Introduction
Bone resorption and formation are coupled to
maintain skeletal mass (1); this
process is tightly regulated to precisely replace any removed bone
with regards to location and quantity (2). However, the process is disrupted by
aging, which leads to net bone loss (2). As bone loss is a risk factor for
fragility fracture, it is essential to clarify the regulation of
bone remodeling at the cellular level. Osteoclasts (OCs) are formed
by the fusion of precursor cells to form the monocyte/macrophage
lineage (3). OC precursors (OCPs)
are drawn from the bloodstream into bone by various factors
released at sites undergoing resorption in the bone
microenvironment, and subsequently differentiate into OCs (3). The factors involved include
macrophage-colony-stimulating factor, receptor activator of NF-κB
ligand (RANKL), and transforming growth factor (TGF)-β1 (4). Sphingosine-1 phosphate controls the
migration of OCPs between bone tissues and the bloodstream
(5,6). Several studies have investigated cell
chemotaxis in the bone microenvironment (5–8);
however, OCP migration in the bone microenvironment has not been
analyzed. The stage at which OCPs differentiate into OCs is unclear
(before, during, or after migration to bone resorption sites in the
bone microenvironment).
TGF-β1 serves major roles in the proliferation,
migration, differentiation and survival of a variety cell types
(9). It is abundantly stored in
the bone matrix and has profound biological functions, including
roles in bone homeostasis (10,11).
The binding of TGF-β1 to its type II receptor leads to the
recruitment and phosphorylation of the type I receptor, thereby
activating downstream signaling pathways, including Smad and
non-Smad pathways (12). TGF-β1
consistently stimulates RANKL-induced OC differentiation in
differentiation models using RAW264.7 cells, which are widely known
as OCPs and are regarded as important for in vitro analyses
(13–15). Fox et al (16) reported that TGF-β directly induces
the expression of nuclear factor of activated T cells c1 (NFATc1),
a key regulator of OC differentiation. Yasui et al (17) demonstrated that TGF-β is essential
for RANKL-induced OC differentiation and has a possible role in the
molecular interaction between tumor necrosis factor
receptor-associated factor 6 and Smad2/3 in osteoclastogenic
RANKL/RANK signal transduction. Several molecules have been shown
to mediate a promoting effect of TGF-β1 on RANKL-induced OC
differentiation. Although the mechanisms by which
monocyte/macrophage-lineage cells differentiate into OCs are
well-defined, the effects of TGF-β1 on the characteristics of OCP
migration in vitro remain unclear. Macrophage migration is
affected by the duration of stimulation with TGF-β1 (18). The aim of the present study was to
determine the effect of TGF-β1 on the migration of OCPs in
vitro.
Materials and methods
Reagents
Recombinant human TGF-β1 was purchased from R&D
Systems, Inc., SB431542 was purchased from Sigma-Aldrich (Merck
KGaA), and recombinant human soluble RANKL was purchased from
Oriental Yeast Co., Ltd. α-minimum essential medium (α-MEM) and
fetal bovine serum (FBS) were purchased from Thermo Fisher
Scientific, Inc.
Cell culture
RAW264.7 cells were obtained from RIKEN BioResource
Center. Cells cultured in α-MEM supplemented with 10% FBS, 100 U/ml
penicillin, and 100 µg/ml streptomycin at 37°C under a humidified
5% CO2 atmosphere were used as control cells. Other
RAW264.7 cells were incubated at 37°C under a humidified 5%
CO2 atmosphere with or without TGF-β1 (2, 5 or 20
ng/ml), SB431542 (10 µM), and/or RANKL (50 ng/ml) for the duration
designated by the following experiments (Table I). Passages three to eight were
used for all experiments.
 | Table I.RAW264.7 cells were incubated with
and without TGF-β1, SB431542, and/or RANKL. |
Table I.
RAW264.7 cells were incubated with
and without TGF-β1, SB431542, and/or RANKL.
|
|
Group |
|---|
|
|
|
|---|
| Treatment | Co | T2 | T5 | T20 | T2S | T5S | R | RT2 | RT5 | RT20 | RT2S | RT5S |
|---|
| TGF-β1 (ng/ml) | − | 2 | 5 | 20 | 2 | 5 | − | 2 | 5 | 20 | 2 | 5 |
| RANKL (50
ng/ml) | − | − | − | − | − | − | + | + | + | + | + | + |
| SB431542 (10
µM) | − | − | − | − | + | + | − | − | − | − | + | + |
Cell proliferation assay
RAW264.7 cells were plated on 96-well culture plates
at 5×103 cells/well. After the addition of reagents at
various concentrations as aforementioned, the WST-8 reagent of a
Cell Counting Kit-8 (Dojindo Molecular Technologies, Inc.) was
added to the cells (10 µl/well) and incubated at 37°C for 2 h on
days 1, 2, 3 and 4. The number of viable cells was determined by
measuring the absorbance at 450 nm with a microplate reader
(Benchmark Plus™ Microplate Spectrophotometer; Bio-Rad
Laboratories, Inc.).
Tartrate-resistant acid phosphatase
(TRAP) staining
RAW264.7 cells were plated on 12-well culture plates
at 2×104 cells/well. After the addition of reagents at
various concentrations, the cultures were maintained for 7 days.
The culture medium, supplemented with reagents, was replaced every
3 days. Subsequently, the cells were stained using a TRAP staining
kit (Cosmobio Co., Ltd.), in accordance with the manufacturer's
instructions. TRAP-positive multinucleated cells were identified as
OCs at a magnification of ×200 by using a light microscope (Ti-E,
Nikon Instech Co., Ltd.).
TRAP activity assay
RAW264.7 cells were plated on 96-well culture plates
at 2.5×103 cells/well. After the addition of reagents at
various concentrations, the cultures were maintained for 2, 3 or 4
days. The cells were then fixed with ethanol/acetone (1:1) as
described previously (19), and
evaluated for TRAP activity using a TRAP solution kit (Oriental
Yeast Co. Ltd.), in accordance with the manufacturer's protocols.
Briefly, 150 µl of 50 mM citrate buffer (pH 4.5) containing 5.5 mM
p-nitrophenol phosphate and 10 mM sodium tartrate was added to each
well. After incubation for 60 min at room temperature, 50 µl of 0.1
N NaOH was added and the absorbance at 405 nm was determined using
a microplate reader (Benchmark Plus™ Microplate
Spectrophotometer).
NFATc1 activation assay
To determine the role of TGF-β1-induced OC
differentiation in RANKL-treated RAW264.7 cells, NFATc1 activation
was assessed. RAW264.7 cells were plated at 2×106 cells,
pretreated at 37°C with reagents (TGF-β1, SB431542, and/or RANKL)
for 24 h in 60-mm culture plates, and prepared nuclear extracts
using a Nuclear Extraction Kit (Abcam) according to the
manufacturer's protocol. NFATc1 activation in the cell lysates was
assessed using the NFATc1 Transcription Factor Kit (Abcam), in
accordance with the manufacturer's protocols. Briefly, 100 µl of
diluted primary antibody (included in the kit) was added to each
well. After incubation for 60 min at room temperature, 100 µl of
diluted horseradish peroxidase (HRP) antibody was added and
incubated for 1 h at room temperature and the absorbance at 450 nm
was determined using a microplate reader (Benchmark Plus™
Microplate Spectrophotometer).
Cell migration assay
Assays were performed with a 96-well cell migration
assay kit (CytoSelect™; Cell Biolabs Inc.). Following pretreatment
with reagents for 24 or 72 h, RAW264.7 cells were suspended in
serum-free medium and plated at 5×104 cells/well in the
upper part of 96-well, 8-µm pore, cell-migration chambers, in
accordance with the manufacturer's protocols. Medium containing 10%
FBS was placed in the lower wells as a chemotactic stimulus. After
incubation for 14 h at 37°C, the migrated cells were dissociated
from the membranes via the addition of cell detachment buffer
(included in the kit) to the lower wells, lysed and incubated with
CyQuant GR dye® (included in the kit) for 20 min at room
temperature. Migrated cells were quantified by determining the
fluorescence at 480/520 nm using a scanning fluorometer
(Infinite® 200 PRO; Tecan Group, Ltd.).
Western blot analysis
RAW264.7 cells were plated at 3×105
cells, pretreated with reagents for 24 or 72 h in 60-mm culture
plates and lysed with RIPA buffer (cat. no. sc24948, Santa Cruz
Biotechnology, Inc.) containing TBS, 1% Nonidet P-40, 0.5% sodium
deoxycholate, 0.1% SDS, 0.004% sodium azide with PMSF, sodium
orthovanadate and protease inhibitor cocktail. The cell lysates
were analyzed by western blotting, as described previously
(20,21). Equal amounts of protein (20 µg) in
each sample were separated by 10% SDS-PAGE for 30 min at a constant
voltage (200 V) and transferred onto a polyvinylidene fluoride
(PVDF) membrane (Bio-Rad Laboratories, Inc.). Membranes were
blocked in TBS + Tween-20 (TBST; 20 mM Tris, 500 mM NaCl pH 7.5 and
0.1% Tween-20) containing 5% non-fat milk for 1 h at room
temperature and then incubated overnight at 4°C with appropriate
primary antibodies. The following primary antibodies were used:
Rabbit polyclonal antibodies against RhoA (1:200; cat. no. sc179;
Santa Cruz Biotechnology, Inc.), Cdc42 (1:200; cat. no. sc87; Santa
Cruz Biotechnology, Inc.), and Rac1/2/3 (1:1,000; cat. no. 2465;
Cell Signaling Technology, Inc.), rabbit monoclonal antibody
against c-Fos (1:1,000; cat. no. 2250; Cell Signaling Technology,
Inc.), and goat polyclonal antibody against β-actin (1:1,000; cat.
no. sc1616; Santa Cruz Biotechnology, Inc.). Subsequently,
membranes were washed and incubated with secondary antibodies at
room temperature for 2 h. The secondary antibodies were
HRP-conjugated anti-goat IgG (1:2,000; cat. no. P0049; Dako;
Agilent Technologies, Inc.), HRP-conjugated anti-mouse IgG
(1:1,000; cat. no. 7076; Cell Signaling Technology, Inc.), and
HRP-conjugated anti-rabbit IgG (1:10,000; cat. no. 458; MBL Co.
Ltd., Nagoya, Japan). Signals were detected by chemiluminescence
using a Pierce SuperSignal Western Blotting Kit (Thermo Fisher
Scientific, Inc.). β-actin was evaluated as an internal control to
confirm that equal amounts of total protein were present.
Cell adhesion assay
Cell adhesion assays were performed as described
previously (22). RAW264.7 cells
were plated at 6×105 cells/well in 24-well culture
plates, and pretreated with reagents for 24 or 72 h. The cells were
then replated at 3×104 cells/well in 96-well culture
plates and incubated for 30 min at 37°C under a humidified 5%
CO2 atmosphere. Non-attached cells were removed by three
washes with PBS(−), and attached cells were evaluated with via an
Cell Counting Kit-8 assay as aforementioned by measuring the
absorbance at 450 nm using a microplate reader (Benchmark Plus™
Microplate Spectrophotometer; Bio-Rad).
Immunocytochemical analysis
After pretreatment of cells with reagents for 24 or
72 h, immunocytochemical analysis was performed. The primary
antibody used was a mouse anti-vinculin antibody (1:800; cat. no.
V9131; Sigma-Aldrich; Merck KGaA). Cultured cells were washed twice
with PBS(−), fixed with 3.7% paraformaldehyde for 20 min at room
temperature, and permeabilized with 0.1% Tween in PBS. After
blocking with 2% bovine serum albumin (Roche Diagnostics) for 1 h
at room temperature, the cells were treated with the primary
antibody at 4°C overnight. They were then washed and incubated with
an Alexa Fluor® 488-conjugated secondary antibody
(1:400, cat. no. A21206; Thermo Fisher Scientific Inc.) and
rhodamine phalloidin (1:200; cat. no. PHDH1; Cytoskeleton Inc.) at
room temperature in the dark for 2 h. After the cells had been
washed twice with PBS(−), they were mounted with Vectashield
containing DAPI (cat. no. H1500; Vector Laboratories Inc.) at room
temperature for 2 h. Fluorescence images were obtained for
evaluation of morphological changes, using a confocal laser
microscope (LSM 780; Zeiss AG).
The total numbers of cells with polygonal or spindle
morphology were counted in five non-overlapping fields at
magnification, ×200.
Statistical analysis
Experiments were repeated independently at least
three times. All data were expressed as mean ± standard deviation.
Statistical analyses were performed using one-way analysis of
variance followed by a Tukey-Kramer post-hoc test for intergroup
comparisons in each of the experiments using JMP statistical
software, Pro14.2 (SAS Institute Inc.). P<0.05 was considered to
indicate statistically significance difference.
Results
Effects of TGF-β1, SB431542 and RANKL
on cell proliferation
RAW264.7 cells were incubated with or without TGF-β1
(2, 5 or 20 ng/ml), SB431542 (10 µM), and/or RANKL (50 ng/ml) for 4
days. Cell proliferation was observed to be significantly inhibited
by TGF-β1 on days 3 and 4, and significantly inhibited by RANKL,
compared with control cells on day 4. The addition of SB431542
appeared to have reversed the inhibition (Fig. 1).
 | Figure 1.Cell proliferation assay. RAW264.7
cells at 5×103 cells/well in 96-well culture plates were
cultured with reagents at various concentrations for 1, 2, 3 or 4
days, and assessed for proliferation using the WST-8 reagent in the
Cell Counting Kit-8. *P<0.05 vs. Co cells. RANKL, receptor
activator of NF-κB ligand; TGF-β1, transforming growth factor-β1;
Co, control group; T2, TGF-β1 (2 ng/ml); T5, TGF-β1 (5 ng/ml); T20,
TGF-β1 (20 ng/ml); T5S, TGF-β1 (5 ng/ml) + SB431542; R, RANKL; RT2,
RANKL + TGF-β1 (2 ng/ml); RT5, RANKL + TGF-β1 (5 ng/ml); RT20,
RANKL + TGF-β1 (20 ng/ml); RT5S, RANKL + TGF-β1 (5 ng/ml) +
SB431542. |
Effects of TGF-β1 on OC
differentiation
To investigate the effects of TGF-β1 on OC
differentiation, we determined TRAP staining and activity. The
level of TRAP enzymatic activity in cultured cell lysates was
reported to be correlated with the relative number of OCs observed
by TRAP staining (23). On day 7,
RANKL induced the formation of large multinucleated (≥5 nuclei)
OC-like cells. Single treatment with TGF-β1 did not induce the
formation of TRAP-positive multinucleated cells, while combined
treatment with TGF-β1 and RANKL increased the number of
TRAP-positive multinucleated cells; the effect was reversed by
addition of SB431542 (Fig. 2A). To
confirm the potential effect of TGF-β1 on RANKL-induced OC
differentiation, we determined TRAP enzymatic activity. TRAP
activity was significantly increased in response to RANKL, and
further increased by combined treatment with RANKL and TGF-β1,
compared with control cells at day 3. The levels of TRAP activity
in RAW264.7 cells treated with TGF-β1 (2 ng/ml) and RANKL were
highest among all groups on day 4. TRAP activity after combined
treatment with TGF-β1 at 5 or 20 ng/ml and RANKL was markedly lower
than that after combined treatment with TGF-β1 at 2 ng/ml and
RANKL. Therefore, TGF-β1 combined with RANKL did not significantly
increase the level of TRAP activity in a dose-dependent manner. The
level of TRAP activity in cells treated with TGF-β1 (5 ng/ml) and
RANKL was significantly decreased by the addition of SB431542
compared with RT5 cells (Fig. 2B).
c-Fos protein expression in RAW264.7 cells was markedly elevated
following treatment with RANKL, compared with that in control cells
(Fig. 2C). The level of NFATc1 in
RAW264.7 cells that were treated with TGF-β1 (2 ng/ml) and RANKL
was significantly increased than that in RAW264.7 cells that were
treated with RANKL alone (Fig.
3).
 | Figure 2.Effect of TGF-β1 on differentiation
of RAW264.7 cells. (A) TRAP staining. After 7 days of culture with
reagents at various concentrations, RAW264.7 cells were stained for
TRAP. TRAP-positive multinucleated cells were observed under a
light microscope as osteoclasts. Scale bars, 100 µm. (B) TRAP
activity. RAW264.7 cells at 5×103 cells/well in 96-well
culture plates were cultured with reagents at various
concentrations for 2, 3 or 4 days, and analyzed for their TRAP
activity. *P<0.05 vs. compared with control cells.
#P<0.05. (C) Expression levels of c-Fos protein
evaluated by western blot analysis. RAW264.7 cells
(3×105 cells) were pretreated with reagents for 24 h in
60-mm culture plates and lysed. The cell lysates were subjected to
western blot analysis. RANKL, receptor activator of NF-κB ligand;
TGF-β1, transforming growth factor-β1; T2, TGF-β1 (2 ng/ml); T5,
TGF-β1 (5 ng/ml); T20, TGF-β1 (20 ng/ml); T5S, TGF-β1 (5 ng/ml) +
SB431542; R, RANKL; RT2, RANKL + TGF-β1 (2 ng/ml); RT5, RANKL +
TGF-β1 (5 ng/ml); RT20, RANKL + TGF-β1 (20 ng/ml); RT5S, RANKL +
TGF-β1 (5 ng/ml) + SB431542; OD, optical density; TRAP,
tartrate-resistant acid phosphatase. |
 | Figure 3.NFATc1 activation assay. RAW264.7
cells were plated at 2×106 cells per 60-mm culture
plate, pretreated with reagents at various concentrations for 24 h,
and lysed. NFATc1 activation was quantified in cell lysates. The
absorbance at 450 nm was determined using a microplate reader.
*P<0.05. RANKL, receptor activator of NF-κB ligand; TGF-β1,
transforming growth factor-β1; R, RANKL; RT2, RANKL + TGF-β1 (2
ng/ml); RT5, RANKL + TGF-β1 (5 ng/ml); RT20, RANKL + TGF-β1 (20
ng/ml); RT5S, RANKL + TGF-β1 (5 ng/ml) + SB431542. NFATc1, nuclear
factor of activated T cells 1. |
Effects of TGF-β1 and RANKL on cell
migration
To address the effect of TGF-β1 and RANKL on the
migration of RAW264.7 cells, we performed a Transwell migration
assay. In the presence of FBS in the lower chambers, pretreatment
with TGF-β1 (2 ng/ml) for 24 h followed by RANKL significantly
stimulated migration compared with control cells (Fig. 4A), while pretreatment with TGF-β1
(5 and 20 ng/ml) for 72 h significantly reduced migration (Fig. 4B). The increased migration
stimulated by TGF-β1 pretreatment for 24 h was notably inhibited by
the addition of SB431542 (Fig.
4A). In contrast, the suppressed migration induced by TGF-β1
for 72 h was markedly reversed by the addition of SB431542
(Fig. 4B). The effects of TGF-β1
treatment on migration were similar to the effects of combined
treatment with TGF-β1 and RANKL under the same durations of
treatment. These findings indicate that TGF-β1 has two different
sequential effects on RAW cell migration: Stimulation, followed by
inhibition. Treatment with RANKL alone did not significantly
enhance the migration of RAW264.7 cells.
 | Figure 4.Cell migration assay. Following
pretreatment with reagents at various concentrations for (A) 24 or
(B) 72 h, cells suspended in serum-free medium were plated in the
upper part of 96-well, 8-µm pore, cell-migration chambers. Medium
containing 10% fetal bovine serum was placed in the lower wells as
a chemotactic stimulus. After 14 h, migrated cells were detached
from the membranes and quantified by measuring the fluorescence at
480/520 nm using a scanning fluorometer. *P<0.05 vs. Co cells.
RANKL, receptor activator of NF-κB ligand; TGF-β1, transforming
growth factor-β1; Co, control group; T2, TGF-β1 (2 ng/ml); T5,
TGF-β1 (5 ng/ml); T20, TGF-β1 (20 ng/ml); T5S, TGF-β1 (5 ng/ml) +
SB431542; R, RANKL; RT2, RANKL + TGF-β1 (2 ng/ml); RT5, RANKL +
TGF-β1 (5 ng/ml); RT20, RANKL + TGF-β1 (20 ng/ml); RT5S, RANKL +
TGF-β1 (5 ng/ml) + SB431542. |
Effects of TGF-β1 and RANKL on
expression of Rho GTPases
Rho GTPases play key roles in the regulation of
cellular responses required for cell migration (24). We investigated whether RAW264.7
cell migration in response to TGF-β1 requires Rho GTPases. When
RAW264.7 cells were treated with TGF-β1 (2 and 5 ng/ml), or TGF-β1
(2 and 5 ng/ml) and RANKL for 24 h, RhoA and Rac protein expression
levels were markedly increased (Fig.
5). Conversely, when RAW264.7 cells were treated with TGF-β1
with or without RANKL for 72 h, RhoA and Rac protein expression
levels were significantly decreased, while that of Cdc42 was
slightly decreased (Fig. 5).
 | Figure 5.Effect of TGF-β1 and RANKL on
expression of Rho GTPases. Expression levels of RhoA, Rac1/2/3, and
Cdc42 proteins were evaluated by western blot analysis. RAW264.7
cells (3×105 cells) were pretreated with reagents at
various concentrations for 24 or 72 h in 60-mm culture plates, and
lysed. The cell lysates were subjected to western blot analysis.
RhoA and Rac1/2/3 data (pretreatment with reagents for 24 h) were
obtained from the same gel. Cdc42, cell division cycle 42; RANKL,
receptor activator of NF-κB ligand; TGF-β1, transforming growth
factor-β1; Co, control group; T2, TGF-β1 (2 ng/ml); T5, TGF-β1 (5
ng/ml); T20, TGF-β1 (20 ng/ml); T5S, TGF-β1 (5 ng/ml) + SB431542;
R, RANKL; RT2, RANKL + TGF-β1 (2 ng/ml); RT5, RANKL + TGF-β1 (5
ng/ml); RT20, RANKL + TGF-β1 (20 ng/ml); RT5S, RANKL + TGF-β1 (5
ng/ml) + SB431542. |
Effects of TGF-β1 and RANKL on cell
adhesion
Pretreatment with TGF-β1 (2 and 5 ng/ml) for 24 h
markedly reduced cell adhesion; this effect was reversed by
addition of SB431542 (Fig. 6A).
Extending the pretreatment with TGF-β1 (2 and 5 ng/ml) from 24 to
72 h induced a marked recovery in cell adhesion (Fig. 6B). Treatment with both TGF-β1 and
RANKL had an effect on cell adhesion similar to that of
pretreatment with TGF-β1 alone.
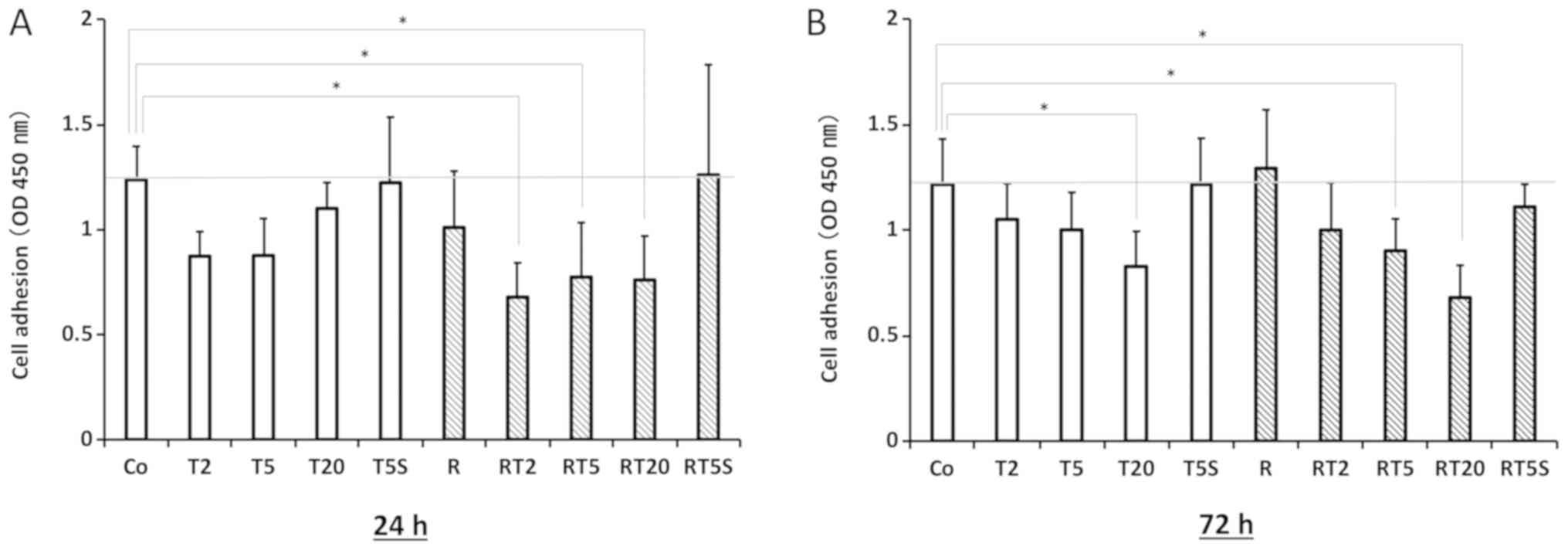 | Figure 6.Cell adhesion assay. RAW264.7 cells
(6×105 cells/well) were pretreated with reagents at
various concentrations for (A) 24 or (B) 72 h in 24-well culture
plates, re-plated in 96-well culture plates at 3×104
cells/well, and incubated for 30 min. Non-attached cells were
removed by three washes with PBS(−). Attached cells were assessed
using the Cell Counting Kit-8 by measuring the absorbance at 450 nm
with a microplate reader. *P<0.05. RANKL, receptor activator of
NF-κB ligand; TGF-β1, transforming growth factor-β1; Co, control
group; T2, TGF-β1 (2 ng/ml); T5, TGF-β1 (5 ng/ml); T20, TGF-β1 (20
ng/ml); T5S, TGF-β1 (5 ng/ml) + SB431542; R, RANKL; RT2, RANKL +
TGF-β1 (2 ng/ml); RT5, RANKL + TGF-β1 (5 ng/ml); RT20, RANKL +
TGF-β1 (20 ng/ml); RT5S, RANKL + TGF-β1 (5 ng/ml) + SB431542. |
Effects of TGF-β1 and RANKL on
morphological changes, the actin cytoskeleton and focal adhesions
of RAW264.7 cells
The actin cytoskeleton and focal adhesion formation
were evaluated by antibody staining (Fig. 7A and B). RAW264.7 cells treated
with TGF-β1 for 24 h exhibited morphological changes from round to
polygonal or spindle morphology (Fig.
7C). TGF-β1 treatment induced protrusions that were positive
for actin and vinculin (Fig. 7A).
The formation of protrusions was notably abolished in response to
TGF-β1 treatment for 72 h; RANKL treatment induced the formation of
multinucleated cells (Fig.
7B).
 | Figure 7.Immunohistochemical staining of actin
and vinculin in cells. (A and B) Cells pretreated with reagents at
various concentrations for (A) 24 or (B) 72 h were evaluated by
immunohistochemical staining. Rhodamine phalloidin-labeled F-actin
(red), Alexa Fluor 488-labeled vinculin (green), DAPI-stained
nuclei (blue), and overlaid fluorescent images of immunostained
cellular components (merged) are shown. Scale bars, 20 µm. (C)
Treatment with TGF-β1 resulted in morphological changes from round
to polygonal cellular morphology. The total numbers of cells with
polygonal or spindle morphology were counted in five
non-overlapping fields at magnification, ×200. *P<0.05. RANKL,
receptor activator of NF-κB ligand; TGF-β1, transforming growth
factor-β1; Co, control group; T5, TGF-β1 (5 ng/ml); R, RANKL; RT5,
RANKL + TGF-β1 (5 ng/ml). |
Discussion
OC differentiation is a key regulatory point for
bone disease therapy. It is important that the mechanism underlying
OC differentiation in the bone microenvironment is elucidated.
Since the discovery of the RANK signaling pathway, several studies
have focused on clarifying the involvement of this pathway in OC
differentiation and function (13,14),
and have provided insight into the mechanisms for OC
differentiation and activation in bone resorption (25). OCP migration ability during the OC
differentiation stage requires further investigation; thus, we
explored the link between OCP migration and differentiation over
time.
TGF-β inhibits the proliferation of epithelial,
endothelial and hematopoietic cell lineages (26–28).
The roles of TGF-β in tumorigenesis are complex and paradoxical.
Specifically, TGF-β has a tumor-suppressive role in normal tissues
and early-stage cancers, but switches to a tumor-promotive role in
late-stage cancers (29). The
switch in TGF-β function is known as the ‘TGF-β paradox’ (29). Understanding of the underlying
mechanisms that determine when and how TGF-β switches from a tumor
suppressor to a tumor promoter remains a great challenge in the
research field. Our study demonstrated the presence of a TGF-β
switch for OCPs in vitro.
In the present study, cell proliferation was
significantly inhibited in the presence of TGF-β1 after 3 days, and
this effect was prevented by the addition of SB431542. Cell
proliferation was also significantly inhibited by RANKL treatment
for 4 days. The TRAP activity in RAW264.7 cells was significantly
increased by RANKL treatment compared with control cells on day 4.
As RAW264.7 cells fused together and form large multinucleated
transparent cells from day 4, RANKL was proposed to inhibit
RAW264.7 cell proliferation to stimulate the fusion of these cells
and the subsequent formation of giant multinucleated transparent
cells. Park et al (30)
reported in a minireview that osteoclasts are bone-resorbing cells
derived from hematopoietic precursors, and require
macrophage-colony-stimulating factor and RANKL for their survival,
proliferation, differentiation, and activation. As RANKL promotes
cell proliferation, it enables partial recovery from the inhibition
of cell proliferation by TGF-β1, as demonstrated on day 4. OC
differentiation of RAW264.7 cells was significantly induced in
vitro by treatment with TGF-β1 and RANKL. TRAP activity on day
4 in RAW264.7 cells treated with both TGF-β1 (2 ng/ml) and RANKL
was the highest among the obtained data. When TGF-β1 signaling was
inhibited by the addition of SB431542 to the medium during culture,
OC differentiation was suppressed. Furthermore, we reported a
significant enhancement of OC differentiation in RANKL-treated
RAW264.7 cells that were exposed to TGF-β1, through the
quantitative measurement of NFATc1 activation. These findings
suggest that TGF-β1 accelerates RANKL-induced OC differentiation,
but does not act in a dose-dependent manner. TGF-β1 is known to
induce apoptosis in mature osteoclasts; Houde et al
(31) reported that this effect of
TGF-β1 is mediated by upregulation of Bim. TGF-β1 can also exhibit
the opposite effect, differentiation. Therefore, TGF-β1 may
influence OC differentiation when present throughout
differentiation; lower doses may promote differentiation, while
higher doses inhibit differentiation.
We analyzed the migration ability of RAW264.7 cells
using a Transwell chamber assay. Cell migration was increased by
TGF-β1 treatment at 24 h, but was decreased at 72 h. We found that
OCP migration was enhanced by TGF-β1 at the early stages of OC
differentiation. At later stages, the differentiated cells
exhibited suppressed migration. Kim et al (18) reported that treatment with TGF-β1
stimulated the migration of macrophages, while long-term exposure
decreased their migration. In the present study, TGF-β1 activated
RhoA at early stages, followed by inactivation, suggesting that
inactivation of RhoA may be the cause of the reduced cell migration
in response to TGF-β1 at later stages. RhoA and Rac expression
levels were increased after 24 h of TGF-β1 treatment, but were
notably reduced after 72 h of treatment. Cell migration was
decreased at 72 h following treatment with both TGF-β1 and RANKL
compared with control cells; TRAP activity had increased 72 h after
treatment with TGF-β1 and RANKL. Therefore, changes in cell
migration may be caused by OC differentiation. Prior to OC
differentiation, the expression levels of RhoA and Rac proteins,
which play key roles in regulating the cellular responses required
for cell migration (24), may
increase in response to TGF-β1, though these levels may be
decreased at the onset of differentiation. In contrast, cell
adhesion was significantly decreased after 24 h of TGF-β1
treatment, but increased after 72 h of treatment, except in
cultures treated with high-dose TGF-β1 (T20 and RT20 cells).
Therefore, adhesion ability was reduced by TGF-β1 prior to OC
differentiation, but was recovered by the onset of
differentiation.
RhoA activity has opposing effects, by decreasing
cell migration through adhesion while increasing migration through
cell-body contraction (24). High
levels of Rho activity are correlated with the potential of
attachment to the extracellular matrix, and also inhibit cell
migration (24,32). However, cell-body contraction of
moving cells was determined to be regulated by RhoA (33). In less adherent cells that lack
focal adhesions, such as macrophages and neutrophils, RhoA does not
affect adhesion, but does induce cell-body contraction (18). OCPs are monocyte/macrophage-lineage
hematopoietic precursor cells (3).
In macrophages, Allen et al (34) reported that Rho and Rac are
required for the cell migration process, while Cdc42 is not
essential.
We analyzed the morphological changes of RAW264.7
cells during differentiation by immunofluorescence staining.
RAW264.7 cells treated with TGF-β1 for 24 h displayed morphological
changes from round to polygonal cellular morphology. Furthermore,
TGF-β1 induced protrusions that were positive for actin and
vinculin. These changes in cells with a motile phenotype were
confirmed by Transwell chamber assays. The protrusions were
completely lost and the cell morphology changed from polygonal to
round after TGF-β1 treatment for 72 h.
Murphy-Ullrich (35) reported that the cell adhesion
process involved a transition from a state of weak adherence,
characterized by the attachment of a round cell to a substrate, to
a strongly-adherent state, which is characterized by focal
adhesions and stress fibers. The strongly-adherent state is
exhibited by differentiated and quiescent cells. There is an
intermediate state, which is characterized by a spread cell shape
and increased motility (35).
In conclusion, we proposed that when RAW264.7 cells
receive signals for OC differentiation, including TGF-β1 and RANKL
signals, TGF-β1 enhances the migration and alters the morphology of
RAW264.7 cells by inducing the production of RhoA and Rac prior to
differentiation. Subsequently, cells starting to differentiate show
reduced migration through decreases in RhoA and Rac production, and
the recovery of adhesion ability. Therefore, OCP migration may be
modified by differentiation in the bone microenvironment; further
studies are required to determine the factors that control these
phenomena. It is possible that clarification of the association
between migration and differentiation of OCPs could improve
understanding of bone-resorptive disorders mediated by OCs.
Acknowledgements
The authors would like to thank Professor Takashi
Daimon (Department of Medical Informatics, Hyogo College of
Medicine, Nishinomiya, Japan) for assistance with the statistical
analyses and Shinobu Osawa M.Sc. for technical assistance. The
authors also thank Dr Alison Sherwin, and Dr Ryan Chastain-Gross
for editing a draft of this manuscript.
Funding
The present study was supported by JSPS KAKENHI
(grant nos. 24593069, 15K11332, and 18K09825 to KT, 26861761 to MY,
and 19K19182 to MU) and by a Grant-in-Aid for Researchers, Hyogo
College of Medicine, 2016 (to KT).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MU, KT, and HK conceived and designed the study. MU,
KT, MY, HM, JT, and YN performed the experiments. MU, KT, and KN
analyzed the data. MU, KT, KN, and HK wrote the paper. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Rodan GA and Martin TJ: Therapeutic
approaches to bone diseases. Science. 289:1508–1514. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ota K, Quint P, Weivoda MM, Ruan M,
Pederson L, Westendorf JJ, Khosla S and Oursler MJ: Transforming
growth factor beta 1 induces CXCL16 and leukemia inhibitory factor
expression in osteoclasts to modulate migration of osteoblast
progenitors. Bone. 57:68–75. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Boyce BF, Zuscik MJ and Xing L: Biology of
bone and cartilageGenetics of bone biology and skeletal disease.
Thakker RV, Eisman J, Igarashi T and Whyte MP: Elsevier; London,
UK: pp. 3–24. 2012
|
|
4
|
Boyce BF: Advances in the regulation of
osteoclasts and osteoclast functions. J Dent Res. 92:860–867. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kikuta J, Iwai K, Saeki Y and Ishii M:
S1P-targeted therapy for elderly rheumatoid arthritis patients with
osteoporosis. Rheumatol Int. 31:967–969. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ishii M and Kikuta J:
Sphingosine-1-phosphate signaling controlling osteoclasts and bone
homeostasis. Biochim Biophys Acta. 1831:223–227. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yu X, Huang Y, Collin-Osdoby P and Osdoby
P: Stromal cell-derived factor-1 (SDF-1) recruits osteoclast
precursors by inducing chemotaxis, matrix metalloproteinase-9
(MMP-9) activity, and collagen transmigration. J Bone Miner Res.
18:1404–1418. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wright LM, Maloney W, Yu X, Kindle L,
Collin-Osdoby P and Osdoby P: Stromal cell-derived factor-1 binding
to its chemokine receptor CXCR4 on precursor cells promotes the
chemotactic recruitment, development and survival of human
osteoclasts. Bone. 36:840–853. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rahimi RA and Leof EB: TGF-beta Signaling:
A tale of two responses. J Cell Biochem. 102:593–608. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Janssens K, ten Dijke P, Janssens S and
Van Hul W: Transforming growth factor-beta1 to the bone. Endocr
Rev. 26:743–774. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tang SY and Alliston T: Regulation of
postnatal bone homeostasis by TGFβ. Bonekey Rep. 9:2552013.
|
|
12
|
Ikushima H and Miyazono K: TGF-b signal
transduction spreading to a wider field: A broad variety of
mechanisms for context-dependent effects of TGF-b. Cell Tissue Res.
347:37–49. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Koseki T, Gao Y, Okahashi N, Murase Y,
Tsujisawa T, Sato T, Yamato K and Nishirara T: Role of TGF-beta
family in osteoclastogenesis induced by RANKL. Cell Signal.
14:31–36. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shui C, Riggs BL and Khosla S: The
immunosuppressant rapamycin, alone or with transforming growth
factor-beta, enhances osteoclast differentiation of RAW264.7
monocyte-macrophage cells in the presence of RANK-ligand. Calcif
Tissue Int. 71:437–446. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chin SL, Johnson SA, Quinn J,
Mirosavljevic D, Price JT, Dudley AC and Thomas DM: A role for
alphaV integrinsubunitin TGF-beta-stimulated osteoclastogenesis.
Biochem Biophys Res Commun. 307:1051–1058. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fox SW, Evans KE and Lovibond AC:
Transforming growth factor-beta enables NFATc1 expression during
osteoclastogenesis. Biochem Biophys Res Commun. 366:123–128. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yasui T, Kadono Y, Nakamura M, Oshima Y,
Matsumoto T, Masuda H, Hirose J, Omata Y, Yasuda H, Imamura T, et
al: Regulation of RANKL-induced osteoclastogenesis by TGF-b through
molecular interaction between Smad3 and Traf6. J Bone Miner Res.
26:1447–1456. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kim JS, Kim JG, Moon MY, Jeon CY, Won HY,
Kim HJ, Jeon YJ, Seo JY, Kim JI, Kim J, et al: Transforming growth
factor-1 regulates macrophage migration via RhoA. Blood.
108:1821–1829. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Takahashi N, Yamana H, Yoshiki S, Roodman
GD, Mundy GR, Jones SJ, Boyde A and Suda T: Osteoclast-like cell
formation and its regulation by osteotropic hormones in mouse bone
marrow cultures. Endocrinology. 122:1373–1382. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hashitani S, Urade M, Nishimura N, Maeda
T, Takaoka K, Noguchi K and Sakurai K: Apoptosis induction and
enhancement of cytotoxicity of anticancer drugs by celecoxib, a
selective cyclooxygenase-2 inhibitor, in human head and neck
carcinoma cell lines. Int J Oncol. 23:665–672. 2003.PubMed/NCBI
|
|
21
|
Hiromoto T, Noguchi K, Yamamura M, Zushi
Y, Segawa E, Takaoka K, Moridera K, Kishimoto H and Urade M:
Up-regulation of neurophil gelatinase-associated lipocalin in oral
squamous cell carcinoma: Relation to cell differentiation. Oncol
Rep. 26:1415–1421. 2013.
|
|
22
|
Baksh D, Song L and Tuan RS: Adult
mesenchymal stem cells: Characterization, differentiation, and
application in cell and gene therapy. J Cell Mol Med. 8:301–316.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Simonet WS, Lacey DL, Dunstan CR, Kelley
M, Chang MS, Lüthy R, Nguyen HQ, Wooden S, Bennett L, Boone T, et
al: Osteoprotegerin: A novel secreted protein involved in the
regulation of bone density. Cell. 89:309–319. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ridley AJ: Rho GTPases and cell migration.
J Cell Sci. 114:2713–2722. 2001.PubMed/NCBI
|
|
25
|
Boyle WJ, Simonet WS and Lacey DL:
Osteoclast differentiation and activation. Nature. 423:337–342.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Blobe GC, Schiemann WP and Lodish HF: Role
of transforming growth factor-beta in human disease. N Engl J Med.
342:1350–1358. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Massague J: TGFbeta in cancer. Cell.
134:215–230. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tian M, Neil JR and Schiemann WP:
Transforming growth factor-β and the hallmarks of cancer. Cell
Signal. 23:951–962. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Morrison CD, Parvani JG and Schiemann WP:
The relevance of the TGF-β paradox to EMT-MET programs. Cancer
Lett. 341:30–40. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Park JH, Lee NK and Lee SY: Current
understanding of RANK signaling in osteoclast differentiation and
maturation. Mol Cells. 40:706–713. 2017.PubMed/NCBI
|
|
31
|
Houde N, Chamoux E, Bisson M and Roux S:
Transforming growth factor-beta1 (TGF-beta1) induces human
osteoclast apoptosis by up-regulating Bim. J Biol Chem.
284:23397–23404. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cox EA, Sastry SK and Huttenlocher A:
Integrin-mediated adhesion regulates cell polarity and membrane
protrusion through the Rho family of GTPases. Mol Biol Cell.
12:265–277. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mitchison TJ and Cramer LP: Actin-based
cell motility and cell locomotion. Cell. 84:371–379. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Allen WE, Zicha D, Ridley AJ and Jones GE:
A role for Cdc42 in macrophage chemotaxis. J Cell Biol.
141:1147–1157. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Murphy-Ulirich JE: The de-adhesive
activity of matricellular proteins: Is intermediate cell adhesion
an adaptive state? J Clin Invest. 107:785–790. 2001. View Article : Google Scholar : PubMed/NCBI
|














