, Pasch T
and Schaub MC: Volatile anesthetics mimic cardiac preconditioning
by priming the activation of mitochondrial K(ATP) channels via
multiple signaling pathways. Anesthesiology. 97:4–14. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Stowe DF and Camara AK: Mitochondrial
reactive oxygen species production in excitable cells: Modulators
of mitochondrial and cell function. Antioxid Redox Signal.
11:1373–1414. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Denninger JW and Marletta MA: Guanylate
cyclase and the .NO/cGMP signaling pathway. Biochim Biophys Acta.
1411:334–350. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Loschen G, Flohe L and Chance B:
Respiratory chain linked H(2)O(2) production in pigeon heart
mitochondria. FEBS Lett. 18:261–264. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lambert AJ and Brand MD: Inhibitors of the
quinone-binding site allow rapid superoxide production from
mitochondrial NADH: Ubiquinone oxidoreductase (complex I). J Biol
Chem. 279:39414–39420. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Turrens JF and Boveris A: Generation of
superoxide anion by the NADH dehydrogenase of bovine heart
mitochondria. Biochem J. 1191:421–427. 1980. View Article : Google Scholar
|
|
33
|
Liu Y, Fiskum G and Schubert D: Generation
of reactive oxygen species by the mitochondrial electron transport
chain. J Neurochem. 80:780–787. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Heinen A, Aldakkak M, Stowe DF, Rhodes SS,
Riess ML, Varadarajan SG and Camara AK: Reverse electron
flow-induced ROS production is attenuated by activation of
mitochondrial Ca2+-sensitive K+ channels. Am J Physiol Heart Circ
Physiol. 293:H1400–H1407. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chen Q, Camara AK, Stowe DF, Hoppel CL and
Lesnefsky EJ: Modulation of electron transport protects cardiac
mitochondria and decreases myocardial injury during ischemia and
reperfusion. Am J Physiol Cell Physiol. 292:C137–C147. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hoyer S and Krier C: Ischemia and aging
brain. Studies on glucose and energy metabolism in rat cerebral
cortex. Neurobiol Aging. 7:23–29. 1986. View Article : Google Scholar : PubMed/NCBI
|