Introduction
Fibrosis is defined by the excessive deposition of
collagenous and non-collagenous extracellular matrix components in
organs and tissues. Previous studies have demonstrated the
importance of aldosterone in the inflammatory and fibrotic
processes of kidney diseases, in which the profibrotic effects of
aldosterone are a consequence of both aldosterone-induced
hypertension and its effect on inflammatory cells and
myofibroblasts (1,2). The promotion of renal lesions caused
by aldosterone is commonly accompanied by a considerable
inflammatory response, primarily involving the release of
inflammatory factors by macrophages during renal fibrosis.
Allograft inflammatory factor 1 (AIF-1) serves an
important role in aldosterone-induced renal fibrosis (3) and is secreted by macrophages,
fibroblasts, endothelial cells and smooth muscle cells in a number
of immune-inflammatory disorders (4,5). As
a modulator of the immune response, AIF-1 promotes the
proliferation and migration of macrophages and human vascular
smooth muscle cells (6). It also
promotes the expression of inflammatory mediators, including
cytokines and chemokines, in macrophages, which in turn inhibits
macrophage apoptosis (7). Previous
findings have indicated that the expression of AIF-1 is a key
component in the pathogenesis of systemic sclerosis (SSc). Elevated
expression of AIF-1 was found in SSc-affected tissues and
peripheral blood mononuclear cells (8), which suggests that AIF-1 may serve an
important role in renal interstitial fibrosis. Therefore, the
present study aimed to investigate the role of AIF-1 in
aldosterone-induced renal interstitial fibrosis.
AIF-1 has been found to protect fibroblast-like
synoviocytes from NO-induced apoptosis by promoting the
phosphorylation of AKT serine/threonine kinase (AKT), and this
effect was subsequently inhibited by pretreatment with a
phosphatidylinositol 3-kinase (PI3K) inhibitor (9). The knockdown of AIF-1 in macrophages
significantly reduced the expression of phosphorylated (p-)AKT
(10), suggesting a close
association between the expression of AIF-1 and signaling
transduction within the PI3K/AKT/mammalian target of rapamycin
(mTOR) pathway. mTOR is a protein kinase regulated by a large
number of signaling molecules, including PI3K and AKT. It has been
highlighted as an important regulator of renal diseases, including
diabetic nephropathy (11) and
acute kidney injury (12); the
phosphorylation of constituents of the PI3K/AKT/mTOR pathway was
significantly increased in the kidneys of unilateral ureteral
obstruction (UUO) rats (13).
Additionally, aldosterone can bind with cytoplasmic
mineralocorticoid receptors in kidney fibroblasts, resulting in the
transactivation of growth factor receptors. This has been shown to
facilitate the rapid activation of PI3K and AKT, whereas the
inhibition of PI3K prevented this aldosterone-induced proliferative
response (14). Therefore, it was
hypothesized that the effect of AIF-1 in aldosterone-induced renal
interstitial fibrosis was considered to be associated with
activation of the PI3K/AKT/mTOR signaling pathway.
Aldosterone-induced renal interstitial fibrosis is
also associated with oxidative stress (15). NADPH oxidase 2 (NOX2), an important
member of the NADPH oxidases (NOXs), is expressed at high levels in
kidney tubular cells (16) and
serves a principal role in renal ischemia-reperfusion injury
(17) and fibrosis (18). Nuclear transcription factor
erythroid-related factor 2 (Nrf2) regulates antioxidative and
anti-inflammatory reactions during tissue injury. Previous studies
have illustrated that silencing of the Nrf2 gene intensified
inflammation, oxidative stress and histological changes. In a rat
model of chronic interstitial nephritis, the activation of Nrf2 was
significantly reduced and expression of its target genes was
downregulated (19). The
overexpression of angiotensinogen in renal proximal tubular cells
resulted in the cytosolic accumulation of Nrf2 and reduced the
translocation of Nrf2 into the nucleus, subsequently limiting the
expression of antioxidant-associated genes (20).
Therefore, aldosterone-induced renal injury is
associated with the upregulation of AIF-1, although it is not clear
whether AIF-1 functions through the PI3K/AKT/mTOR signaling
pathway. Furthermore, NOX2 and Nrf2 are involved in oxidative
stress during renal injury, but how this is affected by AIF-1 under
the influence of aldosterone remains to be fully clarified. The
present study aimed to elucidate the underlying molecular
mechanisms of aldosterone-induced renal fibrosis and to determine
the specific effects of AIF-1 on PI3K/AKT/mTOR signaling, NOX2 and
Nrf2 in this pathogenic process.
Materials and methods
Animals
Male Wistar rats (6–8 weeks of age, 200–250 g) were
purchased from the Experimental Animal Center of the Second
Affiliated Hospital of Harbin Medical University, (Harbin, China).
Animal experiments were performed in accordance with the Guiding
Principles for the Care and Use of Laboratory Animals (updated
2011; National Institutes of Health, Bethesda) and were approved by
the Experimental Animal Usage and Welfare Ethics Committee of
Harbin Medical University. Upon arrival, the animals were allowed
an adjustment period of 1 week and were housed in standard cages in
a quiet room on a 12-h light-dark cycle. The facility was
maintained at 20–22°C with a humidity of 50–60%. All rats were fed
with standard pellet laboratory chow and allowed free access to
water.
The animals were randomly divided into seven groups
(n=10 in each group) as follows: i) Control (sham-surgery and
distilled water infusion as vehicle); ii) aldosterone; iii)
aldosterone + spironolactone; iv) spironolactone; v) UUO; vi) UUO +
aldosterone; and vii) UUO + aldosterone + spironolactone.
Aldosterone (Sigma-Aldrich; Merck KGaA) was administered using an
osmotic pump (0.75 µg/h, subcutaneous infusion) and spironolactone
(Sigma-Aldrich; Merck KGaA) was administered orally (100
mg/kg/day). The experimental rats were anesthetized with 2%
pentobarbital solution at a dose of 3 ml/kg, and UUO induction and
aldosterone administration were conducted as follows: The rats were
placed in the prone position and the retroperitoneal area was
accessed through a skin incision. The left kidney was exposed, and
the proximal ureter was ligated with 4-0 silk thread. At the time
of UUO surgery, an Alzet 2002 osmotic mini-pump (Durect
Corporation) was inserted subcutaneously between the shoulder
blades; 0.75 µg/h aldosterone or vehicle was administered by
subcutaneous infusion. All animals were sacrificed with 5%
pentobarbital solution (3 ml/kg) 14 days after surgery and the
obstructed kidneys were removed for analysis.
In vitro experiments
The RAW264.7 macrophage cell line was purchased from
the American Type Culture Collection. RAW264.7 transduction was
conducted using lentivirus vectors (pGLV3/H1/GFP+Puro Vector and
LV5/EF-1αF/GFP/Puro Vector) encoded with AIF-1 or an AIF-1
silencer, synthesized by Shanghai GenePharma Co. Ltd. Briefly, the
cells were cultured in 24-well plates (5×104/well) in
0.5 ml Dulbecco's modified Eagle's medium (DMEM; 10% FBS, Thermo
Fisher Scientific, Inc.). Following incubation for 24 h at 37°C,
the cells were resuspended in 100 µl fresh DMEM containing
lentivirus vector or pShuttle vector (5 µg/ml per 1×108
cells/l), and incubated for 48 h. Following transduction, the
stable transfectants were isolated using fluorescence and
antibiotic selection (2.5 µg/ml puromycin; Clontech Laboratories,
Inc.). The expression of AIF-1 in the overexpression and knockdown
cells was determined using western blotting and reverse
transcription-quantitative (RT-q)PCR analysis. The pShuttle
vector-transduced RAW264.7 cells (pShuttle), AIF-1-overexpressing
cells and AIF-1/small interfering (s)iRNA cells (5×104;
siRNA sequence: 5′-GGTGAAGTACATGGAGTTTGA-3′) were subsequently
treated with aldosterone (10−6 M; Sigma-Aldrich, Merck
KGaA); untreated RAW264.7 cells were used as a control. The cells
were cultured in 75 cm2 bottles with 15 ml DMEM (10%
FBS), harvested 72 h after transduction and subjected to mRNA and
protein analyses.
Morphological analysis and
immunohistochemistry
To evaluate the severity of tubulointerstitial
inflammatory cell infiltration, kidney sections were processed and
stained with hematoxylin and eosin (H&E). Briefly, the renal
tissue samples (fixed with 4% paraformaldehyde solution or 24 h)
were embedded in paraffin and cut into 4-µm-thick slices. The
paraffinized sections were stained with H&E, analyzed by light
microscopy and images were captured at ×400 magnification. A total
of 10 H&E-stained fields were randomly observed in each group,
and the number of inflammatory cells in the average field was
calculated by three experienced pathologists. Renal collagen
deposition was evaluated using a Masson's trichrome staining kit
(cat. no. BA-4079B; Baso Diagnostics, Inc.), which revealed
collagen as blue-stained lesions in the kidney, and was
semi-quantified using ImageJ 1.8.0 software (National Institutes of
Health). Subsequently, 10 non-overlapping fields were scanned in
each kidney section and the positively-stained areas were
calculated as a percentage of the total area. The specific steps
were as follows: All sections were stained with
Wiegert-iron-hematoxylin (1:1) for 5 min at room temperature.
Following washing in running water for 10 min, the sections were
treated with hydrochloride-ethanol solution (1%) for 5 sec at room
temperature and rinsed under running tap water for a further 20
min. The sections were stained in ponceau (1%) staining solution
for 5–10 min at room temperature (observed under an electron
microscope at ×200 magnification) and washed using phosphomolybdic
acid solution (Baso Diagnostics, Inc.) for 5 min at room
temperature. All sections were stained with aniline blue solution
for 5 min at room temperature (observed under an electron
microscope; magnification, ×400), followed by washing in glacial
acetic acid for 1 min. Subsequent to dehydration with an ethanol
series (95 and 100%, 10 min each, twice) and xylene (5 min each,
twice), all sections were mounted and the collagen tissues were
stained blue. The sections were examined using light microscopy,
and images of 10 non-repeating visual fields were randomly captured
(magnification, ×400). Immunohistochemistry was performed as
described previously (3). For
blocking the antigen, 5% bovine serum albumin was used for 20 min
at room temperature. Then, the slides were incubated overnight at
4°C with the following primary antibody: Polyclonal rabbit anti-rat
AIF-1 (1:150; cat. no. ab153696; Abcam), and then an HRP-conjugated
secondary antibody (1:100; SV0002, Boster Biological Technology,
Ltd.) was used as the secondary antibody for 30 min at room
temperature. Images were captured with a Nikon ECLIPSE 80i
microscope (Nikon Corporation; magnification, ×400), and positive
staining was quantified using ImageJ software 1.8.0 software
(National Institutes of Health); 10 non-overlapping fields were
scanned in each kidney section and the areas of positive staining
were calculated as a percentage of the total area.
RT-qPCR analysis
Total RNA was isolated from the kidney tissues and
cells with the E.Z.N.A. Total RNA Kit II (Omega Bio-tek, Inc.)
according to the manufacturer's protocol. cDNA was acquired by
reverse transcription with the ImProm-II Reverse Transcriptase kit
(Promega Corporation). qPCR was performed with FastStart Universal
SYBR Green Master (ROX; Roche Diagnostics) using the
2−ΔΔcq method (21),
the thermocycling conditions were as follows: 95°C for 10 min for
activating FastStart Taq DNA Polymerase, 95°C for 15 sec, and 60°C
for 1 min (40 cycles for amplification). The expression levels of
the genes of interest were normalized to that of GAPDH. The
following primers were used: AIF-1 forward,
5′-GTTCCCAAGACCCATCTAGAGCTG-3′ and reverse,
5′-AGTTGGCTTCTGGTGTTCTTTGTTT-3′; PI3K forward,
5′-TTATTCTGGTTCTTGCGAAGTGAG-3′ and reverse,
5′-TGCTGCGTGAAGTCCTGTAG-3′; AKT forward,
5′-GAGGAGGAGACGATGGACTTC-3′ and reverse,
5′-GGCATAGTAGCGACCTGTGG-3′; mTOR forward,
5′-TTGTGTCCTGCTGGTCTGAAC-3′ and reverse,
5′-GCTCTTTGTAGTGTAGTGCTTTGG-3′; GAPDH forward,
5′-AGGTCGGTGAACGGATTTG-3′ and reverse,
5′-GGGGTCGTTGATGGCAACA-3′.
Western blotting
The preparation of protein samples from renal
tissues and cells was performed as previously described (3). Briefly, protein was extracted in 1X
SDS sample buffer following centrifugation at 12,000 × g for 15 min
at 4°C. The protein concentration of each sample was measured using
a BCA protein assay kit (cat. no. ab207002; Abcam). Equal
quantities of protein (20 µg) were separated using SDS-PAGE (8, 10
and 12%), transferred onto polyvinylidene difluoride membranes, and
then blocked in Tris-buffered saline/Tween 20 containing 5% non-fat
milk for 1 h at room temperature. The membranes were then incubated
overnight at 4°C with the following primary antibodies: Rabbit
anti-rat polyclonal AIF-1 (1:150; cat. no. ab153696; Abcam), rabbit
anti-rat polyclonal Nrf2 antibody (1:200; cat. no. ab31163, Abcam),
rabbit anti-rat polyclonal NOX2 antibody (1:300; cat. no. bs-3889R;
Bioss Antibodies), rabbit anti-rat monoclonal p-PI3K p85 (1:200;
cat. no. 4257; Cell Signaling Technology, Inc.), rabbit anti-rat
monoclonal p-AKT (Ser473) (1:200; cat. no. 4060; Cell Signaling
Technology, Inc.), rabbit anti-rat monoclonal p-mTOR (Ser2448)
antibody (1:200; cat. no. #5536; Cell Signaling Technology, Inc.)
and anti-β-actin monoclonal antibody (1:2,000; cat. no. A00702;
GenScript). Then the membranes were washed four times for 5 min
with TBST and incubated with horseradish peroxidase
conjugate-secondary antibodies (cat. no. ZB2301; 1:2,000 dilution;
OriGene Technologies, Inc.) for 1 h at 37°C. Imaging was performed
with enhanced chemiluminescence (ECL; Beyotime Institute of
Biotechnology) and the protein band densities were quantified by
Gel-Pro-Analyzer 4.0 software (Media Cybernetics, Inc.). The
results are expressed as an n-fold increase over the value of the
control group.
Statistical analysis
SPSS 17.0 software (SPSS Inc.) was used to perform
statistical analysis, and the results are presented as the mean ±
standard deviation. Differences between the means were assessed
using one-way ANOVA and the differences between multiple groups
were determined using Tukey's post hoc test. P<0.05 was
considered to indicate a statistically significant difference. All
experiments were performed at least three times.
Results
Aldosterone promotes renal
interstitial inflammatory cell infiltration and collagen deposition
in normal rats
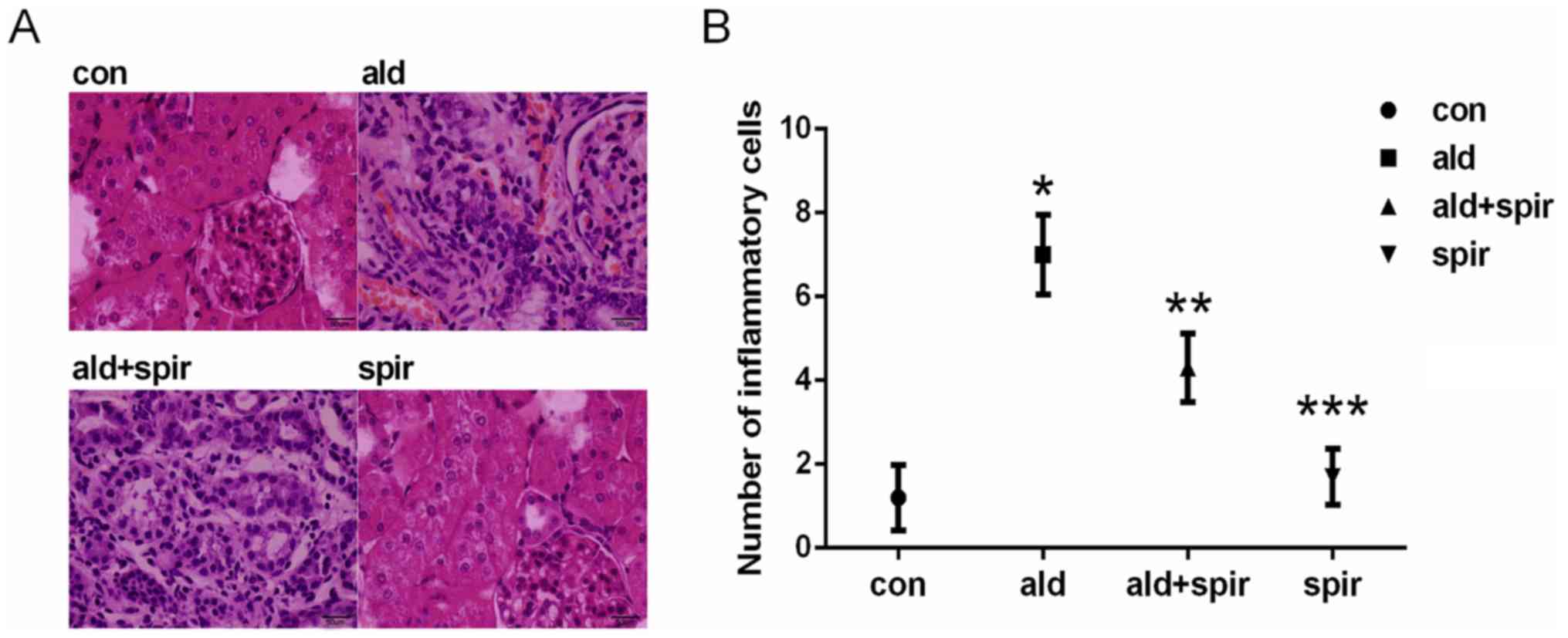
The profibrotic effects of aldosterone in normal
rats were determined using H&E and Masson's trichome staining.
The results showed that aldosterone promoted renal interstitial
inflammatory cell infiltration, tubular dilatation (Fig. 1A and B) and collagen deposition
(Fig. 2A and B) in normal rats.
Subsequent treatment with spironolactone reversed these
pathological changes, indicating that aldosterone promoted renal
interstitial fibrosis in normal rats.
Upregulation of AIF-1, PI3K, AKT, mTOR
and NOX2, and downregulation of Nrf2 is induced by aldosterone in
normal rats
The effects of aldosterone on the protein expression
levels of AIF-1, PI3K, AKT, mTOR, NOX2 and Nrf2 were determined in
normal rats. The results of western blotting revealed that
aldosterone promoted the expression of AIF-1, PI3K, AKT, mTOR and
NOX2, but inhibited the expression of Nrf2, and that spironolactone
inhibited these effects (Fig.
3A-G).
 | Figure 3.Effect of aldosterone on the protein
expression levels of (A) AIF-1, p-PI3K, p-AKT, p-mTOR, NOX2 and
Nrf2 in normal rats using western blotting. Aldosterone (0.75 µg/h
subcutaneous infusion) increased the expression of (B) AIF-1, (C)
p-PI3K, (D) p-AKT, (E) p-mTOR and (F) NOX2 and decreased the
expression of (G) Nrf2 in normal rats, which was attenuated by
spironolactone (100 mg/kg/day). *P<0.05 vs. con group,
**P<0.05 vs. ald group, ***P<0.05 vs. ald + spir group. The
data are presented as the mean ± standard deviation (n=3). AIF-1,
allograft inflammatory factor-1; p-PI3K, phosphorylated
phosphatidylinositol 3-kinase; p-AKT, phosphorylated AKT
serine/threonine kinase; p-mTOR, phosphorylated mammalian target of
rapamycin; NOX2, NADPH oxidase 2; Nrf2, nuclear transcription
factor erythroid-related factor 2; con, control; ald, aldosterone;
spir, spironolactone. |
Aldosterone promotes inflammatory cell
infiltration and collagen deposition in UUO rats
To further investigate the role of aldosterone in
renal interstitial fibrosis, a UUO model of fibrosis was
established in rats and used to examine the effects of aldosterone
on renal interstitial fibrosis. Histopathological changes to the
ureteral obstructed kidney sections were evaluated using H&E
and Masson's trichrome staining. The H&E staining results
revealed considerable interstitial inflammatory cell infiltration
in the aldosterone + UUO group compared with that in the UUO-only
group, and that treatment with spironolactone decreased
inflammatory cell infiltration (Fig.
4A and B). In line with the H&E results, Masson's trichrome
staining showed that, compared with UUO alone, aldosterone also
increased interstitial collagen deposition at 14 days post-UUO,
whereas the control group exhibited normal structure and reduced
collagen expression; spironolactone treatment reversed the effects
of aldosterone treatment (Fig. 5A and
B).
Aldosterone increases the expression
of AIF-1 in UUO rats
The expression of AIF-1 in the kidney tissues was
detected by immunohistochemistry, and semi-quantitative analysis of
AIF-1 was conducted using Image J software. The expression level of
AIF-1 in the normal renal interstitium was low and was notably
increased in the UUO group. Parallel to extracellular inflammatory
cell infiltration and collagen deposition, aldosterone further
increased the expression of AIF-1 compared with that in the UUO
group, which was inhibited by spironolactone (Fig. 6A and B). The mRNA expression level
of AIF-1 was also evaluated in the kidney, and the result was
consistent with the increase induced by aldosterone in UUO rats
observed in the quantitative immunohistochemical results (Fig. 7).
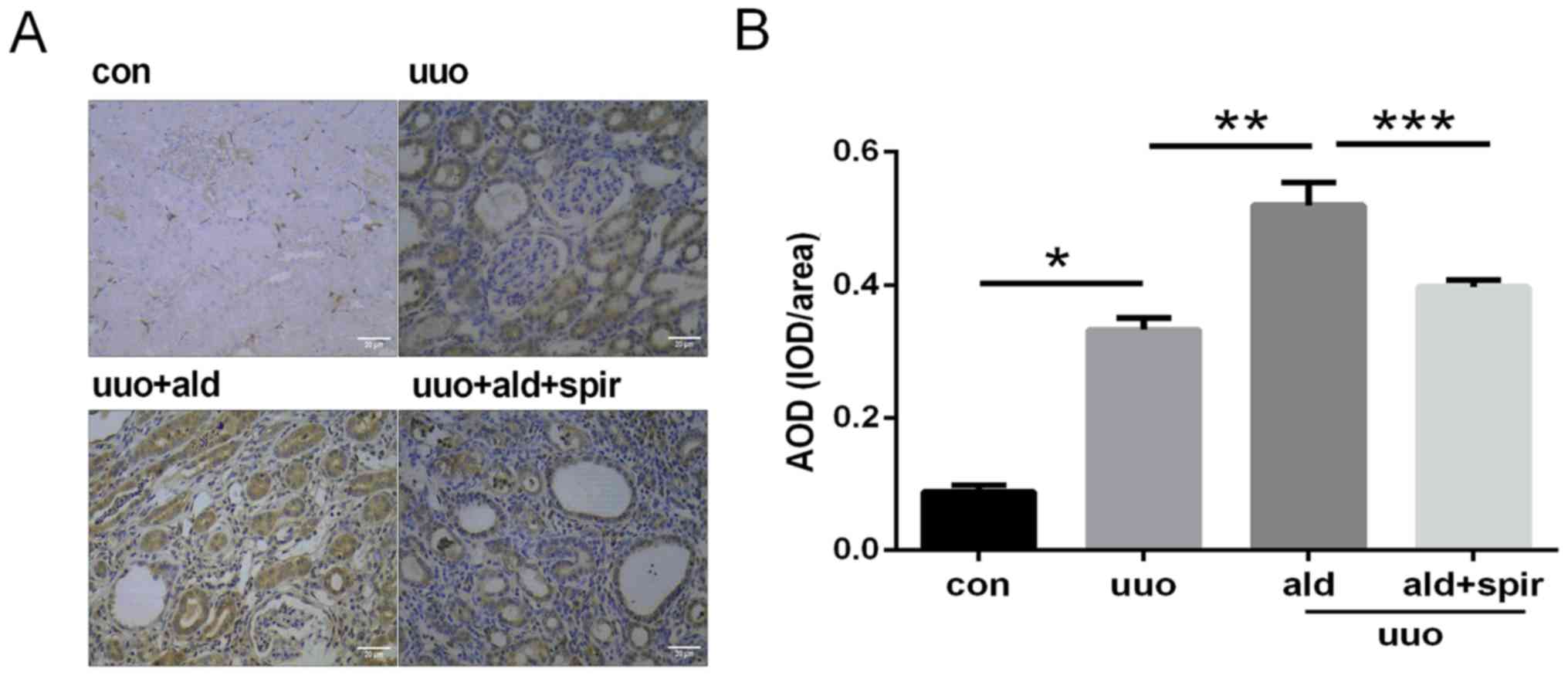 | Figure 6.Expression of AIF-1 in renal sections
by immunohistochemistry and semi-quantitative analysis of
expression levels of AIF-1. Magnification, ×400; scale bar=20 µm.
(A) Expression of AIF-1 was upregulated by aldosterone (0.75 µg/h
subcutaneous infusion) in the UUO rat model of renal fibrosis, and
inhibited by spironolactone treatment (100 mg/kg/day). (B)
Semi-quantitative analysis of expression levels of AIF-1.
*P<0.05 vs. con group, **P<0.05 vs. UUO group, ***P<0.05
vs. UUO + ald group. Data are presented as the mean ± standard
deviation (n=10). AIF-1, allograft inflammatory factor-1; con,
control; UUO, unilateral ureteric obstruction; ald, aldosterone;
spir, spironolactone; AOD, average optical density; IOD, integrated
optical density. |
Aldosterone upregulates the expression
of PI3K, AKT, mTOR, NOX2 and downregulates the expression of Nrf2
in the kidneys of UUO rats
In the ureteral obstructed kidney, the mRNA
expression levels of PI3K, AKT and mTOR were significantly
increased compared with those in the control group. Aldosterone
administration further upregulated the mRNA expression levels of
PI3K, AKT and mTOR in the UUO rats, whereas its effect was
significantly inhibited by spironolactone (Fig. 8A-C). Concurrently, the protein
expression levels of p-PI3K, p-AKT and p-mTOR in the kidney were
increased by aldosterone compared with those in the UUO-only group,
which was subsequently reversed by spironolactone (Fig. 9A-D).
 | Figure 9.(A) Western blot analysis of the
protein expression levels of p-PI3K, p-AKT, p-mTOR, NOX2 and Nrf2
in the rat kidney. Aldosterone (0.75 µg/h subcutaneous infusion)
upregulated the expression levels of (B) p-PI3K, (C) p-AKT, (D)
p-mTOR and (E) NOX2 and downregulated the expression of (F) Nrf2,
which was attenuated by spironolactone (100 mg/kg/day). *P<0.05
vs. con group, **P<0.05 vs. UUO group, ***P<0.05 vs. UUO +
ald group. Data are presented as the mean ± standard deviation
(n=3). p-PI3K, phosphorylated phosphatidylinositol 3-kinase; p-AKT,
phosphorylated AKT serine/threonine kinase; p-mTOR; phosphorylated
mammalian target of rapamycin; UUO, unilateral ureteric
obstruction; NOX2, NADPH oxidase 2; Nrf2, nuclear transcription
factor erythroid-related factor 2; con, control; ald, aldosterone;
spir, spironolactone. |
In order to investigate the influence of aldosterone
on oxidative stress in UUO rats, the protein expression levels of
NOX2 and Nrf2 were also determined by western blot analysis. The
results revealed that aldosterone increased the expression level of
NOX2 and decreased that of Nrf2 in the UUO rats. Treatment with
spironolactone attenuated the effects of aldosterone on the
expression levels of NOX2 and Nrf2 (Fig. 9E and F).
AIF-1 mediates the activation of AKT,
mTOR and aldosterone-induced oxidative stress in vitro
Based on the results of a previous study (3), the RAW264.7 macrophage cell line was
selected to examine the potential effect of AIF-1 on the
aldosterone-induced activation of the PI3K/AKT/mTOR pathway; these
cells constitutively express low levels of AIF-1 and facilitate the
positive selection of stable transfectants for AIF-1 gene
overexpression and knockdown. The results showed that aldosterone
increased the mRNA expression levels of PI3K, AKT and mTOR compared
with those in the untreated cells. With the same concentration of
aldosterone, the expression levels of AKT and mTOR were increased
in the cells overexpressing AIF-1, compared with those in the
control-transfected cells (pShuttle), although there was no obvious
difference in the expression of PI3K. The mRNA expression levels of
AKT and mTOR were decreased in the AIF-1/siRNA-transfected cells
compared with those in the control cells stimulated with
aldosterone; no difference in the mRNA expression level of PI3K was
observed (Fig. 10A-C). The
protein expression levels of p-PI3K, p-AKT and p-mTOR were also
detected by western blotting, and the results showed that
aldosterone increased the expression levels of p-PI3K, p-AKT and
p-mTOR compared with those in the untreated cells. The expression
levels of p-AKT and p-mTOR induced by aldosterone were increased in
cells overexpressing AIF-1, compared with those in the
control-transfected cells (pShuttle), although there was no
significant difference in the expression of p-PI3K. The expression
levels of p-AKT and p-mTOR were decreased in the
AIF-1/siRNA-transfected cells compared with those in the control
cells stimulated with aldosterone; no difference in the expression
level of p-PI3K was observed (Fig.
11A-D).
 | Figure 10.mRNA expression levels of (A) PI3K,
(B) AKT and (C) mTOR in cells. Compared with the control (RAW264.7
cells only), mRNA expression levels of PI3K, AKT and mTOR were
increased by aldosterone (10-6 M) in cells transfected with
pShuttle. The expression levels of AKT and mTOR increased
significantly in cells overexpressing AIF-1 and decreased in
AIF-1/siRNA cells. *P<0.05 vs. control, **P<0.05 vs. ald +
pShuttle. Data are presented as the mean ± standard deviation
(n=3). ald, aldosterone; PI3K, phosphatidylinositol 3-kinase; AKT,
AKT serine/threonine kinase; mTOR, mammalian target of rapamycin;
AIF-1, allograft inflammatory factor-1; siRNA, small interfering
RNA. |
 | Figure 11.Effect of aldosterone on the protein
expression levels of p-PI3K, p-AKT, p-mTOR, NOX2 and Nrf2 in
RAW264.7 cells. Aldosterone (10-6 M) increased the expression
levels of p-AKT, p-mTOR, NOX2 and Nrf2 via AIF-1 in RAW264.7 cells.
(A) Western blot image of p-PI3K, p-AKT, p-mTOR, NOX2 and Nrf2 in
different cells. Western blot analysis of (B) p-PI3K, (C) p-AKT,
(D) p-mTOR, (E) NOX2 and (F) Nrf2. *P<0.05 vs. control,
**P<0.05 vs. ald + pShuttle. Data are presented as the mean ±
standard deviation (n=3). p-PI3K, phosphorylated
phosphatidylinositol 3-kinase; p-AKT, phosphorylated AKT
serine/threonine kinase; p-mTOR, phosphorylated mammalian target of
rapamycin; NOX2, NADPH oxidase 2; Nrf2, nuclear transcription
factor erythroid-related factor 2; AIF-1, allograft inflammatory
factor-1; siRNA, small interfering RNA; over-exp,
overexpression. |
To further investigate the role of AIF-1 in
aldosterone-induced oxidative stress in RAW264.7 cells, the protein
expression levels of NOX2 and Nrf2 were evaluated. It was
demonstrated that aldosterone increased the expression levels of
NOX2 and Nrf2 compared with those in the untreated cells.
Additionally, the levels of NOX2 and Nrf2 were significantly
upregulated in cells overexpressing AIF-1 compared with those in
the control, pShuttle-transfected cells treated with aldosterone,
whereas the expression levels were reduced in the
AIF-1/siRNA-transfected cells compared with those in the control
group cells (Fig. 11E and F).
Discussion
In the present study, aldosterone was found to
promote inflammatory cell infiltration, collagen deposition and
tubular dilatation in normal rat kidney tissues, and these
pathological changes were alleviated following treatment with the
mineralocorticoid receptor antagonist spironolactone. These results
were consistent with those of another study (22) and highlighted the profibrotic
effect of aldosterone.
It is well known that aldosterone is associated with
the development of inflammatory renal fibrosis; the inflammatory
factor AIF-1 is important in the activation and proliferation of
macrophages, T lymphocytes, vascular smooth muscle cells and
endothelial cells (23,24). These cells are all involved in the
response to injury in renal interstitial fibrosis (25). In anti-glomerular basement membrane
nephritis, the expression level of AIF-1 was found to be
upregulated in infiltrating cells (26). In patients with type 2 diabetes,
the serum concentration of AIF-1 was positively correlated with
urinary albumin excretion and inversely correlated with the
estimated glomerular filtration rate (27). The results of the present study
indicated that, accompanied with aldosterone-induced renal
interstitial fibrosis, the expression level of AIF-1 was
simultaneously increased by aldosterone in normal rats, which may
indicate an important role of AIF-1 in kidney disease. However, few
studies have been conducted to confirm the function of
aldosterone-induced AIF-1 in the development of kidney fibrosis.
Therefore, in the present study, the effect of aldosterone on AIF-1
was examined using the UUO model of renal interstitial fibrosis.
The results demonstrated that aldosterone promoted inflammatory
cell infiltration and collagen deposition in the UUO rats. In line
with these pathological findings, the expression level of AIF-1 was
upregulated by aldosterone in UUO rats compared with that in
untreated UUO rats. The pathological changes and elevated
expression levels of AIF-1 were reversed by spironolactone, an
inhibitor of the mineralocorticoid receptor; this indicated that
aldosterone may induce kidney interstitial fibrosis through AIF-1
and its downstream signaling pathway, confirming the key role of
AIF-1 in this pathogenic process.
Other studies have indicated an association between
the expression of AIF-1 and signaling of the PI3K/AKT/mTOR pathway
(14). PI3K promotes the
conversion of phosphatidylinositol (4,5)-bisphosphate to phosphatidylinositol
(3,4,5)-trisphosphate, which results in the
activation of AKT (28). AKT
activation is critically involved in epithelial-mesenchymal
transition (29,30), an important process in kidney
fibrosis. AKT also regulates the expression of fibrotic proteins,
including fibronectin and composition-like collagens (31,32),
and the excessive and persistent accumulation of fibronectin and
composition-like collagens results in renal fibrosis. The
activation of PI3K and AKT further induces the activation of mTOR,
a central regulator of protein synthesis and cell growth in various
cells and organs; the expression of phosphorylated PI3K, AKT and
mTOR were reported to be increased in UUO rats (13), and collectively, these results
indicate a potential role for the PI3K/AKT/mTOR signaling pathway
in renal fibrosis. As supported by RT-qPCR results, aldosterone
upregulated the mRNA expression levels of PI3K, AKT and mTOR in the
UUO rats; concurrently, western blotting confirmed that aldosterone
promoted the phosphorylation of PI3K, AKT and mTOR, and it was
shown that these aldosterone-induced effects were reversed by
spironolactone. This indicated a fibrotic role for the
PI3K/AKT/mTOR signaling pathway in aldosterone-induced renal
interstitial fibrosis.
There is evidence to suggest that inhibition of the
expression of AIF-1 in macrophages suppresses the phosphorylation
of AKT (6). A number of studies
have shown that aldosterone promoted inflammatory cell infiltration
in the kidney, including that of macrophages. The present study
demonstrated considerably increased macrophage infiltration to the
renal interstitium of UUO rats, indicating an important role for
macrophages in renal interstitial fibrosis, in addition to the
localization of AIF-1 in infiltrating macrophages (3). Therefore, the present study selected
RAW264.7 macrophage cells, which constitutively express AIF-1, to
investigate the effect of AIF-1 on the aldosterone-induced
activation of AKT in vitro. This revealed that aldosterone
increased the expression levels of AKT and mTOR in RAW264.7 cells
transduced with the pShuttle vector and that the overexpression of
AIF-1 further promoted the phosphorylation and increased mRNA
expression levels of AKT and mTOR. By contrast, AIF-1 knockdown
counteracted the effects of aldosterone on AKT and mTOR in RAW264.7
cells, indicating that AIF-1 serves an important role in the
aldosterone-induced activation of AKT and mTOR in macrophages.
Aldosterone-induced renal injury is also associated
with oxidative stress. NADPH oxidases, one of a number of sources
of reactive oxygen species, are implicated in numerous
pathophysiologic processes. Of the seven NADPH oxidase isoforms
(Nox1-5, Duox1 and Duox2) identified, NOX2 is expressed at high
levels in kidney tubular cells and endothelial cells and has been
identified as a contributor to oxidative stress in kidney diseases
(33). In diabetic NOX2-knockout
mice, macrophage infiltration and the expression levels of
chemotactic factor MCP-1 were significantly reduced in the kidney
compared with observations in wild-type diabetic mice (34), indicating an association between
inflammation and oxidative stress. In the present study, it was
hypothesized that aldosterone exerted its effects on oxidative
stress through the inflammatory factor AIF-1. The results showed
that, in UUO rats, aldosterone upregulated the expression of
ROS-generating enzyme NOX2, which was in line with the
aldosterone-associated increase in the expression of NOX2 in
interstitial cardiac fibrosis (35). In vitro, aldosterone
increased the expression levels of NOX2 in pShuttle-transduced
cells; this expression was further upregulated in cells
overexpressing AIF-1, and the effect of aldosterone on the
expression of NOX2 was attenuated in AIF-1-knockdown cells. These
results verified the experimental hypothesis and may indicate a
novel mechanism linking aldosterone to oxidative stress via
AIF-1.
The aldosterone-associated upregulation of NOX2
in vivo was associated with the downregulation of Nrf2 in
the UUO kidney. Nrf2 is an important antioxidative transcription
factor, the activation of which ultimately results in
transcriptional regulation of diverse phase II detoxification and
antioxidant enzymes, including heme oxygenase 1 and NADPH quinone
dehydrogenase 1 (36). These
enzymes protect tissues and cells against oxidative stress. The
overexpression of Nrf2 in transforming growth factor-β-treated rat
mesangial cells and renal fibroblast cells decreased the expression
of α-smooth muscle actin, fibronectin and type 1 collagen (37). In the present study, the expression
level of Nrf2 decreased significantly in aldosterone-treated UUO
rats compared with that in the UUO-only group, 14 days after ureter
ligation. Furthermore, the expression level of AIF-1 was
upregulated, and these effects were reversed by spirolactone. These
results indicated a possible correlation between AIF-1 and Nrf2
caused by aldosterone. Notably, in RAW264.7 macrophage cells, 72 h
of aldosterone treatment promoted the expression of Nrf2, which was
further increased in cells overexpressing AIF-1 and decreased in
AIF-1/siRNA RAW264.7 cells. These results indicated that AIF-1 may
influence the expression of Nrf2. It has also been suggested that
the upregulation of Nrf2 may be a transient, adaptive-protective
response to inflammatory cytokines and AIF-1 in the early stages of
aldosterone stimulation; this was supported by the results of
Queisser et al (38) who
also observed an increase in the expression level of Nrf2 following
in vitro aldosterone treatment. However, in UUO rats,
aldosterone may trigger a considerable inflammatory response and
chemotactic effects; this early protective reaction may not be
sufficient to counteract the profibrotic effects of UUO and
aldosterone in vivo, ultimately leading to an imbalance in
the complex interplay between injury and protective factors. This
hypothesis was also supported by Queisser et al, who
hypothesized that Nrf2 was activated rather than inhibited in
aldosterone-induced liver fibrosis and, due to the limited
awareness of liver disease at the time, the reduced expression
level of Nrf2 was only observed in later disease stages (39).
In conclusion, the results of the present study
indicated that aldosterone promoted renal interstitial fibrosis in
UUO rats via AIF-1 and that AIF-1 serves an important role in
AKT/mTOR activation and aldosterone-induced oxidative stress in
macrophages. As a complicated dynamic process, further detailed
investigations are required to elucidate the mechanism of
aldosterone in renal fibrosis.
Acknowledgements
The authors would like to acknowledge the
contributions of Professor Wei Wang and Dr Hui Wang (Molecular and
Image Center laboratory, The Fourth Affiliated Hospital of Harbin
Medical University, Harbin, China) for their technical
assistance.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant. no. 81570638) and the
Scientific Funding Project for the Returned Overseas of Education
Department of Heilongjiang Province (grant. no. 1253HQ006).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
LH and XW were major contributors to the
experimental design. XL established the animal models. SZ performed
the protein analysis. YL and XY were involved in cell culture. XY
was involved in writing the manuscript, and the analysis and
interpretation of data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
All experimental procedures adhered to the
principles stated in the Guide for the Care and Use of Laboratory
Animals (updated 2011; National Institutes of Health, Bethesda, MD,
USA) and were approved by the Experimental Animal Usage and Welfare
Ethics Committee of Harbin Medical University (Harbin, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kang YS, Ko GJ, Lee MH, Song HK, Han SY,
Han KH, Kim HK, Han JY and Cha DR: Effect of eplerenone, enalapril
and their combination treatment on diabetic nephropathy in type II
diabetic rats. Nephrol Dial Transplant. 24:73–84. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Miana M, de Las Heras N, Rodriguez C,
Sanz-Rosa D, Martin-Fernandez B, Mezzano S, Lahera V,
Martinez-Gonzalez J and Cachofeiro V: Effect of eplerenone on
hypertension-associated renal damage in rats: Potential role of
peroxisome proliferator activated receptor gamma (PPAR-γ). J
Physiol Pharmacol. 62:87–94. 2011.PubMed/NCBI
|
|
3
|
Li Y, Wang X, Zhang L, Yuan X, Hao J, Ni J
and Hao L: Upregulation of allograft inflammatory factor-1
expression and secretion by macrophages stimulated with aldosterone
promotes renal fibroblasts to a profibrotic phenotype. Int J Mol
Med. 42:861–872. 2018.PubMed/NCBI
|
|
4
|
Deininger MH, Meyermann R and Schluesener
HJ: The allograft inflammatory factor-1 family of proteins. FEBS
Lett. 514:115–121. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liu G, Ma H, Jiang L and Zhao Y: Allograft
inflammatory factor-1 and its immune regulation. Autoimmunity.
40:95–102. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen X, Kelemen SE and Autieri MV: AIF-1
expression modulates proliferation of human vascular smooth muscle
cells by autocrine expression of G-CSF. Arterioscler Thromb Vasc
Biol. 24:1217–1222. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Watano K, Iwabuchi K, Fujii S, Ishimori N,
Mitsuhashi S, Ato M, Kitabatake A and Onoé K: Allograft
inflammatory factor-1 augments production of interleukin-6, −10 and
−12 by a mouse macrophage line. Immunology. 104:307–316. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Del Galdo F, Artlett CM and Jimenez SA:
The role of allograft inflammatory factor 1 in systemic sclerosis.
Curr Opin Rheumatol. 18:588–593. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu Y, Mei C, Du R and Shen L: Protective
effect of allograft inflammatory factor-1 on the apoptosis of
fibroblast-like synoviocytes in patients with rheumatic arthritis
induced by nitro oxide donor sodium nitroprusside. Scand J
Rheumatol. 42:349–355. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tian Y, Kelemen SE and Autieri MV:
Inhibition of AIF-1 expression by constitutive siRNA expression
reduces macrophage migration, proliferation, and signal
transduction initiated by atherogenic stimuli. Am J Physiol Cell
Physiol. 290:C1083–C1091. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lloberas N, Cruzado JM, Franquesa M,
Herrero-Fresneda I, Torras J, Alperovich G, Rama I, Vidal A and
Grinyó JM: Mammalian target of rapamycin pathway blockade slows
progression of diabetic kidney disease in rats. J Am Soc Nephrol.
17:1395–1404. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen G, Dong Z, Liu H, Liu Y, Duan S, Liu
Y, Liu F and Chen H: mTOR signaling regulates protective activity
of transferred CD4+Foxp3+ T cells in repair of acute kidney injury.
J Immunol. 197:3917–3926. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ma SK, Joo SY, Kim CS, Choi JS, Bae EH,
Lee J and Kim SW: Increased phosphorylation of PI3K/Akt/mTOR in the
obstructed kidney of rats with unilateral ureteral obstruction.
Chonnam Med J. 49:108–112. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Huang LL, Nikolic-Paterson DJ, Ma FY and
Tesch GH: Aldosterone induces kidney fibroblast proliferation via
activation of growth factor receptors and PI3K/MAPK signaling.
Nephron, Exp Nephrol. 120:E115–E122. 2012. View Article : Google Scholar
|
|
15
|
Toba H, Mitani T, Takahashi T, Imai N,
Serizawa R, Wang J, Kobara M and Nakata T: Inhibition of the renal
renin-angiotensin system and renoprotection by pitavastatin in type
1 diabetes. Clin Exp Pharmacol Physiol. 37:1064–1070. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shan Q, Zhuang J, Zheng G, Zhang Z, Zhang
Y, Lu J and Zheng Y: Troxerutin reduces kidney damage against
BDE-47-induced apoptosis via inhibiting NOX2 activity and
increasing Nrf2 activity. Oxid Med Cell Longev. 2017:60346922017.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Karim AS, Reese SR, Wilson NA, Jacobson
LM, Zhong W and Djamali A: Nox2 is a mediator of ischemia
reperfusion injury. Am J Transplant. 15:2888–2899. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fukuda M, Nakamura T, Kataoka K, Nako H,
Tokutomi Y, Dong YF, Ogawa H and Kim-Mitsuyama S: Potentiation by
candesartan of protective effects of pioglitazone against type 2
diabetic cardiovascular and renal complications in obese mice. J
Hypertens. 28:340–352. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Aminzadeh MA, Nicholas SB, Norris KC and
Vaziri ND: Role of impaired Nrf2 activation in the pathogenesis of
oxidative stress and inflammation in chronic tubulo-interstitial
nephropathy. Nephrol Dial Transplant. 28:2038–2045. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chang SY, Lo CS, Zhao XP, Liao MC, Chenier
I, Bouley R, Ingelfinger JR, Chan JS and Zhang SL: Overexpression
of angiotensinogen downregulates aquaporin 1 expression via
modulation of Nrf2-HO-1 pathway in renal proximal tubular cells of
transgenic mice. J Renin Angiotensin Aldosterone Syst.
17:14703203166687372016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak K and Schmittgen T: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Brown NJ: Contribution of aldosterone to
cardiovascular and renal inflammation and fibrosis. Nat Rev
Nephrol. 9:459–469. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Autieri MV and Carbone CM: Overexpression
of allograft inflammatory factor-1 promotes proliferation of
vascular smooth muscle cells by cell cycle deregulation.
Arterioscler Thromb Vasc Biol. 21:1421–1426. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Schluesener HJ, Seid K, Kretzschmar J and
Meyermann R: Allograft-inflammatory factor-1 in rat experimental
autoimmune encephalomyelitis, neuritis, and uveitis: Expression by
activated macrophages and microglial cells. Glia. 24:244–251. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tian Y, Jain S, Kelemen SE and Autieri MV:
AIF-1 expression regulates endothelial cell activation, signal
transduction, and vasculogenesis. Am J Physiol Cell Physiol.
296:C256–C266. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tsubata Y, Sakatsume M, Ogawa A, Alchi B,
Kaneko Y, Kuroda T, Kawachi H, Narita I, Yamamoto T and Gejyo F:
Expression of allograft inflammatory factor-1 in kidneys: A novel
molecular component of podocyte. Kidney Int. 70:1948–1954. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fukui M, Tanaka M, Asano M, Yamazaki M,
Hasegawa G, Imai S, Fujinami A, Ohta M, Obayashi H and Nakamura N:
Serum allograft inflammatory factor-1 is a novel marker for
diabetic nephropathy. Diabetes Res Clin Pract. 97:146–150. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ma Y, Vassetzky Y and Dokudovskaya S:
mTORC1 pathway in DNA damage response. Biochim Biophys Acta Mol
Cell Res. 1865:1293–1311. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kattla JJ, Carew RM, Heljic M, Godson C
and Brazil DP: Protein kinase B/Akt activity is involved in renal
TGF-beta1-driven epithelial-mesenchymal transition in vitro and in
vivo. Am J Physiol Renal Physiol. 295:F215–F225. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Meadows KN, Iyer S, Stevens MV, Wang D,
Shechter S, Perruzzi C, Camenisch TD and Benjamin LE: Akt promotes
endocardial-mesenchyme transition. J Angiogenes Res. 1:22009.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ghosh Choudhury G and Abboud HE: Tyrosine
phosphorylation-dependent PI 3 kinase/Akt signal transduction
regulates TGFbeta-induced fibronectin expression in mesangial
cells. Cell Signal. 16:31–41. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Runyan CE, Schnaper HW and Poncelet AC:
The phosphatidylinositol 3-kinase/Akt pathway enhances
Smad3-stimulated mesangial cell collagen I expression in response
to transforming growth factor-beta1. J Biol Chem. 279:2632–2639.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Asaba K, Tojo A, Onozato ML, Goto A, Quinn
MT, Fujita T and Wilcox CS: Effects of NADPH oxidase inhibitor in
diabetic nephropathy. Kidney Int. 67:1890–1898. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
You YH, Okada S, Ly S, Jandeleit-Dahm K,
Barit D, Namikoshi T and Sharma K: Role of Nox2 in diabetic kidney
disease. Am J Physiol Renal Physiol. 304:F840–F848. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Johar S, Cave AC, Narayanapanicker A,
Grieve DJ and Shah AM: Aldosterone mediates angiotensin II-induced
interstitial cardiac fibrosis via a Nox2-containing NADPH oxidase.
FASEB J. 20:1546–1548. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Numazawa S and Yoshida T: Nrf2-dependent
gene expressions: A molecular toxicological aspect. J Toxicol Sci.
29:81–89. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Oh CJ, Kim JY, Choi YK, Kim HJ, Jeong JY,
Bae KH, Park KG and Lee IK: Dimethylfumarate attenuates renal
fibrosis via NF-E2-related factor 2-mediated inhibition of
transforming growth factor-β/Smad signaling. PLoS One.
7:e458702012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Queisser N, Oteiza PI, Link S, Hey V,
Stopper H and Schupp N: Aldosterone activates transcription factor
Nrf2 in kidney cells both in vitro and in vivo. Antioxid Redox
Signal. 21:2126–2142. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Queisser N, Happ K, Link S, Jahn D, Zimnol
A, Geier A and Schupp N: Aldosterone induces fibrosis, oxidative
stress and DNA damage in livers of male rats independent of blood
pressure changes. Toxicol Appl Pharmacol. 280:399–407. 2014.
View Article : Google Scholar : PubMed/NCBI
|