Introduction
Breast cancer is the most frequently diagnosed
malignancy in women worldwide and the second leading cause of
cancer-associated mortality in women after lung cancer; breast
cancer is responsible for over one million of the estimated 10
million neoplasms diagnosed worldwide each year in both sexes
(1,2). Breast cancer is commonly treated with
anti-estrogens, surgical resection, radiotherapy and chemotherapy
(3,4). Tamoxifen, aromatase inhibitors,
metformin, 5-fluorouracil (5-FU) and cisplatin are widely used in
the treatment of breast cancer (5). However, these drugs not only kill
cancer cells, but also affect human normal cells. Thus, there is an
imperative need to develop more effective and less toxic antitumor
drugs.
Inducing cancer cell apoptosis via chemotherapy is a
commonly used method in the treatment of various different types of
cancer. Apoptosis targets that are currently being investigated for
chemotherapy include the mitogen-activated protein kinases (MAPK),
signal transducer and activator of transcription-3 (STAT3) and
NF-κB signaling pathways (6,7). The
MAPK signaling pathways include extracellular-signal-regulated
kinase (ERK), c-Jun N-terminal kinase (JNK) and p38, which regulate
a variety of cellular behaviors (8). JNK and p38 are activated in response
to several stress signals and are associated with the induction of
apoptosis. ERK can antagonize apoptosis by phosphorylating
pro-apoptotic Bcl-2-associated death promoter (Bax) and
anti-apoptotic Bcl-2 proteins, and inhibiting their functions
(9). Numerous studies have
revealed that STAT3 expression is higher in tumor tissues compared
with in normal tissues, and its prolonged activation is associated
with a number of different types of malignancy (10). NF-κB, a family of signal-responsive
transcription factors, can be maintained in an inactive state
within the cytoplasm through interactions and binding to inhibitor
of κB (i-κB) proteins in normal cells, and has been demonstrated to
be activated in cancer cells, including prostate and lung cancer
(11,12). These pathways may be triggered in
response to extra- or intracellular stimuli, such as reactive
oxygen species (ROS) (13).
ROS is an important second messenger in apoptosis
and cell signaling (14), and high
ROS levels have been suggested to activate intrinsic pathways and
induce cell apoptosis (15). A
number of studies have used oxidation therapy to treat patients
with cancer through increasing ROS generation to induce cancer cell
apoptosis (16–19) Therefore, ROS are highly promising
drug targets for cancer therapy.
Quinalizarin is an anthraquinone component isolated
from Rubiaceae; its anthraquinone ring is similar to the
nuclei of antitumor drugs such as doxorubicin and daunorubicin
(20). Previous studies have
demonstrated that it promotes apoptosis in A549 lung cancer cells,
AGS gastric cancer cells, and Huh 7 hepatoma cells via the MAPK and
STAT3 signaling pathways (21,22).
However, to the best of our knowledge, there are currently no
detailed reports describing the effects of quinalizarin in human
breast cancer.
In the present study, in order to determine whether
quinalizarin induced human breast cancer cell mortality and
decreased normal cells toxicity, the cytotoxic effects, apoptotic
effects, cell cycle, ROS effects and key molecular signaling
proteins involved in regulation of apoptosis were investigated in
human breast cancer cells.
Materials and methods
Chemicals and reagents
In the present study, 5-fluorouracil (5-FU; MedChem
Express) was dissolved in 20 mM 100% dimethyl sulfoxide (DMSO).
Quinalizarin (Sigma-Aldrich; Merck KGaA) was prepared as a stock
solution in DMSO and stored at −20°C until use.
Cell cultures
Human breast cancer estrogen-dependent cell lines
(MCF-7, T47D and MDA-MB231) were purchased from the American Type
Culture Collection and normal human cells (L-02, IMR-90 and 293-T)
were obtained from Sai Qi (Shanghai) Biological Engineering Co.,
Ltd. The human normal liver cells (L-02), human lung fibroblast
cells (IMR-90) and normal kidney epithelial cells (293-T) were used
as controls as the liver and kidneys are major target organs for
drug toxicity testing, and IMR-90 cells have not transformed from
embryonic cells, which can directly reflect the effects of toxicity
in toxicity studies. MCF-7, T47D and MDA-MB231 cells were grown in
Dulbecco's modified Eagle's medium (DMEM; Gibco; Thermo Fisher
Scientific, Inc.). L-02, IMR-90 and 293-T cells were grown in
RPMI-1640 medium (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% FBS (Gibco; Thermo Fisher Scientific, Inc.),
100 U/ml penicillin (Gibco; Thermo Fisher Scientific, Inc.) and 100
µg/ml streptomycin (Gibco; Thermo Fisher Scientific, Inc.), and
incubated at 37°C with 5% CO2 for 24 h.
MTT assay
MCF-7, T47D, MDA-MB231, L-02, IMR-90 and 293-T cells
were harvested in the logarithmic growth phase and then seeded in
96-well culture plates at a density of 6×103 cells/per
well. After 24-h incubation at 37°C, the cells were treated with
different concentrations (1, 3, 10, 30 and 100 µmol/l) of 5-FU or
quinalizarin for 24 h. Cells in the control group were treated with
DMSO. The cells were then incubated with 20 µl MTT (5 mg/ml) for 2
h at 37°C. The intracellular formazan crystals were solubilized
with 100 µl DMSO and incubated for 15 min at 37°C, after which the
absorbance of the solutions was measured at 490 nm (BioTek
Instruments, Inc.). The half maximal inhibitory concentration
(IC50) values were calculated using GraphPad Prism
version 5.01 (GraphPad Software, Inc.).
Hoechst 33324 staining
Apoptosis was analyzed using Hoechst 33324 (Beyotime
Institute of Biotechnology) staining. MCF-7 cells were seeded onto
cell slides in 6-well plates (1×105 cells/per well) and
treated with 10 µmol/l 5-FU or 10 µmol/l quinalizarin for 3, 6, 12
and 24 h at 37°C. After washing twice with PBS, cells were
resuspended in 5 µl Hoechst 33324 binding buffer, and dual staining
was performed with 5 µl propidium iodide (PI; Beyotime Institute of
Biotechnology) for 5 min at 37°C. Cells were incubated with Hoechst
33324 stain for 5 min at 37°C and observed under a fluorescence
microscope DM 2500 (Leica Microsystems GmbH) at magnification,
×400.
Annexin V-fluorescein isothiocyanate
(FITC)/PI double staining and flow cytometry
Early and late apoptosis were analyzed by flow
cytometry. MCF-7 cells were seeded onto cell slides in 6-well
plates (1×105 cells/well) and treated with 10 µmol/l
5-FU or 10 µmol/l quinalizarin at 3, 6, 12 and 24 h, as
aforementioned. Cells were centrifuged at 5,000 × g for 5 min at
4°C and washed three times with PBS. A total of 10 µl FITC and 5 µl
PI were incubated with the cell suspension for 20 min at 37°C in
the dark and analyzed by flow cytometry (Beckman Coulter, Inc.
Brea, CA, USA). CytExpert software 2.0 (Beckman Coulter, Inc.) was
used for analysis.
Cell cycle analysis
MCF-7 cells were treated with 10 µmol/l quinalizarin
for 3, 6, 12 and 24 h at a density of 1×105 cells/per
well. Subsequently, the cell culture RPMI-1640 medium (10% FBS; 100
U/ml penicillin; 100 µg/ml streptomycin) was removed, and the cells
were trypsinized (0.05% trypsin-EDTA in PBS), washed twice with
cold PBS and fixed in 70% ethanol for 12 h at −20°C. Cell
suspensions were then incubated with RNase A and PI (both Beyotime
Institute of Biotechnology) for 30 min at 37°C in the dark. The
stained cells were analyzed for DNA content using flow cytometry
(Beckman Coulter, Inc.). The cell cycle was analyzed using
CytExpert software 2.0 (Beckman Coulter, Inc.).
Western blot analysis
MCF-7 cells were pre-incubated for 1 h with FR180204
(ERK inhibitor, 12 µmol/liter) or SP600125 (JNK inhibitor, 12
µmol/liter) or SB203580 (p38 inhibitor, 12 µmol/liter) (MedChem
Express) dissolved in PBS and then treated with 10 µmol/l
quinalizarin for 3, 6, 12 or 24 h. Proteins were extracted with
cell lysis buffer (1 M HEPES, pH 7.0; 5 M NaCl; 0.5% Triton X-100;
10% glycerol; 20 mM β-mercaptoethanol; 20 mg/ml AEBSF; 0.5 mg/ml
pepstatin; 0.5 mg/ml leupeptin; and 2 mg/ml aprotinin) for 30 min
at 37°C and centrifuged at 12,000 × g for 30 min at 4°C. The
protein concentration was quantified using Coomassie blue staining.
Equivalent proteins (30 µg) were separated via SDS-PAGE (8–12% gel)
and transferred onto nitrocellulose membranes, which were incubated
in blocking solution [fresh 5% non-fat milk in 10 mM Tris-HCl
containing 150 mM NaCl and Tris-buffered saline (TBS); pH 7.5] and
TBS+0.2% Tween-20 (TBST) for 1 h at room temperature. The membranes
were incubated for 12 h at 4°C with the following primary
antibodies: Mouse monoclonal antibodies against α-tubulin (1:2,500;
cat. no. sc-8035; internal control), Bax (1:1,500; cat. no.
sc-493), Bcl-2 (1:1,500; cat. no. sc-7382), caspase-3 (1:1,500;
cat. no. sc-373730), cleaved (cle)-poly (ADP-ribose) polymerase
(PARP; 1:1,500; cat. no. sc-8007), phosphorylated (p)-JNK
(Tyr183 and Tyr185; 1:1,500; cat. no.
sc-6254), JNK (1:1,500; cat. no. sc-7345), p-p38
(Tyr182; 1:1,500; cat. no. sc-7973), p-ERK
(Tyr204; 1:1,500; cat. no. sc-8059), p-STAT3
(Tyr705; 1:1,500; cat. no. sc-8059) and STAT3 (1:1,500;
cat. no. sc-8019); and rabbit polyclonal antibodies against CDK1/2
(1:2,500; cat. no. sc-163), cyclin B1 (1:2,500; cat. no. sc-4073),
p27 (1:2,500; cat. no. sc-528), p21 (1:1,500; cat. no. sc-397),
ERK2 (1:1,500; cat. no. sc-154), p38α/β (1:1,500; cat. no.
sc-7194), NF-κB (1:2,500; cat. no. sc-1190) and IκB (1:2,500; cat.
no. sc-7977; all from Santa Cruz Biotechnology, Inc.). The
membranes were incubated with horseradish peroxidase conjugated
anti mouse (1:5,000; cat. no. ZB 2301) and anti rabbit (1:5,000;
cat. no. ZB 2305; both from OriGene Technologies, Inc.) secondary
antibodies for 2 3 h at room temperature followed by washing with
TBST. Proteins were visualized using Pierce ECL Western Blotting
Substrate (Thermo Fisher Scientific, Inc.) and the AI600
chemiluminescence imager (GE Healthcare), and were semi-quantified
using ImageJ software (version 1.46r; National Institutes of
Health). Protein levels were normalized to the matching
densitometry value of α-tubulin as the internal control. The change
in the expression levels of p-p38, p-JNK, p-ERK and p-STAT3 was
based on the expression levels of p38, JNK, ERK and STAT3.
Measurement of intracellular ROS
levels
MCF-7 cells were seeded in 6-well culture plates
(1×105 cells/per well) and incubated for 24 h, and then
treated with 10 µmol/l quinalizarin for 3, 6, 12 and 24 h, as
aforementioned. MCF-7 cells were pretreated with
N-acetyl-L-cysteine (NAC; Beyotime Institute of Biotechnology) for
30 min at 37°C and then treated with 10 µmol/l quinalizarin for 12
h at 37°C. ROS levels were estimated using a fluorescent probe
comprising 2′,7′-dichlorofluorescin diacetate (DCFH-DA; Beyotime
Institute of Biotechnology). The cells were harvested and
centrifuged at 5,000 × g for 5 min at room temperature and
incubated with DCFH-DA for 30 min at 37°C in the dark. The
substrate solution was subsequently removed and the cells were
washed three times with PBS. Flow cytometric analysis was used to
determine the levels of ROS and CytExpert software (version 1.2;
Beckman Coulter, Inc.) was used to analyze the data.
Statistical analysis
Statistical analyses were performed using SPSS
software (version 21.0; IBM Corp.). Data are expressed as the mean
± standard deviation. Multiple comparisons between groups were
performed using one-way ANOVA followed by Tukey's post hoc test.
All experiments were replicated three times. P<0.05 was
considered to indicate a statistically significant difference.
Results
Quinalizarin inhibits the viability of
human breast cancer cells but not normal cells
In order to evaluate the cytotoxic effects of
quinalizarin in human breast cancer cells, MCF-7, T47D and
MDA-MB231 cells were treated with different concentrations of
quinalizarin or 5-FU (1, 3, 10, 30 and 100 µmol/l) for 24 h, after
which the MTT assay was performed to measure cell viability.
Quinalizarin inhibited MDA-MB231 (estrogen-receptor-negative,
IC50 value 30.11 µmol/l) cell viability and
significantly inhibited the viability of the
estrogen-receptor-positive cell lines in a dose-dependent manner,
more so than 5-FU. The IC50 values for quinalizarin in
MCF-7 and T47D cells were 12.66 and 15.21 µmol/l, respectively
(Fig. 1A). Due to the MCF-7 cells
having the lowest IC50 value (12.66 µmol/l) and being
the most sensitive to quinalizarin, this cell line was selected as
the model system to investigate the effects of quinalizarin on
apoptosis and cell cycle arrest. Furthermore, there were no
effective cytotoxic effects of quinalizarin when compared with the
5-FU-treated group in the L-02, IMR-90 and 293-T cell lines
(Fig. 1B). These results
demonstrated that quinalizarin had cytotoxic effects in MCF-7, T47D
and MDA-MB231 cell lines, but not in the L-02, IMR-90 and 293-T
cell lines, which may provide initial evidence that the
pro-apoptotic effects of quinazirilin are specific to
estrogen-receptor-positive breast cancer cells.
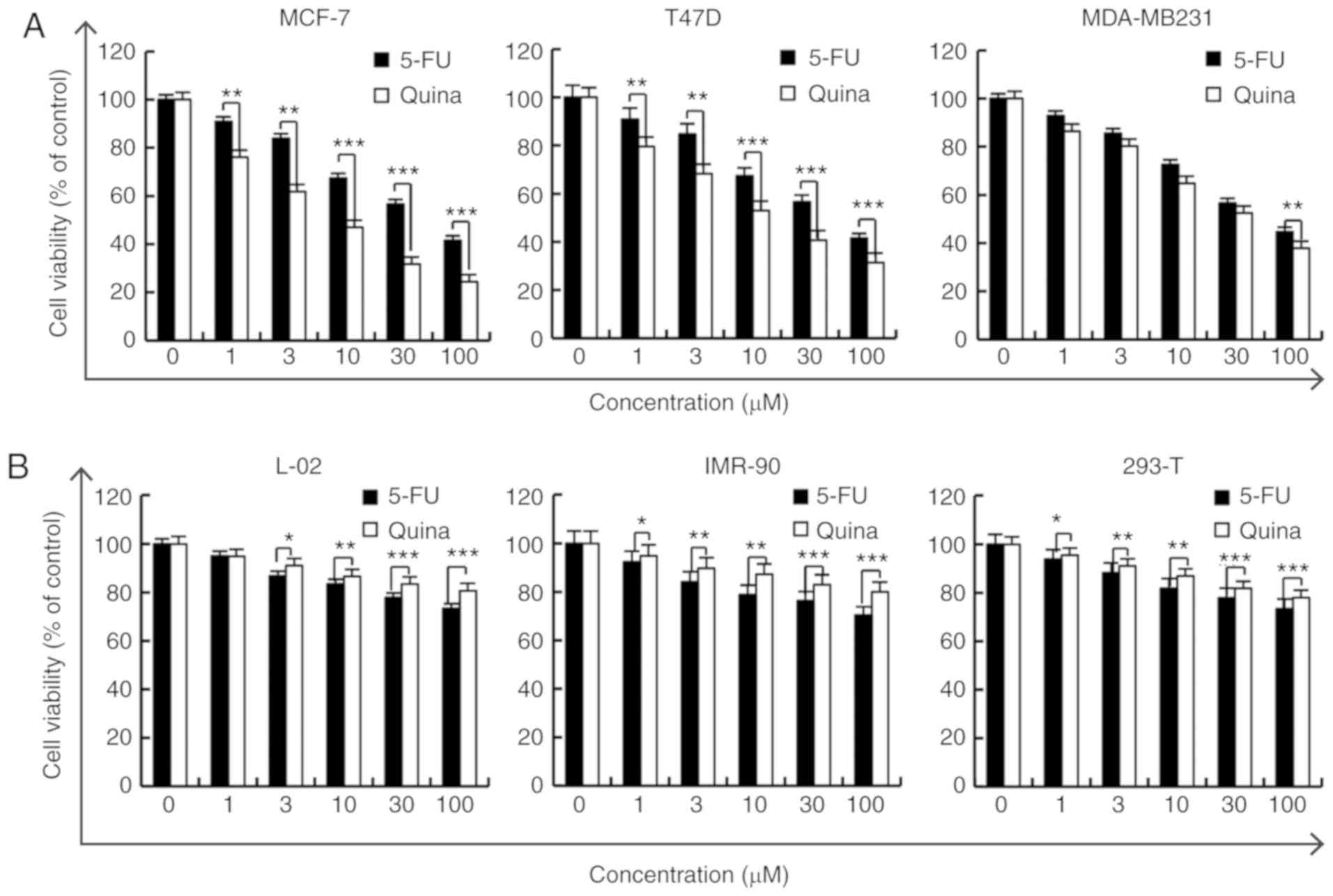 | Figure 1.Quinalizarin inhibits the viability
of human breast cancer cells. (A) MCF-7, T47D and MDA-MB231 cells,
and (B) L-02, IMR-90 and 293-T cells, were treated with 1, 3, 10,
30 and 100 µmol/l 5-FU or quinalizarin for 24 h, and then cell
viability was measured via a MTT assay. The results are presented
as the mean ± standard deviation of three independent experiments.
*P<0.05, **P<0.01 and ***P<0.001, as indicated. 5-FU,
5-fluorouracil; Quina, quinalizarin. |
Quinalizarin induces apoptosis in
MCF-7 cells
To determine whether quinalizarin induces apoptosis
in human breast cancer cells, MCF-7 cells were treated with 10
µmol/l quinalizarin or 10 µmol/l 5-FU for 3, 6, 12 and 24 h,
followed by Hoechst staining, flow cytometric analyses and western
blotting to measure cell apoptosis. As presented in Fig. 2A, MCF-7 cells treated with
quinalizarin for 24 h exhibited cell shrinkage and chromatin
condensation, as demonstrated by a strong bright red fluorescence.
The flow cytometric analysis results revealed that treatment with
quinalizarin for 24 h markedly increased the rate of apoptosis to
29.61%, and significantly increased cell apoptosis between 3 and 24
h compared with 5-FU (Fig. 2B). In
addition, quinalizarin also significantly increased the expression
levels of Bax, caspase-3 and PARP proteins in a time-dependent
manner, and decreased the expression levels of Bcl-2 protein
(Fig. 2C). These results suggested
that quinalizarin-induced apoptosis was partially mediated by the
mitochondrial pathway and caspase activation in MCF-7 cells.
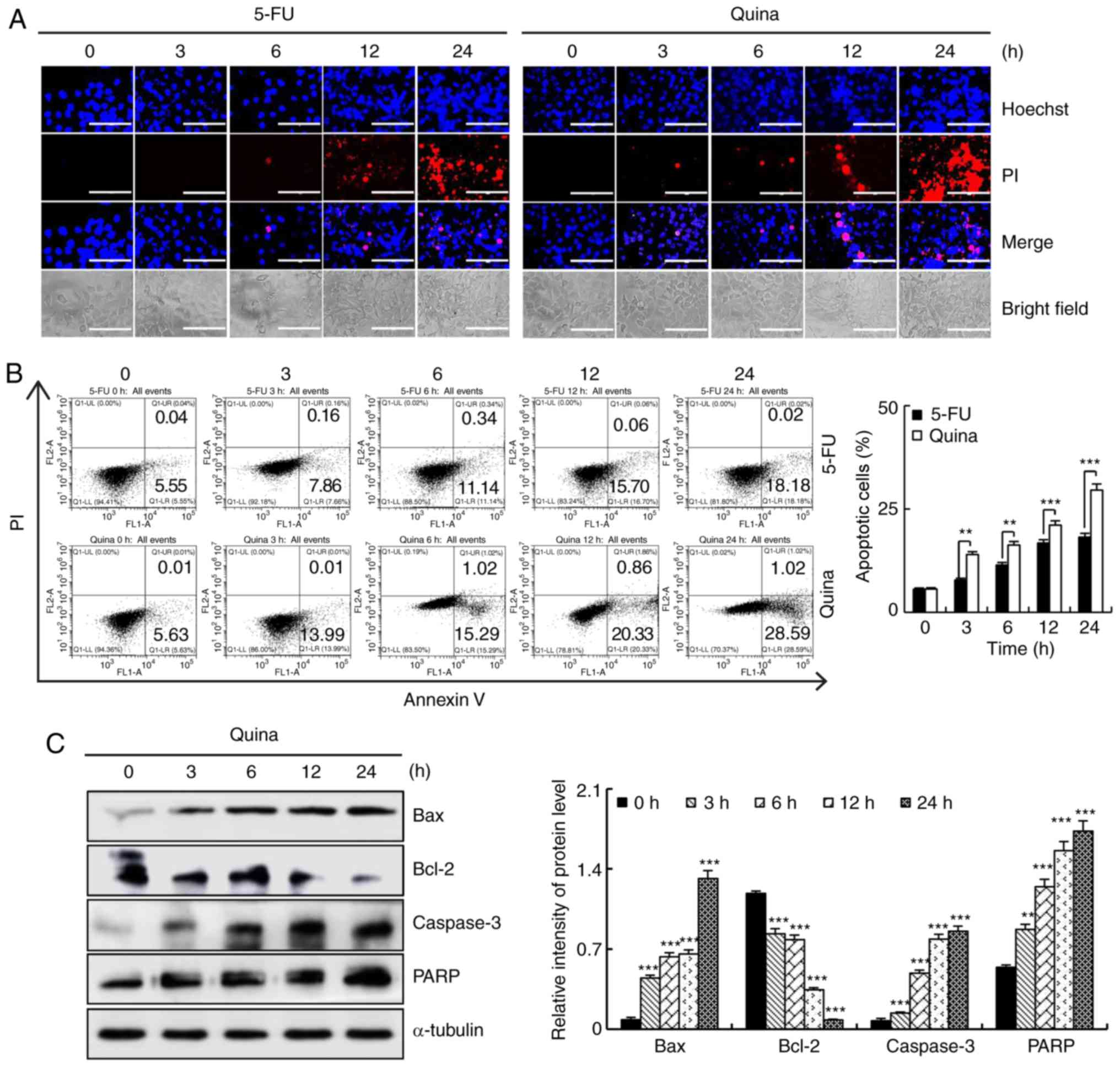 | Figure 2.Quinalizarin induces apoptosis in
human breast cancer cells. (A) Cells were treated with 10 µmol/l
5-FU or 10 µmol/l quinalizarin for 3, 6, 12 and 24 h, after which
the cell morphology and fluorescence intensity were observed by
fluorescence microscopy. Scale bars, 200 µm. (B) Cells were treated
with 10 µmol/l quinalizarin for 3, 6, 12 and 24 h, and apoptosis
was analyzed by flow cytometric analysis. **P<0.01 and
***P<0.001, as indicated. (C) The expression levels of Bax,
Bcl-2, caspase-3 and PARP in MCF-7 cells were analyzed via western
blotting. Error bars indicate the mean ± standard deviation of
three independent experiments. **P<0.01 and ***P<0.001 vs. 0
h. 5-FU, 5-fluorouracil; Quina, quinalizarin; PI, propidium iodide;
PARP, poly (ADP-ribose) polymerase. |
Quinalizarin induces cell cycle arrest
in MCF-7 cells
To investigate whether quinalizarin induced cell
cycle arrest in human breast cancer cells, MCF-7 cells were treated
with 10 µmol/l quinalizarin for 3, 6, 12 and 24 h, followed by flow
cytometric analyses and western blotting to evaluate cell cycle
arrest. Quinalizarin significantly increased the number of cells in
the G2/M phase and decreased the number of cells in the
G0/G1 and S phases in a time-dependent manner
(Fig. 3A). The western blotting
results revealed that the expression levels of the G2/M
phase-associated proteins CDK1/2 and cyclin B1 were decreased, and
the expression levels of p21 and p27 were increased over time
(Fig. 3B). These results suggested
that quinalizarin caused G2/M phase cell cycle arrest
through alterations in p21, p27 and G2/M phase cell
cycle-associated protein expression in MCF-7 cells.
Quinalizarin induces apoptosis via
MAPK, STAT3 and NF-κB signaling pathways in MCF-7 cells
In order to determine whether quinalizarin-induced
MCF-7 cell apoptosis involved the MAPK, STAT3 and NF-κB signaling
pathways, MCF-7 cells were treated with 10 µmol/l quinalizarin for
3, 6, 12 and 24 h, followed by western blotting to measure protein
expression levels. Quinalizarin suppressed the level of p-ERK,
p-STAT3 and NF-κB in a time-dependent manner, and increased the
levels of p-JNK, p-p38 and i-κB (Fig.
4A). In order to investigate whether the MAPK signaling pathway
is involved in the regulation of the STAT3 and NF-κB signaling
pathways, MCF-7 cells were treated with the p38 inhibitor
(SB203580), JNK inhibitor (SP600125) and ERK inhibitor (FR180204).
After 24 h, the decreased protein expression levels of p-STAT3 and
NF-κB induced by quinalizarin were blocked by the addition of the
p38 and JNK inhibitors. p-STAT3 and NF-κB protein expressions were
further suppressed following the addition of the ERK inhibitor and
quinalizarin, when compared with quinalizarin treatment alone
(Fig. 4B). These results
demonstrated that the MAPK signaling pathway can regulate the
expression levels of STAT3 and NF-κB, and quinalizarin induced
MCF-7 cell apoptosis via the MAPK, STAT3 and NF-κB signaling
pathways.
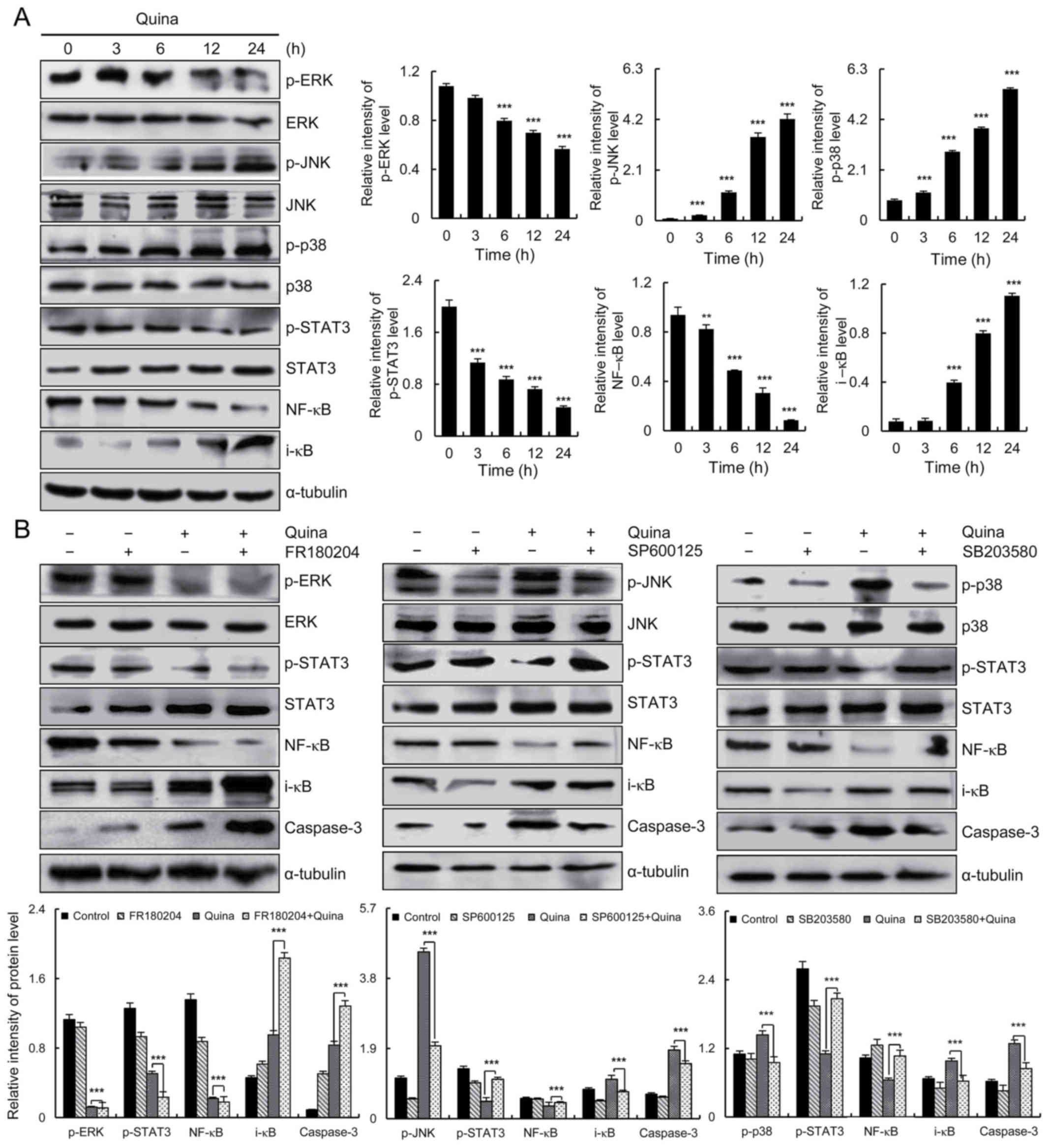 | Figure 4.Quinalizarin induces cell apoptosis
in human breast cancer cells via the MAPK, STAT3 and NF-κB
signaling pathways. (A) Expression levels of ERK, JNK, p38, STAT3,
NF-κB, and i-κB proteins were analyzed via western blotting.
**P<0.01 and ***P<0.001 vs. 0 h. (B) Cells were pre-incubated
for 1 h with 12.1 µmol/l SB203580 and then treated with 10 µmol/l
quinalizarin. The expression levels of p38, STAT3, NF-κB, i-κB and
caspase-3 were analyzed via western blotting. The expression levels
of p-JNK, STAT3, NF-κB, i-κB and caspase-3 were analyzed via
western blotting. Cells were pre-incubated for 1 h with 12.1 µmol/l
FR180204 and then treated with 10 µmol/l quinalizarin. The
expression levels of p-ERK, STAT3, NF-κB, i-κB and caspase-3 were
analyzed via western blotting. Error bars indicate the mean ±
standard deviation of three independent experiments. ***P<0.001,
as indicated. MAPK, mitogen-activated protein kinase; STAT3, signal
transducer and activator of transcription-3; ERK, extracellular
signal-regulated kinase; JNK, c-Jun N-terminal kinase; p-,
phosphorylated; quina, quinalizarin. |
Quinalizarin induces apoptosis by
accelerating ROS generation in MCF-7 cells
To determine whether quinalizarin-induced cell
apoptosis was preceded by ROS generation, MCF-7 cells were treated
with quinalizarin (10 µmol/l) for 24 h, followed by flow cytometric
analyses and western blotting to measure ROS levels. Quinalizarin
increased the levels of intracellular ROS accumulation in MCF-7
cells in a time-dependent manner (Fig.
5A), but pre-incubation with NAC for 24 h partially prevented
the quinalizarin-induced accumulation of ROS. Following
pretreatment with NAC, the quinalizarin-induced apoptosis was
reversed (Fig. 5B). NAC blocked
the quinalizarin-induced decrease in the expression levels of
p-ERK, p-STAT3 and NF-κB, and also blocked the quinalizarin-induced
increase in the expression levels of p-JNK, p-p38, i-κB, caspase-3
and PARP (Fig. 5C). These results
demonstrated that ROS generation is a major regulator of
quinalizarin-induced mitochondrial-dependent apoptosis and
G2/M cell cycle arrest through ROS-mediated MAPK, STAT3
and NF-κB signaling pathways in MCF-7 cells (Fig. 6).
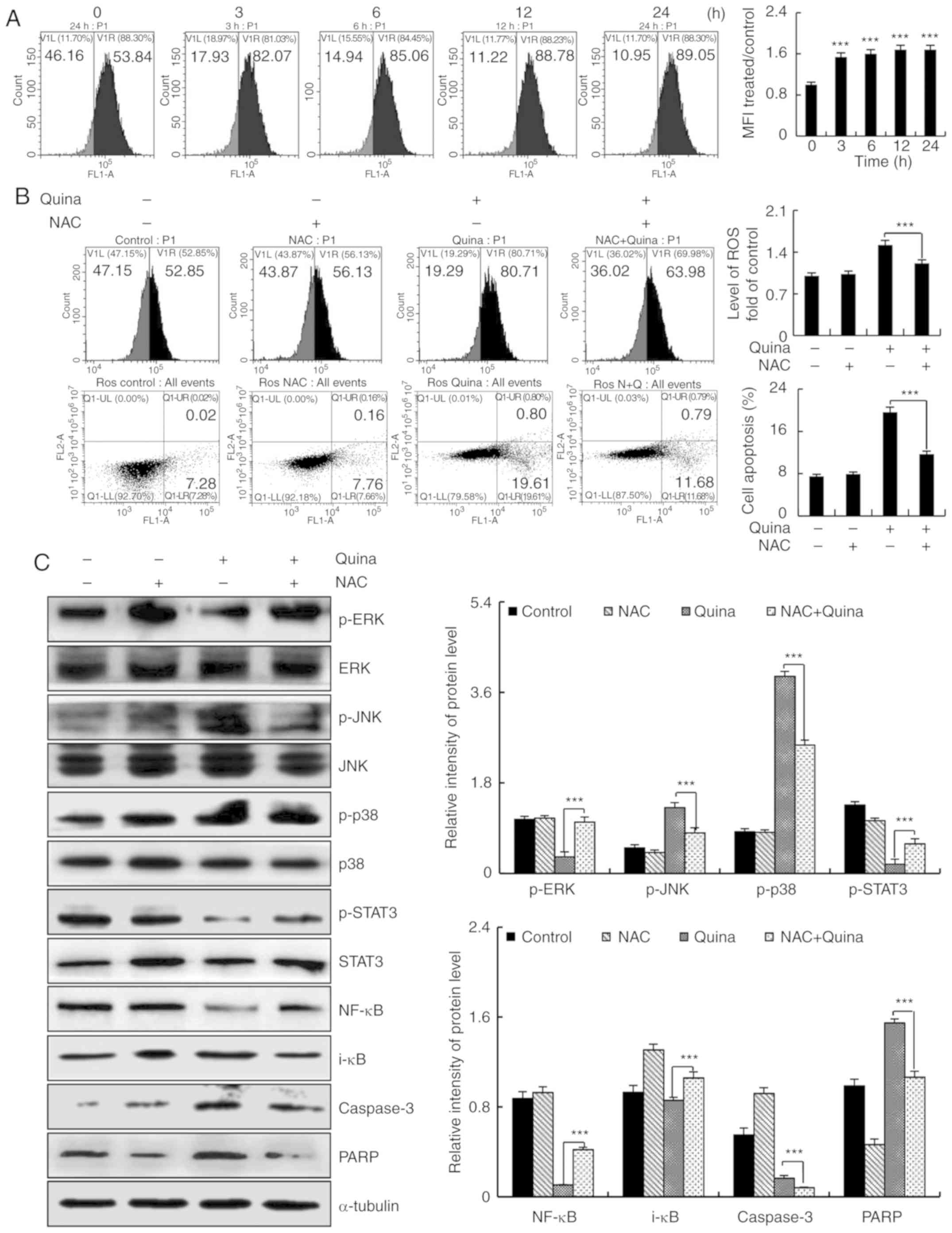 | Figure 5.Quinalizarin induces ROS-mediated
apoptosis in human breast cancer cells. (A) Cells were treated with
10 µmol/l quinalizarin for 3, 6, 12 and 24 h, and the intracellular
ROS levels were analyzed via flow cytometric analyses.
***P<0.001 vs. 0 h. (B) Cells were treated with NAC or
quinalizarin for 24 h, the generation of ROS and cell apoptosis
were analyzed via flow cytometric analyses. ***P<0.001, as
indicated. (C) The expression levels of ERK, JNK, p38, STAT3,
NF-κB, i-κB, caspase-3 and PARP were analyzed via western blotting.
Error bars indicate the mean ± standard deviation of three
independent experiments. ***P<0.001, as indicated. ROS, reactive
oxygen species; NAC, N-acetyl-L-cysteine; ERK, ERK, extracellular
signal-regulated kinase; JNK, c-Jun N-terminal kinase; STAT3,
signal transducer and activator of transcription-3; p-,
phosphorylated; PARP, poly (ADP-ribose) polymerase. |
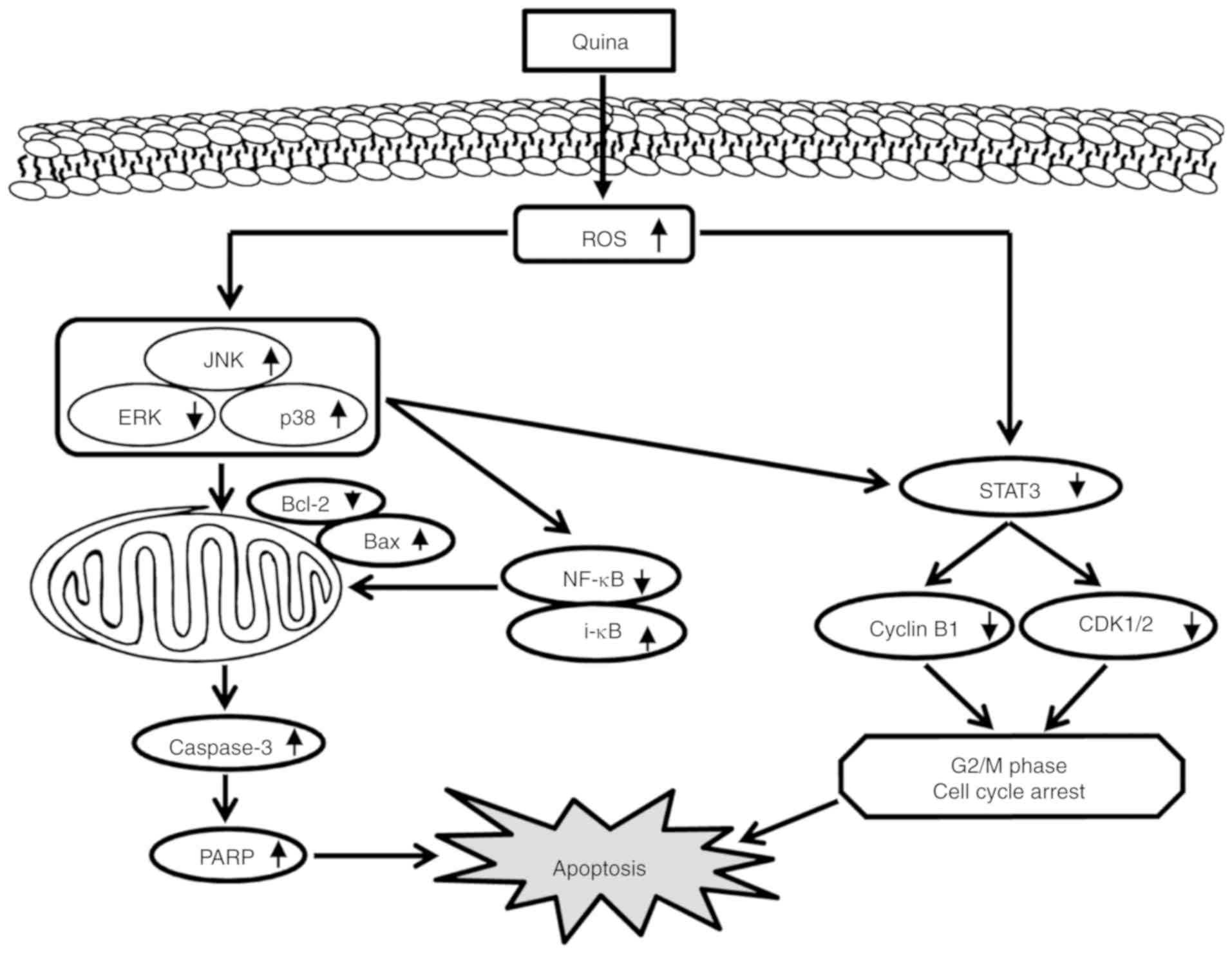 | Figure 6.Schematic diagram of the signaling
pathways in MCF-7 cells affected by quinalizarin. Quinalizarin
induced apoptosis in the human breast cancer cells. The anticancer
effects of quinalizarin induced ROS-mediated mitochondrial
apoptosis by the MAPK, STAT3, and NF-κB signaling pathways. ROS,
reactive oxygen species; MAPK, mitogen-activated protein kinase;
STAT3, signal transducer and activator of transcription-3; quina,
quinalizarin; JNK, c-Jun N-terminal kinase; ERK, ERK, extracellular
signal-regulated kinase; PARP, poly (ADP-ribose) polymerase. |
Discussion
Anthraquinone compounds, an important class of
natural and synthetic compounds, include emodin, rhein,
aloe-emodin, apigenin and quinalizarin (23). Numerous studies have reported that
rhein, apigenin, aloe-emodin and emodin affect the cell
proliferation, migration and apoptosis of different pathological
and genetic types of human cancer cell lines (24,25).
In addition, studies have also reported that anthraquinone
derivatives possess a number of identical activities as they have
an identical mother nucleus (26,27).
Similarly, the present study demonstrated that quinalizarin, an
anthraquinone compound, significantly inhibited MCF-7
(IC50, 12.66 µmol/l) and T47D (IC50 15.21
µmol/l) cell viability as determined by the MTT assay, and had less
toxicity in L-02, IMR-90 and 293-T cells. Thus, quinalizarin may
possess effective anti-tumor activities and decrease cytotoxicity
in normal human cells compared with 5-FU. In order to confirm this
theory, the present study investigated the anti-tumor mechanisms of
quinalizarin in human breast cancer cell lines.
Apoptosis, a form of programmed cell death, is a
critical defense mechanism in inhibiting the development of cancer
(28). The mitochondrial pathway
is one form of apoptosis signaling pathway. Bax and Bcl-2 are Bcl-2
family members that are fundamental for the balance between cell
survival and death in the mitochondrial pathway (29). In the mitochondrial pathway,
caspase-3 is primarily responsible for PARP activation during cell
apoptosis (30). A recent study
demonstrated that emodin, an anthraquinone, decreased the
expression levels of Bcl-2; increased the levels of caspase-3, PARP
and Bax; and induced apoptosis by modulating the expression of
apoptosis-associated genes in human breast cancer cells (31). Similarly, the results from the
present study revealed that quinalizarin significantly induced the
apoptosis of MCF-7 cells by promoting the expression levels of Bax,
downregulating Bcl-2 and activating caspase-3 and PARP. These
findings suggested that quinalizarin induced apoptosis via the
mitochondrial pathway in the human breast cancer cell line,
MCF-7.
In addition to the activation of the mitochondrial
pathway checkpoint, cell cycle control is another primary
regulatory mechanism that controls cell growth and induces cell
apoptosis (32). In eukaryotic
cells, CDK1/2 and cyclin B1 proteins control entry into mitosis
(G2/M) and are involved in regulatory and structural
processes required for mitosis, such as formation of the spindle
and attachment of chromosomes to the spindle. However, these
CDK/cyclin complexes are negatively regulated by p21 and p27. Thus,
tumor-associated G2/M phase arrest is often mediated by
alterations in cyclin B1/CDK complex protein activity and the
expression levels of p21 and p27 (33–36).
Furthermore, previous studies have demonstrated that the MAPK and
STAT3 signaling pathways regulate the eukaryotic cell cycle
(37–38). Aloe-emodin, a type of
anthraquinone, induces cell death through S-phase arrest and
caspase-dependent pathways in human tongue squamous cancer SCC-4
cells, which involves ROS generation (39). The results from the present study
coincide with evidence suggesting that quinalizarin causes
G2/M phase arrest, as the expression levels of p21 and
p27 were significantly increased, and the cyclin B1 and CDK1/2
complex protein, which is required for progression through the
G2 and M phases, were inhibited. These results suggested
that quinalizarin-mediated inhibition of MCF-7 cell proliferation
may involve G2/M phase arrest and expression of
G2/M phase-associated proteins.
The present study also focused on numerous crucial
protein kinases that play roles in regulating the cell cycle and
apoptosis, including JNK, p38, ERK, STAT3 and NF-κB. JNK and p38
induce apoptosis, while ERK promotes cell survival (40–42).
Rhein, a type of anthraquinone, induced apoptosis in human and rat
glioma cells by blocking ERK kinase activity (43). The activation of and interactions
between STAT3 and NF-κB have been demonstrated in a number of
different types of cancer, such as colon, gastric and liver cancer
(44). Apigenin, another type of
anthraquinone, induces apoptosis by decreasing the expression
levels of p-STAT3 and blocking the activation of the STAT3 and
NF-κB signaling pathway in MCF-7 cells (45). The results from the present study
demonstrated that quinalizarin could upregulate the expression
levels of p-p38, p-JNK and i-κB, and inhibit p-ERK, p-STAT3 and
NF-κB activity. The association between the MAPK, STAT3 and NF-κB
signaling pathways were also investigated by inhibiting p-JNK,
p-p38 and p-ERK activation. It was important to note that the
protein levels of p-STAT3 and NF-κB increased, and caspase-3 and
i-κB decreased following treatment with JNK or p38 inhibitors.
Inhibition of ERK activation further decreased the protein levels
of p-STAT3 and NF-κB, and caspase-3 and i-κB levels increased in
MCF-7 cells. These data demonstrate that p-STAT3 and NF-κB were
decreased in quinalizarin-treated cells and were regulated by MAPK
signaling. Therefore, MAPK is considered to be an important factor
for the quinalizarin-mediated induction of apoptosis in MCF-7
cells. These results also demonstrated that quinalizarin-induced
apoptosis was mediated through the MAPK, STAT3 and NF-κB signaling
pathways. In addition, it has been reported that copper chelating
peptide-anthraquinones have the redox effects of a quinone ring to
generate free radical species capable of damaging DNA (46). Quinone-containing drugs undergo a
one-electron reduction to the corresponding semiquinone radicals
which, in the presence of molecular oxygen, produces a superoxide
anion radical, hydrogen peroxide (47). The quinone ring structures of
copper chelating peptide-anthraquinones are similar to that of
quinalizarin (46,48). Thus, it was hypothesized that
quinalizarin caused cell oxidative stress, and induced MCF-7 cell
apoptosis by regulating the MAPK, STAT3 and NF-κB signaling
pathways.
ROS, which serve as redox messengers, are usually
maintained at tolerable levels under normal physiological
conditions, but high ROS levels elicit oxidative stress, leading to
cell death via apoptosis (49–52).
The overproduction of intracellular ROS has been associated with
the apoptotic response induced by several anticancer agents
(53), and excessive ROS
accumulation promotes cell apoptosis through various mechanisms,
including prolonged MAPK activation and inhibition of p-STAT3 and
NF-κB (54,55). A recent study revealed
emodin-induced apoptosis of colon cancer cells in a ROS-dependent
manner (56). It has been reported
that emodin-mediated ROS production acts as an early and upstream
change in the cell death cascade to antagonize cytoprotective ERK
and AKT signaling, Bcl-2 and Bax modulation, and caspase
activation, consequently leading to apoptosis in A549 cells
(57). The results from the
present study coincide with the aforementioned studies. DCFH-DA
staining, one of the most straightforward techniques for directly
measuring the redox state of cells (58), demonstrated that
quinalizarin-induced apoptosis was accompanied by ROS accumulation.
In order to determine the association between quinalizarin-induced
apoptosis and ROS, the NAC antioxidant was used to pretreat MCF-7
cells in the present study. The results revealed that quinalizarin
did not directly regulate the MAPK, STAT3 and NF-κB signaling
pathways, but rather induced their indirect regulation by
stimulating ROS accumulation, thereby promoting apoptosis in MCF-7
cells.
In conclusion, the present study demonstrated that
quinalizarin induced ROS generation and subsequently inhibited the
activation of the MAPK, STAT3 and NF-κB signaling pathways, which
increased G2/M cell cycle arrest and caspase-dependent
apoptosis in human breast cancer cells. These data are indicative
of the value of further studies regarding the application of
quinalizarin as a potential treatment for breast cancer.
Acknowledgements
Not applicable.
Funding
The present study was funded by the Multigrain
Production and Processing Characteristic Discipline Construction
Project (grant no. 2042070010), the Postdoctoral Scientific
Research Foundation of Heilongjiang Province of China (grant no.
LBH-Q13132) and the Heilongjiang Bayi Agricultural University
Support Program for ‘San Zong’ (grant no. TDJH201805).
Availability of data and materials
The datasets used or analyzed during the present
study are available from the corresponding author upon reasonable
request.
Authors' contributions
CHJ and CQY conceived and designed the study. YaQZ,
YYF, YHL, YuQZ, XYJ, YCF, YNS and JRW performed the experiments.
YaQZ, YYF and YHL analyzed the data. YaQZ and YYF wrote the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Elzawawy A: Breast cancer systemic
therapy: The need for more economically sustainable scientific
strategies in the world. Breast Care (Base). 3:434–438. 2008.
View Article : Google Scholar
|
|
2
|
Bray F, Mccarron P and Parkin DM: The
changing global patterns of female breast cancer incidence and
mortality. Breast Cancer Res. 6:229–239. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Van Agthoven T, Sieuwerts AM, Meijer D,
Meijer-van Gelder ME, van Agthoven TL, Sarwari R, Sleijfer S,
Foekens JA and Dorssers LC: Selective recruitment of breast cancer
anti-estrogen resistance genes and relevance for breast cancer
progression and tamoxifen therapy response. Endocr Relat Cancer.
17:215–230. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Feiten S, Dünnebacke J, Heymanns J,
Köppler H, Thomalla J, Van Roye C, Wey D and Weide R: Breast cancer
morbidity: Questionnaire survey of patients on the long term
effects of disease and adjuvant therapy. Dtsch Arztebl Int.
111:537–544. 2014.PubMed/NCBI
|
|
5
|
Van Cutsem E, Moiseyenko VM, Tjulandin S,
Majlis A, Constenla M, Boni C, Rodrigues A, Fodor M, Chao Y, Voznyi
E, et al: Phase III study of docetaxel and cisplatin plus
fluorouracil compared with cisplatin and fluorouracil as first-line
therapy for advanced gastric cancer: A report of the V325 study
group. J Clin Oncol. 24:4991–4997. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fesik SW: Promoting apoptosis as a
strategy for cancer drug discovery. Nat Rev Cancer. 5:876–885.
2005. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yu I and Cheung WY: Comparison of 5-FU
versus capecitabine in combination with mitomycin or cisplatin in
the treatment of anal cancer. J Clin Oncol. 35:680. 2017.
View Article : Google Scholar
|
|
8
|
Haagenson KK and Wu GS: Mitogen activated
protein kinase phosphatases and cancer. Cancer Biol Ther.
9:337–340. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shao Y and Aplin AE: ERK2 phosphorylation
of serine 77 regulates Bmf pro-apoptotic activity. Cell Death Dis.
3:e2532012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Inoue J, Gohda J, Akiyama T and Semba K:
NF-kappaB activation in development and progression of cancer.
Cancer Sci. 98:268–274. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tang X, Soch E, Shishodia S, Diane LJ,
Jack L, Waun KH, Bharat A and Ignacio IW: Immunohistochemical
analysis indicates that nuclear factor-κB (NF-alysis frequently
activated in lung cancer. Cancer Res. 46:1–5. 2005.
|
|
12
|
Grivennikov SI and Karin M: Dangerous
liaisons: STAT3 and NF-kappaB collaboration and crosstalk in
cancer. Cytokine Growth Factor Rev. 21:11–19. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang J, Wang X, Vikash V, Ye Q, Wu D, Liu
Y and Dong W: ROS and ROS-mediated cellular signaling. Oxid Med
Cell Longev. 2016:43509652016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jacquemin G, Margiotta D, Kasahara A,
Bassoy EY, Walch M, Thiery J, Lieberman J and Martinvalet D:
Granzyme B-induced mitochondrial ROS are required for apoptosis.
Cell Death Differ. 22:862–874. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Redzadutordoir M and Averillbates DA:
Activation of apoptosis signalling pathways by reactive oxygen
species. Biochim Biophys Acta. 1863:2977–2992. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wen L, Lu X, Wang R, Jin X, Hu L and You
C: Pyrroloquinoline quinone induces chondrosarcoma cell apoptosis
by increasing intracellular reactive oxygen species. Mol Med Rep.
17:7184–7190. 2018.PubMed/NCBI
|
|
17
|
Bu HQ, Cai K, Shen F, Bao XD, Xu Y, Yu F,
Pan HQ, Chen CH, Du ZJ and Cui JH: Induction of apoptosis by
capsaicin in hepatocellular cancer cell line SMMC-7721 is mediated
through ROS generation and activation of JNK and p38 MAPK pathways.
Neoplasma. 62:582–591. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Vogel J, Bumgarner J, Espinosa N and Young
W: An evaluation of the use of resistivity counters in the touchet
river in washington and the imnaha river in oregon. Am Fish Soc.
33:6953–6960. 2011.
|
|
19
|
Piska K, Koczurkiewicz P, Bucki A,
Wójcik-Pszczoła K, Kołaczkowski M and Pękala E: Metabolic carbonyl
reduction of anthracyclines-role in cardiotoxicity and cancer
resistance. Reducing enzymes as putative targets for novel
cardioprotective and chemosensitizing agents. Invest New Drugs.
35:375–385. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cozza G, Venerando A, Sarno S and Pinna
LA: The selectivity of CK2 inhibitor quinalizarin: A reevaluation.
Biomed Res Int. 2015:1–9. 2015. View Article : Google Scholar
|
|
21
|
Meng LQ, Liu C, Luo YH, Piao XJ, Wang Y,
Zhang Y, Wang JR, Wang H, Xu WT, Liu Y, et al: Quinalizarin exerts
an anti-tumour effect on lung cancer A549 cells by modulating the
Akt, MAPK, STAT3 and p53 signaling pathways. Mol Med Rep.
17:2626–2634. 2018.PubMed/NCBI
|
|
22
|
Zhou Y, Li K, Zhang S, Li Q, Li Z, Zhou F,
Dong X, Liu L, Wu G and Meng R: Quinalizarin, a specific CK2
inhibitor, reduces cell viability and suppresses migration and
accelerates apoptosis in different human lung cancer cell lines.
Indian J Cancer. 52 (Suppl 2):e119–e124. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Malik EM and Müller CE: Anthraquinones as
pharmacological tools and drugs. Med Res Rev. 36:705–748. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lordan S, O'Neill C and O'Brien NM:
Effects of apigenin, lycopene and astaxanthin on 7
beta-hydroxycholesterol-induced apoptosis and Akt phosphorylation
in U937 cells. Br J Nutr. 100:287–296. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen YY, Chiang SY, Lin JG, Ma YS, Liao
CL, Weng SW, Lai TY and Chung JG: Emodin, aloe-emodin and rhein
inhibit migration and invasion in human tongue cancer SCC-4 cells
through the inhibition of gene expression of matrix
metalloproteinase-9. Int J Oncol. 36:1113–1120. 2010.PubMed/NCBI
|
|
26
|
Zhang YY, Li ZC, Zhu JK, Yang ZY, Wang QJ,
He PG, Hang YZ, Pin JW, Yu GH and Fang Z: Simultaneous
determination of flavonoids and anthraquinones in chrysanthemum by
capillary electrophoresis with amperometry detection. Chin Chem
Lett. 21:1231–1234. 2010. View Article : Google Scholar
|
|
27
|
Zong JR, Chao ZM, Liu ZL and Wang J:
Review about structure-function relationships of anthraquinone
derivatives from Radix et Rhizoma Rhei. Zhongguo Zhong Yao Za Zhi.
33:2424–2427. 2008.(In Chinese). PubMed/NCBI
|
|
28
|
Martin KR: Using nutrigenomics to evaluate
apoptosis as a preemptive target in cancer prevention. Curr Cancer
Drug Targets. 7:438–446. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zheng JH, Viacava Follis A, Kriwacki RW
and Moldoveanu T: Discoveries and controversies in BCL-2
proteins-mediated apoptosis. FEBS J. 283:2690–2700. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Alenzi FQ, Lotfy M and Wyse R: Swords of
cell death: Caspase activation and regulation. Asian Pac J Cancer
Prev. 11:271–280. 2010.PubMed/NCBI
|
|
31
|
Zu C, Zhang M, Xue H, Cai X, Zhao L, He A,
Qin G, Yang C and Zheng X: Emodin induces apoptosis of human breast
cancer cells by modulating the expression of apoptosis-related
genes. Oncol Lett. 10:2919–2924. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Antonsson A and Persson JL: Induction of
apoptosis by staurosporine involves the inhibition of expression of
the major cell cycle proteins at the G(2)/m checkpoint accompanied
by alterations in Erk and Akt kinase activities. Anticancer Res.
29:2893–2898. 2009.PubMed/NCBI
|
|
33
|
Gao SY, Li J, Qu XY, Zhu N and Ji YB:
Downregulation of Cdk1 and cyclinB1 expression contributes to
oridonin-induced cell cycle arrest at G2/M phase and growth
inhibition in SGC-7901 gastric cancer cells. Asian Pac J Cancer
Prev. 15:6437–6441. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nasheuer HP, Smith R, Bauerschmidt C,
Grosse F and Weisshart K: Initiation of eukaryotic DNA replication:
Regulation and mechanisms. Prog Nucleic Acid Res Mol Biol.
72:41–70. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Santamaria D and Ortega S: Cyclins and
CDKS in development and cancer: Lessons from genetically modified
mice. Front Biosci. 11:1164–1188. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang Z, Leonard SS, Huang C, Castranova V
and Shi X: Role of reactive oxygen species and MAPKs in
vanadate-induced G(2)/M phase arrest. Free Radic Biol Med.
34:1333–1342. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang XH, Liu BR, Qu B, Xing H, Gao SL, Yin
JM, Wang XF and Cheng YQ: Silencing STAT3 may inhibit cell growth
through regulating signaling pathway, telomerase, cell cycle,
apoptosis and angiogenesis in hepatocellular carcinoma: Potential
uses for gene therapy. Neoplasma. 58:158–171. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Clotet J, Vendrell J and Escoté X: Control
of the cell cycle progression by the MAPK Hog1. MAP Kinase.
2:e32013. View Article : Google Scholar
|
|
39
|
Chiu TH, Lai WW, Hsia TC, Yang JS, Lai TY,
Wu PP, Ma CY, Yeh CC, Ho CC, Lu HF, et al: Aloe-emodin induces cell
death through S-phase arrest and caspase-dependent pathways in
human tongue squamous cancer SCC-4 cells. Anticancer Res.
29:4503–4511. 2009.PubMed/NCBI
|
|
40
|
Su Y, Li G, Zhang X, Gu J, Zhang C, Tian Z
and Zhang J: JSI-124 inhibits glioblastoma multiforme cell
proliferation through G(2)/M cell cycle arrest and apoptosis
augment. Cancer Biol Ther. 7:1243–1249. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Guo Y, Lin D, Zhang M, Zhang X, Li Y, Yang
R, Lu Y, Jin X, Yang M, Wang M, et al: CLDN6-induced apoptosis via
regulating ASK1-p38/JNK signaling in breast cancer MCF-7 cells. Int
J Oncol. 48:2435–2444. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Cook SJ, Stuart K, Gilley R and Sale MJ:
Control of cell death and mitochondrial fission by ERK1/2 MAP
Kinase signaling. FEBS J. 284:4177–4195. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Tang N, Chang J, Lu HC, Zhuang Z, Cheng
HL, Shi JX and Rao J: Rhein induces apoptosis and autophagy in
human and rat glioma cells and mediates cell differentiation by ERK
inhibition. Microb Pathog. 113:168–175. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Degoricija M, Situm M, Korać J, Miljković
A, Matić K, Paradžik M, Marinović Terzić I, Jerončić A, Tomić S and
Terzić J: High NF-κB and STAT3 activity in human urothelial
carcinoma: A pilot study. World J Urol. 32:1469–1475. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Seo HS, Choi HS, Kim SR, Choi YK, Woo SM,
Shin I, Woo JK, Park SY, Shin YC and Ko SG: Apigenin induces
apoptosis via extrinsic pathway, inducing p53 and inhibiting STAT3
and NFκB signaling in HER2-overexpressing breast cancer cells. Mol
Cell Biochem. 366:319–334. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Morier-Teissier E: Effect of a
copper-chelating peptide on the anticancer activity of
anthraquinones. J Pharm Belg. 45:347–354. 1990.(In French).
PubMed/NCBI
|
|
47
|
Morier-Teissier E, Bernier JL, Lohez M,
Catteau JP and Hénichart JP: Free radical production and DNA
cleavage by copper chelating peptide-anthraquinones. Anticancer
Drug Des. 5:291–305. 1990.PubMed/NCBI
|
|
48
|
Cozza G, Mazzorana M, Papinutto E, Bain J,
Elliott M, di Maira G, Gianoncelli A, Pagano MA, Sarno S, Ruzzene
M, et al: Quinalizarin as a potent, selective and cell-permeable
inhibitor of protein kinase CK2. Biochem J. 421:387–395. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Prosperini A, Juan-García A, Font G and
Ruiz MJ: Beauvericin-induced cytotoxicity via, ROS production and
mitochondrial damage in Caco-2 cells. Toxicol Lett. 222:204–211.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Conway GE, Casey A, Milosavljevic V, Liu
Y, Howe O, Cullen PJ and Curtin JF: Non-thermal atmospheric plasma
induces ROS-independent cell death in U373MG glioma cells and
augments the cytotoxicity of temozolomide. Br J Cancer.
114:435–443. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wang CH, Wu SB, Wu YT and Wei YH:
Oxidative stress response elicited by mitochondrial dysfunction:
Implication in the pathophysiology of aging. Exp Biol Med
(Maywood). 238:450–460. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Loor G, Kondapalli J, Schriewer JM,
Chandel NS, Vanden Hoek TL and Schumacker PT: Menadione triggers
cell death through ROS-dependent mechanisms involving PARP
activation without requiring apoptosis. Free Radic Biol Med.
49:1925–1936. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Lo YL and Wang W: Formononetin potentiates
epirubicin-induced apoptosis via ROS production in HeLa cells in
vitro. Chem Biol Interact. 205:188–197. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Kim KY, Park KI, Kim SH, Yu SN, Lee D, Kim
YW, Noh KT, Ma JY, Seo YK and Ahn SC: Salinomycin induces reactive
oxygen species and apoptosis in aggressive breast cancer cells as
mediated with regulation of autophagy. Anticancer Res.
37:1747–1758. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
He G and Karin M: NF-κB and STAT3-key
players in liver inflammation and cancer. Cell Res. 21:159–168.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Wang Y, Luo Q, He X, Wei H, Wang T, Shao J
and Jiang X: Emodin induces apoptosis of colon cancer cells via
induction of autophagy in a ROS-Dependent manner. Oncol Res.
26:889–899. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Su YT, Chang HL, Shyue SK and Hsu SL:
Emodin induces apoptosis in human lung adenocarcinoma cells through
a reactive oxygen species-dependent mitochondrial signaling
pathway. Biochem Pharmacol. 70:229–241. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Eruslanov E and Kusmartsev S:
Identification of ROS using oxidized DCFDA and flow-cytometry.
Methods Mol Biol. 594:57–72. 2010. View Article : Google Scholar : PubMed/NCBI
|















