Introduction
Inflammatory bowel disease (IBD), which includes
ulcerative colitis (UC) and Crohn's disease (CD), is a common
gastrointestinal disease characterized by intestinal inflammation
and intestinal mucosal injury (1).
Although infection, environmental factors, heredity, and
immunological abnormalities have been proposed as possible causes,
the pathogenesis of IBD remains unclear (2). Generally, aminosalicylic acid,
corticosteroids, immunomodulators, and antibiotics may cause
temporary remission of IBD; however, the curative effect of these
therapies is not evident (3).
Moreover, some adverse effects may arise during treatment.
Therefore, it is necessary to investigate the pathogenesis and
treatments of IBD.
Leptin is a satiety hormone primarily produced by
adipose tissue and exerts many biological effects by binding to its
receptor, ob-R. The primary function of leptin is controlling
appetite and adiposity (4).
Recently, leptin was revealed to be involved in inflammatory
responses in diseases such as IBD (5). The effects of leptin on IBD, however,
are controversial. For example, Singh et al (6) reported that the level of leptin was
increased in a rodent colitis model and that colitis was
ameliorated by a leptin antagonist, which suggests that leptin has
proinflammatory action. However, some studies have revealed that
leptin has anti-inflammatory effects in rats with colitis (7,8). In
previous clinical studies, the level of leptin in patients with IBD
was reported to be increased (9),
decreased (10) or unaltered
(11). Therefore, further studies
on leptin and IBD in the clinical setting and in experimental
animals are required.
The aim of the present study was to investigate the
role of leptin and the leptin receptor ob-R, as well as the
underlying mechanism of this role, in patients with UC and in a rat
colitis model.
Materials and methods
Patients and samples
Blood samples and colonic mucosa biopsy specimens
were obtained from 11 Chinese Han patients with active UC (22–83
years old; female 45.5%; from the inpatient and outpatient clinics
of the Second Hospital of Hebei Medical University) and 19 healthy
Chinese Han volunteers (26–67 years old; female 47.4%) between
March 2019 and May 2019. The diagnosis of UC was based on standard
clinical, endoscopic, and histological criteria (12). Patients with any other
inflammation, metabolic disease, and pregnancy were excluded from
the study. The patients did not receive any steroid treatments at
least 1 month before enrolment. The healthy control (HC) volunteers
had no gastrointestinal discomfort and had normal colonic mucosa
under colonoscopy. The procedures and study were approved by the
Ethics Committee of the Second Hospital of Hebei Medical University
and written informed consent was obtained from all patients.
The level of serum leptin was assessed using ELISA
(cat. no. EK197-96; LiankeBio). Furthermore, the protein expression
of the leptin receptor ob-R in the colonic mucosa was determined
using the western blot analysis method.
Animals and induction of colitis
Adult male leptin receptor-deficient (LR-D) Zucker
rats (weighing 500–600 g; 15 weeks old; purchased from Charles
River Laboratories) and age-matched wild-type (WT) control Zucker
rats (weighing 300–400 g) were used in the present study. The
animals were housed in a temperature-controlled room (22±1°C) with
a 12-h light/dark cycle and had free access to water and food. All
experiments were conducted in accordance with the Guide for the
Care and Use of Laboratory Animals (National Research Council,
1996), and all techniques and procedures were reviewed and approved
by the Hebei Medical University Institutional Animal Care and Use
Committee.
Experimental colitis was induced in a rat model
according to a previously described procedure (13). Briefly, after a 24-h fast, the rats
were anesthetized with isoflurane and a medical-grade polyurethane
cannula (external diameter, 2 mm) was inserted into the anus and
advanced to 8 cm proximal to the anal verge. 2,4,6-Trinitrobenzene
sulfonic acid (TNBS; Sigma-Aldrich; Merck KGaA), dissolved in 50%
ethanol, was instilled into the colon (100 mg/kg) to induce
colitis. At the end of instillation, the animals were maintained in
a head-down position for 2 min to prevent leakage of the instilled
TNBS. The body weight was measured daily. After the experiments,
all rats were euthanized using an overdose of sodium pentobarbital
(66 mg/kg, intraperitoneal).
Assessment of colitis
The disease activity index (DAI) was determined
based on stool consistency (0= firm, 1= loose, 2= diarrhea) and
occult blood (0= no blood, 1= occult blood, 2= gross rectal
bleeding) (14). The full colon
was removed and the length was measured. After being cut
longitudinally, colon tissues were rinsed in physiological saline
to remove fecal residue. The gross appearance of colitis was
evaluated based on adhesions (score 0–2), ulcer, and inflammation
(score 0–8) (15).
Colon histology
Sections of inflamed colon were collected and
immersed in 4% paraformaldehyde at 4°C for 48 h and then dehydrated
in gradient ethanol step by step. After embedding in wax, tissues
were sectioned at 6-µm thickness using a microtome and then stained
with hematoxylin-eosin at room temperature for 10 min. Pathological
changes in the colon were imaged with a light microscope digital
camera (magnification, ×50). Microscopic scores were evaluated
based on morphological features according to the severity of
inflammation (score 0–3), extent of inflammation (0–3), and crypt
damage (0–4) (16).
Colon cytokine and myeloperoxidase
(MPO) analysis
Sections of inflamed colon were homogenized with
saline, and the supernatant was obtained after centrifugation for
15 min at 1,004 × g at 4°C. The interleukin-1β (IL-1β; cat. no.
EK201B/3-96), IL-6 (cat. no. EK306/3-96) and tumor necrosis
factor-α (TNF-α; cat. no. EK382/3-96) levels, and the MPO (cat. no.
EK2133/2-96) activity were detected using commercial ELISA kits
(LiankeBio).
Western blot analysis
The collected colon tissues were homogenized in a
lysis buffer (Beyotime Institute of Biotechnology), and the protein
level was determined using the Bradford assay. Protein samples (50
µg) were separated by SDS-PAGE on 10% gels, transferred to a
polyvinylidene fluoride membrane that was blocked for 1 h with 5%
(w/v) non-fat milk in TBS, and incubated with primary antibodies
against the leptin receptor ob-R (1:50; cat. no. sc-8391; Santa
Cruz Biotechnology, Inc.), signal transducer and activator of
transcription 3 (STAT3; 1:1,000; cat. no. 9139; Cell Signaling
Technology, Inc.), phosphorylated STAT3 (pSTAT3; 1:2,000; cat. no.
9145; Cell Signaling Technology, Inc.), nuclear factor (NF)-κB-p65
(1:2,000; cat. no. ab16502; Abcam) and Ras homolog gene family
member A (RhoA; 1:2,000; cat. no. ab219371; Abcam) overnight at
4°C. The same membrane was stripped and re-blotted with an
anti-β-actin antibody (1:5,000; cat. no. T0022; Affinity
Biosciences) for normalization. The membranes were then incubated
with corresponding secondary antibodies (1:5,000, Goat Anti-Rabbit,
cat. no. ab6721; 1:5,000, Rabbit Anti-Mouse, cat. no. ab6728; both
Abcam) for 1 h at room temperature. Blots were developed using the
chemiluminescent detection method (Immobilon Western HRP; EMD
Millipore). The protein blots were quantified by densitometry using
ImageJ (version 1.46; National Institutes of Health) and normalized
to β-actin.
Statistical analysis
All statistical analyses were performed with
GraphPad Prism, version 6 (GraphPad Software, Inc.). The sex ratio
with respect to clinical characteristics was assessed using the
χ2 test. Differences in measurement data were assessed
using one-way analysis of variance followed by Tukey's test or
two-tailed Student's t-test. Data are presented as the mean ±
standard error of mean, unless indicated otherwise. P<0.05 was
considered to indicate a statistically significant difference.
Results
Leptin receptor is elevated in
patients with UC
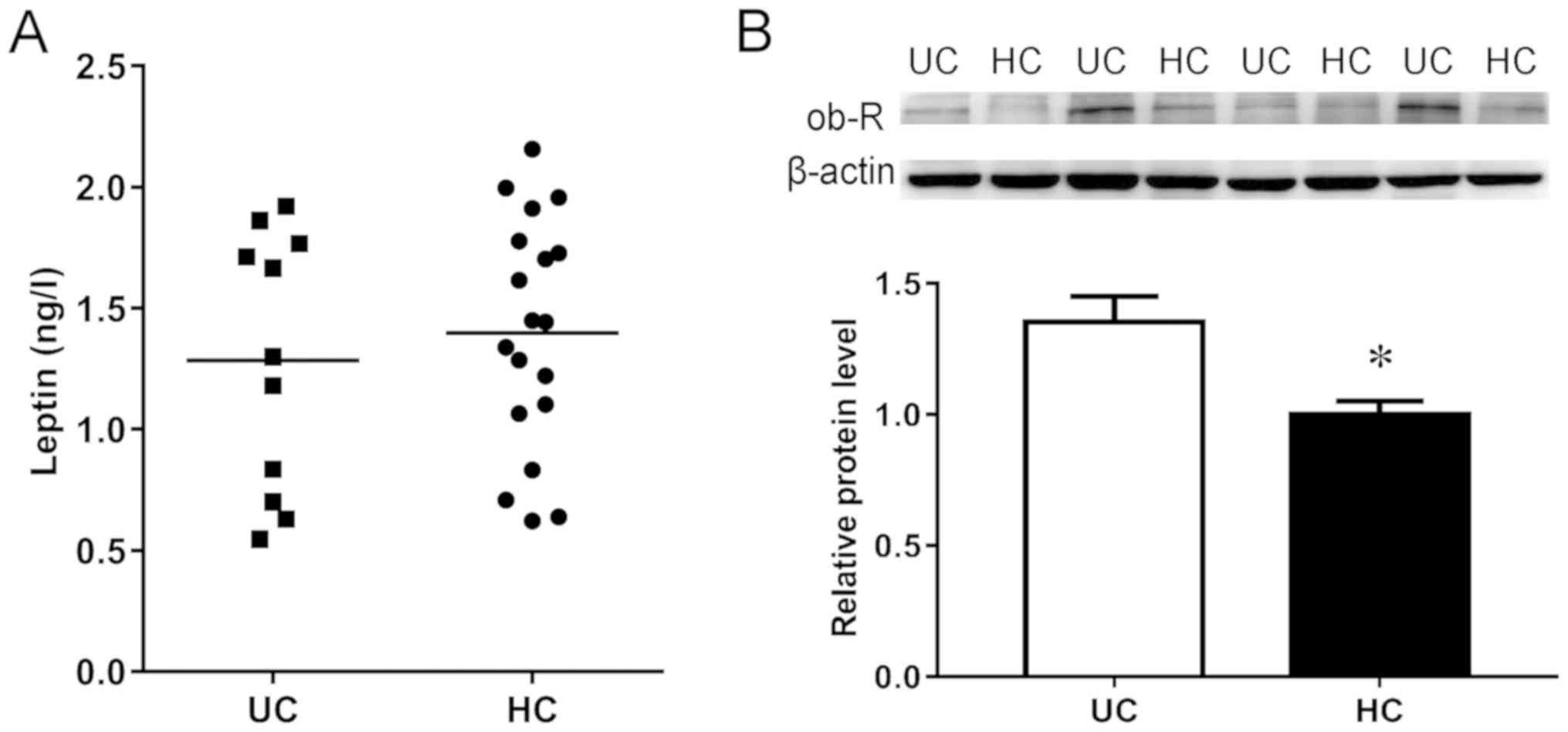
There were no significant differences in age, sex,
body mass index (BMI), and serum leptin level between patients with
UC and the HC group (Table I;
Fig. 1A). However, the expression
of the leptin receptor ob-R in the colonic mucosa was increased in
patients with UC compared with the HC group (Fig. 1B), indicating that leptin signaling
was enhanced in patients with UC.
 | Table I.Clinical characteristics. |
Table I.
Clinical characteristics.
| Characteristic | Ulcerative colitis
(n=11) | Healthy control
(n=19) | P-value |
|---|
| Female, % | 45.5 | 47.4 | 0.919 |
| Age, years | 43.5±5.0 | 44.6±2.8 | 0.847 |
| BMI,
kg/m2 | 20.6±1.3 | 20.9±0.7 | 0.778 |
Experimental colitis was ameliorated
in LR-D rats
After the instillation of TNBS in the colon, all
rats exhibited decreased body weight; however, the weight decrease
was much more severe in WT rats than in LR-D rats (Fig. 2A). In addition, LR-D rats exhibited
low DAI scores, long colon lengths, and low scores in the gross
appearance of the colon compared with the WT control rats (Fig. 2). These results indicated that the
general condition was improved in LR-D rats than in WT rats after
TNBS instillation.
Colonic histological appearance is
improved in LR-D rats
Histological analysis revealed an intact colonic
mucosal epithelium and neatly arranged glands before TNBS
instillation in both LR-D and WT rats (Fig. 3A). After TNBS treatment, the rats
exhibited typical colitis features, such as increased inflammation
in the mucosa, thickening and edema with loss of crypts in the
submucosa, and inflammatory infiltration in the muscularis
(Fig. 3A). However, the
microscopic inflammation scores were higher in WT rats than in LR-D
rats (Fig. 3B), indicating that
LR-D rats had improved histological outcomes than WT rats.
Inflammation is reduced in LR-D
rats
The expression of inflammatory cytokines including
IL-1β, TNF-α, and IL-6 was significantly lower in LR-D rats than in
WT rats (Fig. 4A-C). The reduced
inflammation in LR-D rats was also confirmed by the lower activity
of MPO, a marker of neutrophil infiltration, in the colon of LR-D
rats than that in WT rats (Fig.
4D). This indicated that leptin receptor deficiency could
reduce the level of inflammation in experimental colitis.
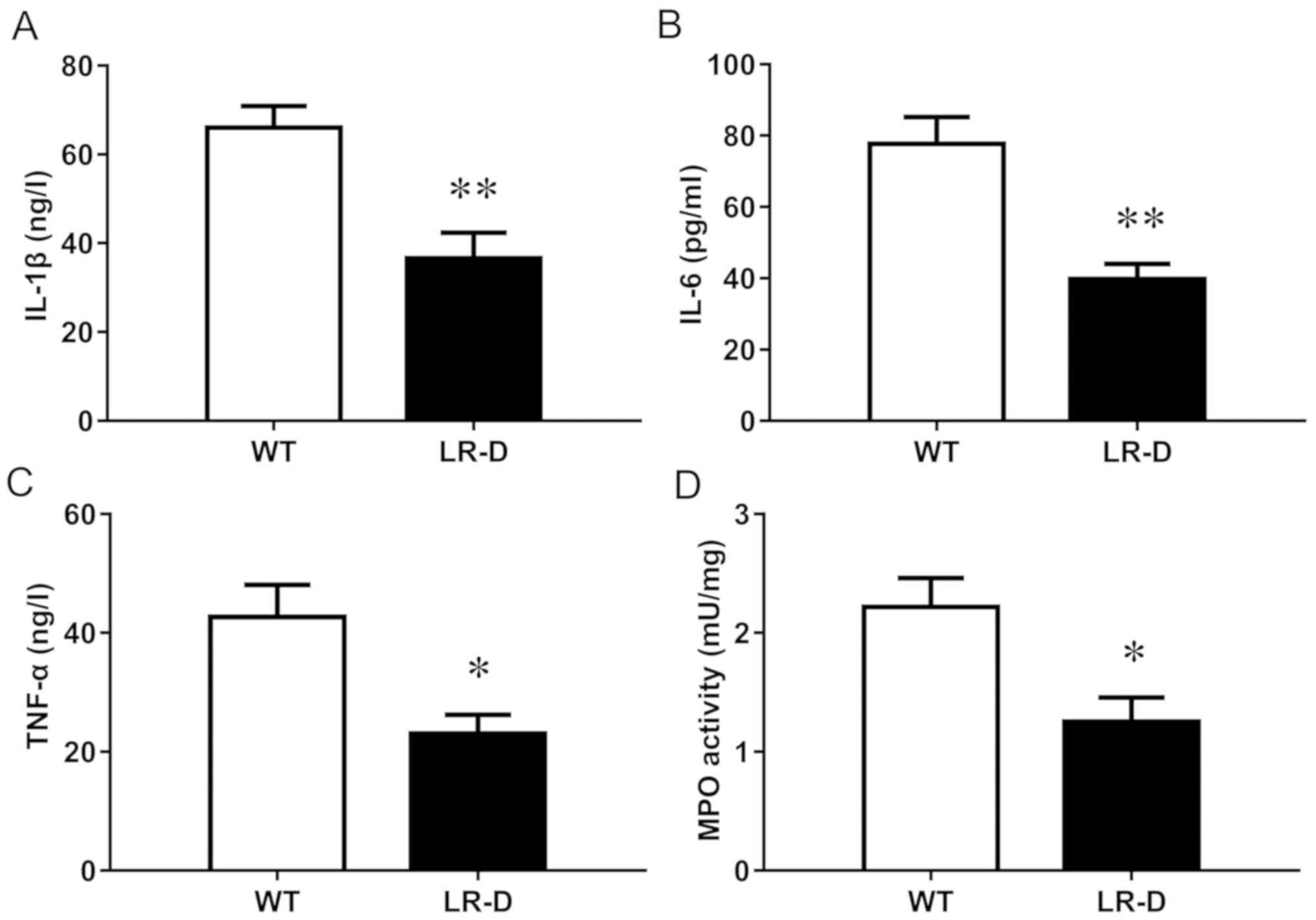 | Figure 4.Inflammatory markers in colon tissues
after treatment with TNBS. The levels of (A) IL-1β, (B) IL-6 and
(C) TNF-α, and the (D) MPO activity are presented. n=6 in each
group. *P<0.05, **P<0.01 vs. the WT group. TNBS,
2,4,6-trinitrobenzene sulfonic acid; IL, interleukin; TNF, tumor
necrosis factor; MPO, myeloperoxidase; WT, wild-type. |
Protein expression in colon
tissue
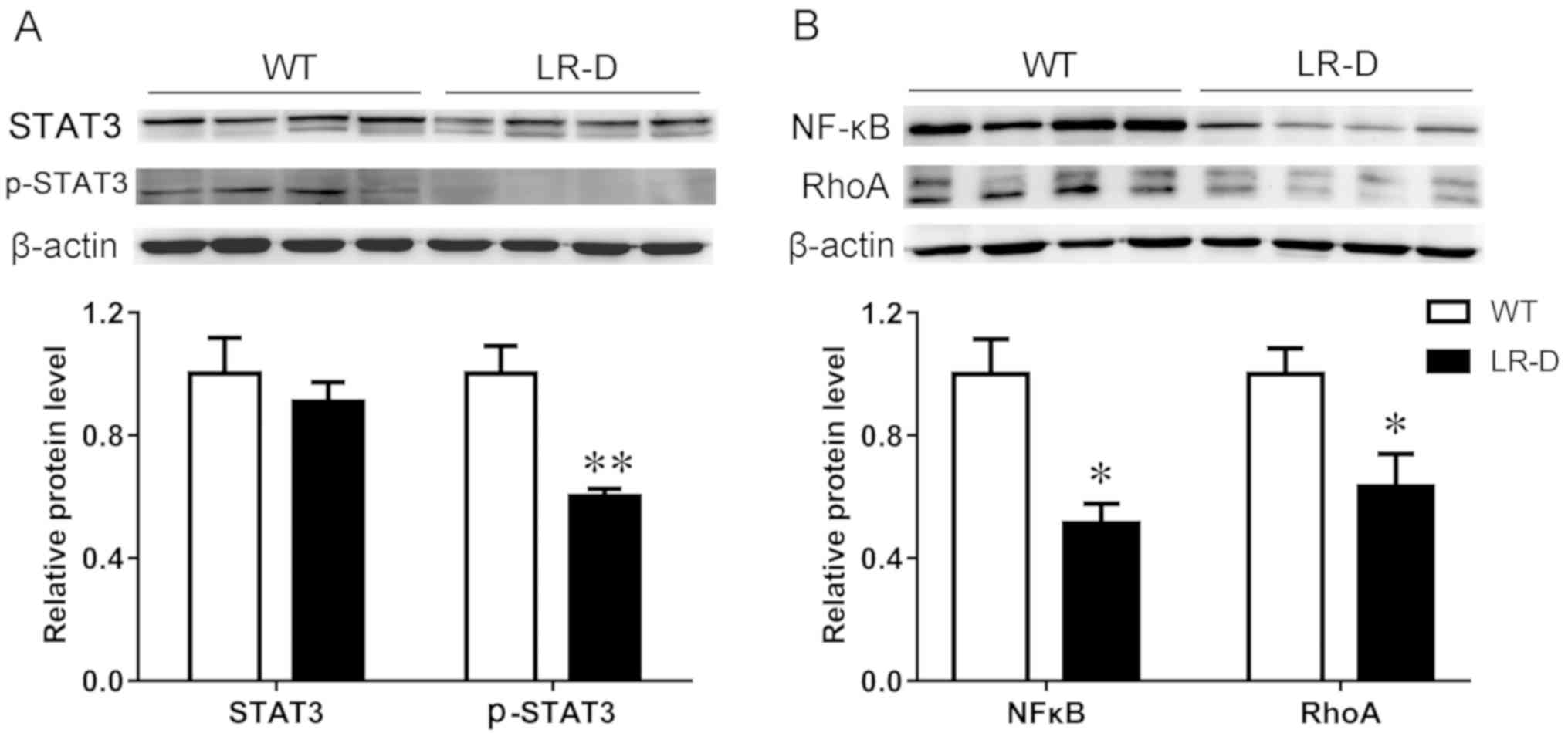
pSTAT3/STAT3 signaling has been suggested to mediate
the major effects of ob-R. Western blot analysis was used to assess
the expression of STAT3 protein in the two groups. pSTAT3 (Fig. 5A) was significantly downregulated
in the colon tissues of LR-D rats compared with that in WT rats,
which reliably suggested the correlation of pSTAT3 with ob-R
deficiency.
The expression of NF-κB-p65, a member of the NF-κB
transcription factor family that is crucial in inflammatory
diseases, was downregulated in TNBS-induced colitis tissues of LR-D
rats compared with that in WT rats (Fig. 5B). Furthermore, the expression of
RhoA protein was downregulated in colon tissues of LR-D rats
compared with that in WT rats (Fig.
5B).
Discussion
In the present study, leptin signaling was examined
in patients with UC. The results revealed that the expression of
the leptin receptor ob-R was increased in the colonic mucosa of
patients with UC compared with that in healthy volunteers, whereas
no difference was revealed in the serum leptin level between the
two groups. To clarify the role of the leptin receptor in UC (a
type of IBD), an experimental colitis model was developed in LR-D
Zucker rats. The results indicated that LR-D rats exhibited a
resistance to TNBS-induced colitis compared with WT rats.
Recent studies have revealed that leptin is a
pro-inflammatory factor (5,17).
However, the serum leptin level in patients with IBD was not
consistently reported in clinical studies [increased (9), decreased (10), or unaltered (11)]. The reason for this discrepancy is
still unclear. It was reported that inflammatory cytokines
stimulate the elevation of the level of leptin with anorexia and
body weight loss, which are common findings due to inflammation in
patients with IBD (18). There is
a close relationship among body weight, plasma leptin and
inflammatory cytokines. For example, body weight loss is often
associated with a decrease in plasma leptin (19) and inflammatory cytokines enhance
the leptin expression. We speculate that the interaction among the
three factors determines the level of plasma leptin in different
stages of the disease. Thus, the leptin level in the circulatory
system cannot reflect the local inflammation status.
Leptin exerts its biological effects by binding to
its receptor, ob-R. Unfortunately, most previous studies on leptin
signaling focused on leptin and few studies paid attention to ob-R.
In the present study, although no difference in the level of serum
leptin was revealed between patients with IBD and the HC group,
this does not exclude the possibility that the leptin signaling
pathway is altered in IBD. Therefore, the expression of the leptin
receptor ob-R in the local inflammatory colonic mucosa of patients
with UC was assessed. On the basis of the clinical result that the
expression of the leptin receptor ob-R was increased in the colonic
mucosa of patients with UC, it was speculated that the leptin
signaling pathway is upregulated in IBD. To further confirm whether
the leptin receptor contributed to the pathogenesis of IBD, an
experimental animal study was performed. An experimental colitis
model in LR-D Zucker rats was developed, which is a good model of
ob-R deficiency and manifests relatively early-onset obesity
(20). The results indicated that
LR-D rats were resistant to TNBS-induced colitis compared with WT
rats. It was speculated that the resistance to colitis of LR-D rats
was due to the leptin receptor deficiency itself rather than
obesity, because obesity exacerbates colitis (21). Therefore, the role of the leptin
receptor in the pathogenesis of IBD was confirmed in both human and
animal models.
Furthermore, the mechanism underlying the
alleviation of colitis in LR-D rats was investigated. NF-κB is a
potent pro-inflammatory nuclear transcription factor and a central
mediator of immune and inflammatory responses (22). It is well known that NF-κB can
upregulate major inflammatory factors and can induce cellular and
DNA damage (23). For example,
NF-κB activity is increased in inflamed intestinal mucosa.
Moreover, cytokines that are implicated in IBD, such as TNF-α and
IL-1β, are potent activators of NF-κB (24). Additionally, many therapies for IBD
act, at least in part, through the inhibition of NF-κB (25,26).
Researchers have demonstrated that RhoA and its downstream effector
Rho-associated kinase (ROCK) are activated in CD and in
TNBS-induced colitis, and that blockade of ROCK can inhibit
proinflammatory cytokine production (27). RhoA-dependent signaling plays an
important role in inflammatory diseases. It was reported that the
blockade of Rho kinase prevents inflammation through NF-κB
inhibition in experimental colitis (28). In the present study, consistent
with the change in histological and inflammatory factors, NF-κB and
RhoA were both decreased in inflamed colon tissues of LR-D rats,
which indicated that the amelioration of inflammation in LR-D rats
with UC may be accomplished by inhibiting the NF-κB and RhoA
signaling pathways. Further studies are required to clarify the
relationship between the leptin receptor and NF-κB and RhoA
signaling.
There are also several unresolved issues in the
present study. Leptin exerts its biological actions by binding to
its long-form receptor ob-R and transmits extracellular signals
through the Janus kinase and STAT signaling pathway. However, it is
unclear which pathway has a role in the pathogenesis of colitis.
Furthermore, the causal relationship between increased expression
of leptin receptor ob-R and colonic inflammation was not
demonstrated.
In summary, the results of the present study
indicate that the activation of the leptin receptor ob-R is
important in the pathogenesis of IBD, and leptin receptor
deficiency may provide resistance against TNBS-induced colitis by
inhibiting the NF-κB and RhoA signaling pathways.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 31671184).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YMT performed the experiments and drafted the
manuscript. SYT performed the experiments and drafted the figures.
DW interpreted the results of the experiments. FC performed part of
the experiments. XJZ analyzed the data. YZ was responsible for the
conception and design of the research, and revised and approved the
final manuscript. All authors read and approved the manuscript and
agree to be accountable for all aspects of the research in ensuring
that the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
The procedures and study were approved by the Ethics
Committee of the Second Hospital of Hebei Medical University and
written informed consent was obtained from all patients. All animal
experiments were conducted in compliance with the Guide for the
Care and Use of Laboratory Animals (National Research Council,
1996), and all techniques and procedures were reviewed and approved
by the Hebei Medical University Institutional Animal Care and Use
Committee.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ng SC, Shi HY, Hamidi N, Underwood FE,
Tang W, Benchimol EI, Panaccione R, Ghosh S, Wu JCY, Chan FKL, et
al: Worldwide incidence and prevalence of inflammatory bowel
disease in the 21st century: A systematic review of
population-based studies. Lancet. 390:2769–2778. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhou M, He J, Shen Y, Zhang C, Wang J and
Chen Y: New frontiers in genetics, gut microbiota and immunity: A
Rosetta stone for the pathogenesis of inflammatory bowel disease.
Biomed Res Int. 2017:82016722017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Coskun M, Vermeire S and Nielsen OH: Novel
targeted therapies for inflammatory bowel disease. Trends Pharmacol
Sci. 38:127–142. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Münzberg H and Morrison CD: Structure,
production and signaling of leptin. Metabolism. 64:13–23. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Abella V, Scotece M, Conde J, Pino J,
Gonzalez-Gay MA, Gómez-Reino JJ, Mera A, Lago F, Gómez R and
Gualillo O: Leptin in the interplay of inflammation, metabolism and
immune system disorders. Nat Rev Rheumatol. 13:100–109. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Singh UP, Singh NP, Guan H, Busbee B,
Price RL, Taub DD, Mishra MK, Fayad R, Nagarkatti M and Nagarkatti
PS: Leptin antagonist ameliorates chronic colitis in
IL-10−/− mice. Immunobiology. 218:1439–1451.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cakir B, Bozkurt A, Ercan F and Yeğen BC:
The anti-inflammatory effect of leptin on experimental colitis:
Involvement of endogenous glucocorticoids. Peptides. 25:95–104.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bozkurt A, Cakir B, Ercan F and Yeğen BC:
Anti-inflammatory effects of leptin and cholecystokinin on acetic
acid-induced colitis in rats: Role of capsaicin-sensitive vagal
afferent fibers. Regul Pept. 116:109–118. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kahraman R, Calhan T, Sahin A, Ozdil K,
Caliskan Z, Bireller ES and Cakmakoglu B: Are adipocytokines
inflammatory or metabolic mediators in patients with inflammatory
bowel disease? Ther Clin Risk Manag. 13:1295–1301. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Trejo-Vazquez F, Garza-Veloz I,
Villela-Ramirez GA, Ortiz-Castro Y, Mauricio-Saucedo P,
Cardenas-Vargas E, Diaz-Baez M, Cid-Baez MA, Castañeda-Miranda R,
Ortiz-Rodriguez JM, et al: Positive association between leptin
serum levels and disease activity on endoscopy in inflammatory
bowel disease: A case-control study. Exp Ther Med. 15:3336–3344.
2018.PubMed/NCBI
|
|
11
|
Ghomraoui FA, Alotaibi ST, Alharthi MA,
Asiri SS, Almadi MA, Alharbi OR, Azzam NA, Aljebreen AM, Saeed M,
Hajkhder B, et al: Plasma ghrelin and leptin in patients with
inflammatory bowel disease and its association with nutritional
status. Saudi J Gastroenterol. 23:199–205. 2017.PubMed/NCBI
|
|
12
|
Nikolaus S and Schreiber S: Diagnostics of
inflammatory bowel disease. Gastroenterology. 133:1670–1689. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lin H, Honglang L, Weifeng L, Junmin C,
Jiantao Y and Junjing G: The mechanism of alopolysaccharide
protecting ulceralive colitis. Biomed Pharmacother. 88:145–150.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang J, Chen H, Wang Y, Cai X, Zou M, Xu
T, Wang M, Wang J and Xu D: Therapeutic efficacy of a mutant of
keratinocyte growth factor-2 on trinitrobenzene sulfonic
acid-induced rat model of Crohn's disease. Am J Transl Res.
8:530–543. 2016.PubMed/NCBI
|
|
15
|
Zhao HM, Wang Y, Huang XY, Huang MF, Xu R,
Yue HY, Zhou BG, Huang HY, Sun QM and Liu DY: Astragalus
polysaccharide attenuates rat experimental colitis by inducing
regulatory T cells in intestinal Peyer's patches. World J
Gastroenterol. 22:3175–3185. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lu Y, Lin H, Zhang J, Wei J, Sun J and Han
L: Sijunzi Decoction attenuates 2, 4, 6-trinitrobenzene sulfonic
acid (TNBS)-induced colitis in rats and ameliorates TNBS-induced
claudin-2 damage via NF-κB pathway in Caco2 cells. BMC Complement
Altern Med. 17:352017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Flatow EA, Komegae EN, Fonseca MT, Brito
CF, Musteata FM, Antunes-Rodrigues J and Steiner AA: Elucidating
the role of leptin in systemic inflammation: A study targeting
physiological leptin levels in rats and their macrophages. Am J
Physiol Regul Integr Comp Physiol. 313:R572–R82. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mowat C, Cole A, Windsor A, Ahmad T,
Arnott I, Driscoll R, Mitton S, Orchard T, Rutter M, Younge L, et
al: Guidelines for the management of inflammatory bowel disease in
adults. Gut. 60:571–607. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pan WW and Myers MG Jr: Leptin and the
maintenance of elevated body weight. Nat Rev Neurosci. 19:95–105.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang B, Chandrasekera PC and Pippin JJ:
Leptin- and leptin receptor-deficient rodent models: Relevance for
human type 2 diabetes. Curr Diabetes Rev. 10:131–145. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Paik J, Fierce Y, Treuting PM, Brabb T and
Maggio-Price L: High-fat diet-induced obesity exacerbates
inflammatory bowel disease in genetically susceptible Mdr1a-/- male
mice. J Nutr. 143:1240–1247. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sun SC: The non-canonical NF-κB pathway in
immunity and inflammation. Nat Rev Immunol. 17:545–58. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fan Y, Mao R and Yang J: NF-κB and STAT3
signaling pathways collaboratively link inflammation to cancer.
Protein Cell. 4:176–185. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hayden MS and Ghosh S: Regulation of NF-κB
by TNF family cytokines. Semin Immunol. 26:253–266. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
El-Salhy M and Umezawa K: Effects of AP-1
and NF-κB inhibitors on colonic endocrine cells in rats with
TNBS-induced colitis. Mol Med Rep. 14:1515–1522. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lee C, Kim BG, Kim JH, Chun J, Im JP and
Kim JS: Sodium butyrate inhibits the NF-kappa B signaling pathway
and histone deacetylation, and attenuates experimental colitis in
an IL-10 independent manner. Int Immunopharmacol. 51:47–56. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Segain JP, Raingeard de la Blétière D,
Sauzeau V, Bourreille A, Hilaret G, Cario-Toumaniantz C, Pacaud P,
Galmiche JP and Loirand G: Rho kinase blockade prevents
inflammation via nuclear factor kappa B inhibition: Evidence in
Crohn's disease and experimental colitis. Gastroenterology.
124:1180–1187. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zou Y, Ma L, Zhao Y, Zhang S, Zhou C and
Cai Y: Inhibition of Rho kinase protects against colitis in mice by
attenuating intestinal epithelial barrier dysfunction via MLC and
the NF-κB pathway. Int J Mol Med. 41:430–438. 2018.PubMed/NCBI
|