Introduction
Autosomal recessive polycystic kidney disease
(ARPKD) is a rare hereditary form of polycystic kidney disease that
typically presents in the perinatal period and occurs in 1 in
20,000-40,000 live births worldwide (1). The polycystic kidney and hepatic
disease 1 (PKHD1) gene is the only gene that has been
identified to cause ARPKD. The longest PKHD1 transcript
encodes a 4,074 amino acid protein called fibrocystin or polyductin
(1–3). This protein is present in fetal and
adult kidney cells, and is also expressed in the liver and pancreas
(3), albeit at lower levels.
Similar to other cystoproteins, polyductin is localized to the
basal body and primary cilia of renal and bile duct epithelial
cells (1,4). Defects in polyductin may disrupt the
normal function of renal cilia. ARPKD is caused by mutations in the
PKHD1 gene, located at the chromosome 6p12.2 (1,2). The
largest PKHD1 gene mutation database contains 748 different
mutations (http://www.humgen.rwth-aachen.de). The most common
mutation is a missense mutation in exon 3 (c.107C>T), accounting
for 20% of all cases (http://www.humgen.rwth-aachen.de). Most patients with
ARPKD present compound heterozygosity, carrying two mutations in
two different alleles. Due to the diversity of PKHD1 gene
mutations, it is difficult to associate the genotype with the
phenotype of ARPKD. At present, it is not understood how genetic
alterations in PKHD1 lead to the formation of numerous cysts
characteristic of polycystic kidney disease.
Recently, targeted exome sequencing has been
successfully used to identify genes that cause Mendelian disorders
(5). Together, DNA capture
technology and next-generation sequencing (NGS) analysis allow for
the rapid and cost-effective parallel sequencing of specific genes
of interest. In previous studies, targeted exome sequencing has
been used as a tool for the molecular diagnosis of ARPKD (1,5,6).
Using this approach, the present study identified two novel
compound heterozygous mutations in the PKHD1 gene causing
ARPKD in a Chinese family.
Materials and methods
Subjects
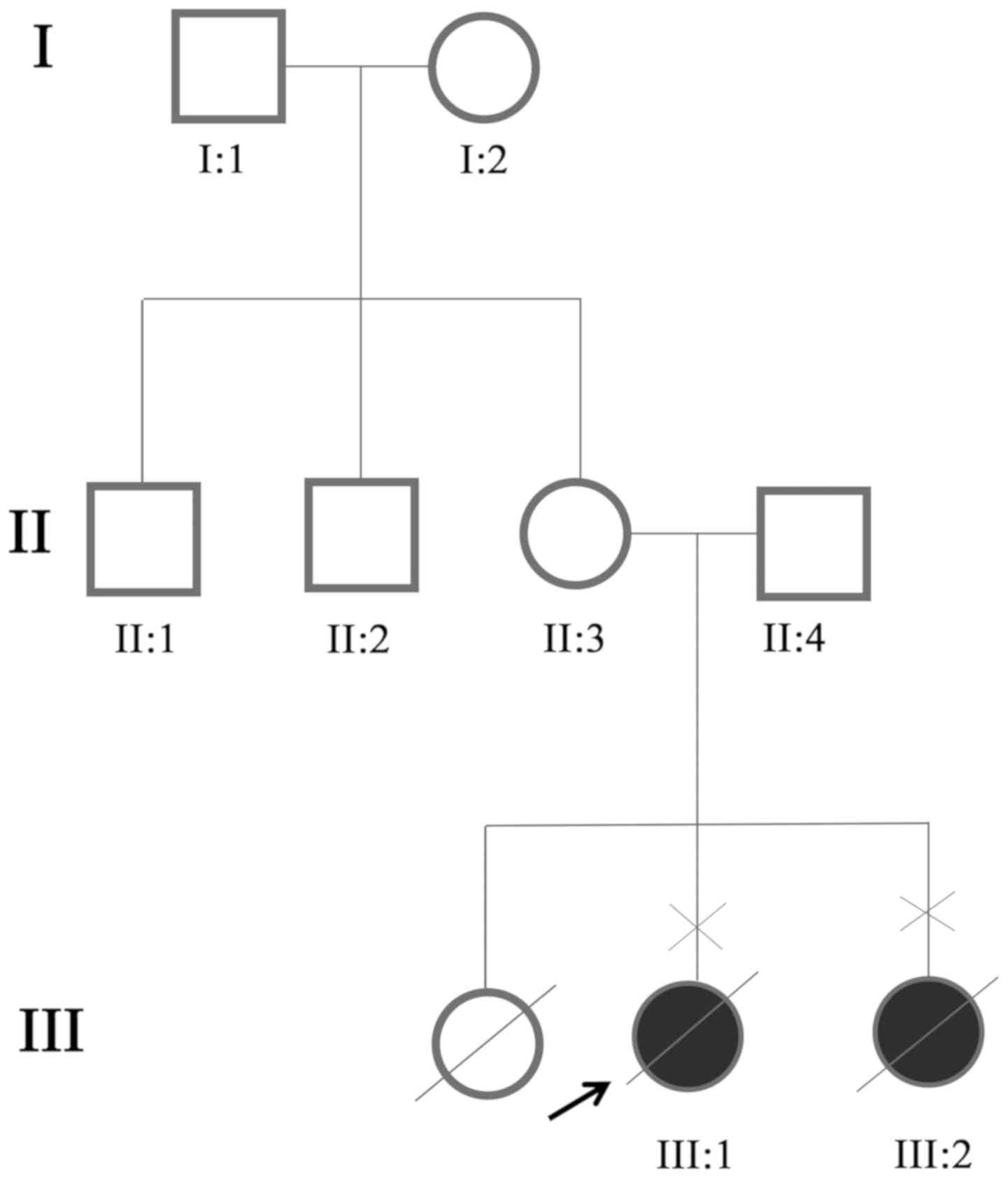
A couple (age, 26 and 27) that underwent prenatal
genetic diagnosis for ARPKD were recruited from Sichuan Provincial
People's Hospital for the present study (Fig. 1). The patients, one male and one
female, were recruited in July 2016. All the subjects underwent
comprehensive examinations and had no other related diseases. The
present study was approved by the Institutional Review Board of
Sichuan Provincial People's Hospital. Written informed consent was
obtained from all subjects prior to the present study. There was no
history of genetic diseases in the other family members recruited
for the present study. Renal ultrasonography in the father (II:4)
and mother (II:3) returned normal findings.
DNA extraction
Genomic DNA was collected from the amniotic fluid of
the fetus (proband, III:1) and from peripheral blood of all other
subjects. Genomic DNA was extracted using a QIAamp DNA Blood Midi
kit (Qiagen GmbH), according to the manufacturer's protocol. DNA
samples were stored at −20°C until use. DNA integrity was evaluated
using a NanoDrop spectrophotometer (NanoDrop Technologies; Thermo
Fisher Scientific, Inc.).
Targeted NGS
DNA samples from the couple and the proband were
analyzed using targeted NGS as reported previously (5). A customdesigned gene panel,
synthesized using the Agilent SureSelect Target Enrichment
technique (Agilent Technologies, Inc.), was used to capture the
coding regions of 356 genes, including their exons and exon-intron
boundaries (1.285 Mbp in total). The variants detected were
annotated and filtered based on public and inhouse databases: i)
Variants within intergenic, intronic and untranslated regions, and
synonymous mutations were excluded from downstream analysis; ii)
variants in dbSNP138 (www.ncbi.nlm.nih.gov/projects/SNP), the 1000 Genomes
Project (ftp.1000genomes.ebi.ac.uk/vol1/ftp), and HapMap
Project (ftp.ncbi.nlm.nih.gov/hapmap) were excluded. Homozygous
and compound heterozygous gene variations with autosomal recessive
heredity were regarded as likely causative variations. Validation
and parental origin analysis were performed for the identified
variations using conventional Sanger sequencing. Causative
mutations were verified using direct sequencing in all family
members and 576 normal controls. Primers [PKHD1-exon
27-forward (F), 5′-ggggcagagaaggaacatttg-3′, and reverse (R),
5′-agaccctccccagattacca-3′; PKHD1-exon 36-F,
5′-caatacttatactatcccgccca-3′ and R, 5′-aagtttccctcctccatccc-3′]
were designed based on genomic sequences from the Human Genome
database (www.ncbi.nlm.nih.gov/genome) and were synthesized by
Invitrogen (Thermo Fisher Scientific, Inc). Direct sequencing was
performed by Sanger sequencing according to the ABI Big Dye
sequencing protocols (cat. no. 4376484; Applied Biosystems; Thermo
Fisher Scientific, Inc.) and processed samples were sequenced using
an ABI3130XL genetic analyzer (Applied Biosystems; Thermo Fisher
Scientific, Inc.). Finally, the possible damaging effects of the
mutations were predicted using SIFT (http://provean.jcvi.org/index.php) and PolyPhen
(http://genetics.bwh.harvard.edu/pph2). The human PKHD1
protein was aligned with other PKHD1 proteins to examine the
conservation of the residues using homologene (www.ncbi.nlm.nih.gov/homologene).
Results
Clinical data
A three-generation family was recruited from Sichuan
Provincial People's Hospital (Fig.
1). Ultrasound examinations identified two affected individuals
(III:1 and III:2) among the eight examined family members. The two
affected members in this family exhibited similar clinical features
and both died before birth. The first pregnancy of the couple (for
the proband, III:1) ended with an induced abortion at 26 weeks.
Ultrasound examination for the bilateral kidneys of the fetus
(III:1) exhibited enlargement of the kidneys, and polycystic
alterations were identified by an abdominal B scan. The echo of the
renal parenchyma was increased diffusely, the demarcation of the
cortex and medulla was not clear, and the volume of amniotic fluid
was also decreased (Fig. 2). In
the current pregnancy (for III:2), repeated prenatal ultrasound
examinations during weeks 22–26 of gestation exhibited enlargement
of the kidneys and polycystic alterations; in addition, the
parenchymal echo was enhanced and the boundary of the cortex and
medulla was not clear (Table I and
Fig. S1). The size of the left
kidney was 48.3×30.6 mm, the left renal parenchymal thickness was
uneven and ~1.7 mm thinner than normal. The left kidney also
revealed renal sinus separation with a diameter of 18 mm and
caliectasis. The right kidney was 35.1×22.5 mm in size, renal sinus
separation was observed with a diameter of ~8.4 mm with
caliectasis. Furthermore, the thickness of the right renal
parenchymal was 3.7 mm. Double renal parenchymal echogenicity was
obvious. Since ultrasound examinations could not produce conclusive
results, the couple was referred for genetic counseling and
molecular prenatal diagnosis.
 | Table I.Pregnancy and fetal growth data of the
fetus (III:2). |
Table I.
Pregnancy and fetal growth data of the
fetus (III:2).
| Gestational age
(based on LMP) | Biometric
measurements | Composite gestational
age | Amniotic fluid
index | Fetal kidney
measurements | Comments |
|---|
| 22.5 weeks | BPD, 54 mm; HC, 206
mm; | 23.1 weeks | 44 mm,
oligohydramnios | Right kidney,
35.1×22.5×3.7 mm | i) Bilateral enlarged
echogenic fetal kidneys; |
|
| AC, 186 mm; Unit, 38
mm; |
|
| (>97%); left
kidney, 48.3×30.6× | ii)
Oligohydramnios |
|
| HL, 36 mm; TCD, 25
mm |
|
| 1.7 mm (>97%) |
|
| 24.6 weeks | N/A | 25.2 weeks | 114 mm,
oligohydramnios | N/A | i) Bilateral enlarged
echogenic fetal kidneys; |
|
|
|
|
|
| ii) Oligohydramnios;
ii) Right cleft lip and palate |
| 26.2 weeks | BPD: 65 mm HC: 241
mm | 27.1 weeks | 120 mm,
oligohydramnios | Right kidney,
44×28×8.1 mm (>97%); | i) Bilateral enlarged
echogenic fetal kidneys; |
|
| AC: 220 mm Unit: 45
mm |
|
| left kidney,
45.5×25×4.4 mm (>97%) | ii)
Oligohydramnios |
|
| HL: 42 mm CER:
N/A |
|
|
|
|
Mutation analysis
The percentage of readable bases and the coverage
depth in the targeted region were used to assess the quality and
reliability of the targeted NGS data, and to ensure complete
sequencing coverage of all coding regions in candidate genes. The
coverage depth was <200X and 100% of bases in coding regions
were readable. On average, 645 variations within the 356 genes were
covered in the samples analyzed. In this family, the disease
exhibited a pattern of recessive inheritance. Under the autosomal
recessive model, the filtered data were reduced to two compound
heterozygous variants, c.2876C>T (p.S959F) and c.5772C>A
(p.F1924L) in the PKHD1 gene (NM_138694.3), which exhibited
complete co-segregation with the disease. The two compound
heterozygous variants were further validated using the Sanger
sequencing method in other family members and 576 normal controls.
Finally, it was revealed that the uncle (II:1), mother (II:3),
father (II:4) and grandmother (I:2) were heterozygous carriers
(Table II). The two mutations
were absent in the unaffected family member (I:1) and the normal
controls. Therefore, the two mutations co-segregated with the
phenotype in this family. The results demonstrated that the
compound heterozygous mutations c.2876C>T (p.S959F) and
c.5772C>A (p.F1924L) in the PKHD1 gene are responsible
for ARPKD (Fig. 3). Comparative
amino acid sequence alignment of other PKHD1 proteins across
different species revealed that the mutations occurred at highly
conserved positions in exons 27 and 36 (data not shown).
Furthermore, the two compound heterozygous mutations were predicted
to be damaging by the SIFT and PolyPhen tools (Table II). The substituted amino acid was
predicted to alter the hydrophobicity of the PKHD1 protein.
For c.2876C>T (p.S959F) mutation, the hydrophilic serine is
changed to a hydrophobic phenylalanine at position 959, while the
phenylalanine is changed to leucine at position 1,924, potentially
affecting the structure of the protein, for c.5772C>A (p.F1924L)
mutation.
| Table II.Two novel polycystic kidney and
hepatic disease 1 mutations identified in a family with autosomal
recessive polycystic kidney disease. |
Table II.
Two novel polycystic kidney and
hepatic disease 1 mutations identified in a family with autosomal
recessive polycystic kidney disease.
| Positiona | Exon | Nucleotide
change | Amino acid
change | SIFT
score/PolyPhen | Mutant type | Mutation present |
|---|
| 51907878 | 27 | c.2876C>T | S959F | 0.03/1.0
(Damaging) | Het | II:4, III:1,
III:2 |
| 51824804 | 36 | c.5772C>A | F1924L | 0.20/1.0
(Damaging) | Het | I:2, II:1, II:2,
II:3, III:1, III:2 |
Discussion
As increased echogenicity and renal enlargement are
the main indicators of ARPKD from an ultrasound (1,7);
notably, prenatal diagnosis of ARPKD using ultrasound alone is not
reliable (8). For 35% of patients
with only medullary abnormalities, the standard ultrasound exhibits
a normal signal (9). The family in
the present study underwent repeated antenatal ultrasound
examinations and fetal diagnosis of ARPKD was confirmed at week
23.
Considering the phenotypic variability (1), clinicians should consider the risk of
predicting the clinical outcome of a child whose parents had
already suffered abnormal pregnancies. The use of PKHD1
sequencing data for clinical decision-making requires caution,
particularly in cases where only novel or rare missense variants
are identified. In the present study, ultrasound results of the
first pregnancy of the couple (for the proband, III:1) exhibited
separation of the left renal sinus, enhancement of the right renal
parenchymal echo, more echo-free zones in the right kidney, which
indicated a number of cystic renal dysplasia, and the right cleft
lip and palate.
Multiple lines of evidence support that PKHD1
gene mutations serve a pathogenic role in ARPKD (1,10).
All of the identified mutations in PKHD1 are distributed
throughout the gene without evidence of clustering at specific
sites. The majority of these mutations are compound heterozygous
mutations, which consist of 332 missenses (252 possibly/probably
pathogenic and 80 single nucleotide polymorphisms), 55 nonsense, 86
frame shift and 58 silent mutations (1,9,11–13).
The two mutations identified in the present study, to the best of
our knowledge, have not previously been reported. Patients with two
truncation mutations exhibit a grievous phenotype resulting in
perinatal or neonatal demise, whereas patients escaping from
neonatal lethality usually possess no more than one amino acid
substitution (14). The proband
(with two mutations) in the present study died, resulting in a
miscarriage, whereas the parents, with only one of the two gene
mutations, had no phenotype. It is hypothesized that the expression
of only a minimal amount of functional protein is required for
survival during the neonatal period in ARPKD (7). Since there are 67 exons in the
PKHD1 gene, it is not efficient to screen mutations for this
gene by Sanger sequencing. Therefore, targeted NGS which may be of
specific interest to identify mutations (2), was employed to identify causative
mutations in PKHD1. In the present study, two newly
identified mutations (c.2876C>T and c.5772C>A), which caused
moderate alterations in the genotype and no changes in the
phenotype of the parents, were identified. These observations were
consistent with previous reports that patients who survive the
neonatal period usually have no more than one missense mutation
(8–10).
In conclusion, the present study emphasized the
importance of prenatal diagnosis of renal abnormalities by routine
fetal anatomic ultrasound and genetic testing for ARPKD.
Furthermore, the present study provided details of the changes in
fetal biometry over the prenatal and perinatal course of ARPKD with
two novel PKHD1 mutations.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
National Natural Science Foundation of China (grant no. 81300618 to
XZ), the Sichuan Provincial Planning Commission Fund (grant no.
17PJ058 to XZ) and the Sichuan Provincial People's Hospital
Research Foundation (grant no. 2016LY07 to XZ).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
JW, DQ, JY, DZ, QW and XZ designed the study,
conducted the experiments and analyzed the data. JW and XZ wrote
the paper. XJ and XZ recruited the participants and collected
clinical information. All of the authors contributed to writing of
the paper and approved the final version.
Ethics approval and consent to
participate
The present study was approved by the Medical Ethics
Committee of the Sichuan Academy of Medical Sciences & Sichuan
Provincial People's Hospital [no. 2013 Natural Science (04)]. The
patients provided written informed consent for participation in the
study at Sichuan Provincial People's Hospital.
Patient consent for publication
The patients received all information regarding this
study. Written informed consent for publication in Molecular
Medicine Reports was obtained from the patients.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ARPKD
|
autosomal recessive polycystic kidney
disease
|
|
PKHD1
|
polycystic kidney and hepatic disease
1
|
|
NGS
|
next-generation sequencing
|
References
|
1
|
Onuchic LF, Furu L, Nagasawa Y, Hou X,
Eggermann T, Ren Z, Bergmann C, Senderek J, Esquivel E, Zeltner R,
et al: PKHD1, the polycystic kidney and hepatic disease 1 gene,
encodes a novel large protein containing multiple
immunoglobulin-like plexin-transcription-factor domains and
parallel beta-helix 1 repeats. Am J Hum Genet. 70:1305–1317. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tavira B, Gomez J, Malaga S, Santos F,
Fernandez-Aracama J, Alonso B, Iglesias S, Benavides A, Hernando I,
Plasencia A, et al: A labor and cost effective next generation
sequencing of PKHD1 in autosomal recessive polycystic kidney
disease patients. Gene. 561:165–169. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dorn L, Menezes LF, Mikuz G, Otto HF,
Onuchic LF and Sergi C: Immunohistochemical detection of polyductin
and co-localization with liver progenitor cell markers during
normal and abnormal development of the intrahepatic biliary system
and in adult hepatobiliary carcinomas. J Cell Mol Med.
13:1279–1290. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ward CJ, Hogan MC, Rossetti S, Walker D,
Sneddon T, Wang X, Kubly V, Cunningham JM, Bacallao R, Ishibashi M,
et al: The gene mutated in autosomal recessive polycystic kidney
disease encodes a large, receptor-like protein. Nat Genet.
30:259–269. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pang M, Liu Y, Hou X, Yang J, He X, Hou N,
Liu P, Liang L, Fu J, Wang K, et al: A novel APC mutation
identified in a large Chinese family with familial adenomatous
polyposis and a brief literature review. Mol Med Rep. 18:1423–1432.
2018.PubMed/NCBI
|
|
6
|
Gunay-Aygun M, Font-Montgomery E, Lukose
L, Tuchman Gerstein M, Piwnica-Worms K, Choyke P, Daryanani KT,
Turkbey B, Fischer R, Bernardini I, et al: Characteristics of
congenital hepatic fibrosis in a large cohort of patients with
autosomal recessive polycystic kidney disease. Gastroenterology.
144:112–121.e2. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Frank V, Zerres K and Bergmann C:
Transcriptional complexity in autosomal recessive polycystic kidney
disease. Clin J Am Soc Nephrol. 9:1729–1736. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xu Y, Xiao B, Jiang WT, Wang L, Gen HQ,
Chen YW, Sun Y and Ji X: A novel mutation identified in PKHD1 by
targeted exome sequencing: Guiding prenatal diagnosis for an ARPKD
family. Gene. 551:33–38. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gunay-Aygun M, Font-Montgomery E, Lukose
L, Tuchman M, Graf J, Bryant JC, Kleta R, Garcia A, Edwards H,
Piwnica-Worms K, et al: Correlation of kidney function, volume and
imaging findings, and PKHD1 mutations in 73 patients with autosomal
recessive polycystic kidney disease. Clin J Am Soc Nephrol.
5:972–984. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bitarafan F and Garshasbi M: Molecular
genetic analysis of PKHD1 mutations in pedigrees with autosomal
recessive polycystic kidney disease. Iran J Kidney Dis. 12:350–358.
2018.PubMed/NCBI
|
|
11
|
Gunay-Aygun M, Tuchman M, Font-Montgomery
E, Lukose L, Edwards H, Garcia A, Ausavarat S, Ziegler SG,
Piwnica-Worms K, Bryant J, et al: PKHD1 sequence variations in 78
children and adults with autosomal recessive polycystic kidney
disease and congenital hepatic fibrosis. Mol Genet Metab.
99:160–173. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Datta R, Shah GN, Rubbelke TS, Waheed A,
Rauchman M, Goodman AG, Katze MG and Sly WS: Progressive renal
injury from transgenic expression of human carbonic anhydrase IV
folding mutants is enhanced by deficiency of p58IPK. Proc Natl Acad
Sci USA. 107:6448–6452. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
O'Meara CC, Hoffman M, Sweeney WE, Tsaih
SW, Xiao B, Jacob HJ, Avner ED and Moreno C: Role of genetic
modifiers in an orthologous rat model of ARPKD. Physiol Genomics.
44:741–753. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Koga T, Duan H, Urabe K and Furue M:
IFN-gamma-positive immunostaining in psoriatic lesional
keratinocytes-reply to the comments of McKenzie. et al Eur J
Dermatol. 13:992003.PubMed/NCBI
|