Introduction
The proliferation of mesangial cells (MCs) is a
characteristic feature of glomerular diseases, including lupus
nephritis, IgA nephropathy, membranoproliferative
glomerulonephritis and diabetic nephropathy (1). Furthermore, in animal models of
nephritis, MC proliferation often occurs before mesangial sclerosis
and the glomerulosclerosis, and is associated with the increase in
extracellular matrix synthesis (2). Regulation of MC proliferation may
therefore represent a potential treatment option for numerous
kidney diseases (3). Transforming
growth factor (TGF)-β1 is a cytokine that promotes fibrosis in
chronic renal diseases, and may trigger and regulate various types
of pathophysiological process. For example, TGF-β contributes to
glomerular extracellular matrix accumulation by stimulating MCs to
produce type I, III and IV collagen, laminin, fibronectin and
heparan sulphate proteoglycans, as well as by inhibiting matrix
degradation, which contributes to the development of
glomerulosclerosis (4). However,
TGF-β1 also has anti-inflammatory properties in certain
pathological conditions. For example, TGF-β blockade increased
kidney inflammation, which was characterized by the upregulation of
MCP1, proinflammatory chemokines and the infiltration of
monocytes/macrophages in the kidneys (5). Considering these situations, TGF-β1
may not be an optimal therapeutic target for kidney disease
(5). It is therefore crucial to
explore the TGF-β1-associated mechanisms in order to identify its
therapeutic targets.
Long non-coding (lnc) RNAs are defined as
transcripts of >200 nucleotides in length that are not
translated into proteins (6).
Compared with protein-coding genes, lncRNAs are more conserved and
specific to organs, tissues, cell types, developmental stages and
disease conditions. In addition, lncRNAs are considered as better
therapeutic targets in kidney diseases compared with protein-coding
transcriptomes (7). It has been
reported that lncRNAs may be more responsible for the early
development of diabetic nephropathy compared with protein-coding
genes (8). For example, the lncRNA
LINC01619 contributes to the development of diabetic nephropathy,
as it can regulate microRNA (miR)-27a/forkhead box protein O1 and
endoplasmic reticulum stress, resulting in podocyte injury
(9). Furthermore, lncARSR was
reported to promote the expression of AXL receptor tyrosine kinase
and c-MET in renal cell carcinoma cells by competitively binding to
miR-34/miR-449, which results in cell resistance to sunitinib
(10). lncRNAs may therefore
represent potential therapeutic targets in kidney diseases.
Our previous study revealed that lncRNA uc.412 is
differentially expressed in TGF-β1-treated MCs (11). In the present study, in order to
elucidate the downstream of roles of lncRNA uc.412, the gene
expression profiles in MCs overexpressing lncRNA uc.412 were
determined using high-throughput sequencing analysis and verified
by reverse-transcription-quantitative (RT-q) PCR. Bioinformatics
analysis of the identified differentially expressed genes (DEGs)
was achieved to predict the function of uc.412. Results from the
present suggested an association between uc.412 and kidney
diseases. These data may aid our understanding of the role of
uc.412 in mesangial cells and in elucidating the mechanism involved
in abnormal MC proliferation.
Materials and methods
Cell culture
The rat mesangial cell line RMC (cat. no. ATCC
CRL-2573) was purchased from the American Type Culture Collection.
RMCs were cultured in DMEM (HyClone; GE Healthcare Life Sciences)
containing 10% FBS (Crystalgen, Inc.) and 1%
penicillin/streptomycin (Gibco; Thermo Fisher Scientific, Inc.) and
placed at 37°C in a humidified incubator containing 5%
CO2. The Medical Ethics Committee of the Second
Affiliated Hospital of Nanjing Medical University approved the
protocol of this study. The proliferation of MCs is a
characteristic feature of glomerular diseases. The TGF-β signaling
pathway is the main pathway for MC proliferation. The cells were
treated with TGF-β1 at a concentration of 10 ng/ml for 24 h and
then MCs were used in further experiments to detect the expression
of lncRNA uc.412 (11).
Cell transfection
MCs were transfected with uc.412 plasmid (oeuc.412;
VectorBuilder) or empty vector control (oeVec) with Lipofectamine
2000® (Invitrogen; Thermo Fisher Scientific, Inc.).
Briefly, 2.5 µg plasmid was diluted into 125 µl Opti-MEM serum-free
medium (Gibco; Thermo Fisher Scientific, Inc.), and 6 µl
Lipofectamine 2000 was diluted with 125 µl Opti-MEM serum-free
medium and incubated room temperature for 5 min. The two solutions
were incubated at room temperature for 20 min and then added to the
cells seeded in 6-well plates at a density of 2×105
cells/well. Culture medium was replaced after 8 h. After 24 h,
puromycin was used to select cells that were puromycin resistant
following transfection. After a further 24 h, these cells were used
for further experiment. The transfection efficacy was detected by
RT-qPCR.
Cell proliferation assay
Transfected cells were seeded in 96-well plates at
the density of 1×104 cells/well in sextuplicate and
cultured in 100 µl DMEM containing 10% FBS. At 0, 24, 48 and 72 h
following seeding, 10 µl Cell Counting Kit-8 reagent (Dojindo
Molecular Technologies, Inc.) was added in each well. Following 30
min incubation, the absorbance was measured at 450 nm using a
microplate reader. The relative cell proliferation rate was
normalized to the optical density value at 0 h.
Total RNA extraction and RT-qPCR
TRIzol® (Thermo Fisher Scientific, Inc.)
was used to extract total RNA from MCs according to the
manufacturers' instructions. Total RNA (500 ng) was transcribed
into cDNA using PrimeScript™ RT Reagent kit (Takara Bio, Inc.),
according to the manufacturers' instructions. qPCR analysis was
performed with the ABI StepOne Real-Time PCR system (Applied
Biosystems; Thermo Fisher Scientific, Inc.) using the
SYBR® Premix Ex Taq™ kit (Takara Bio, Inc.) and the
following thermocycling conditions: 95°C for 30 sec, followed by 40
cycles of 95°C for 5 sec and 60°C for 30 sec. StepOne software v2.0
(Applied Biosystems; Thermo Fisher Scientific, Inc.) was used to
analyze the data based on the 2−ΔΔCq method and GAPDH
was used as the internal control for normalization (12). The primer sequences are listed in
Table SI.
High-throughput sequencing
Total RNA was extracted as aforementioned and
processed by oligo DT enrichment (rRNA removed), and KAPA Stranded
RNA-Seq Library Prep kit (Illumina, Inc.) was used to construct an
RNA library. In the library construction process, a chain-specific
RNA sequencing library was constructed using double-stranded cDNA
synthesis using the dUTP method and subsequently high-fidelity PCR
polymerases (13). The quality of
the constructed library was verified by the Agilent 2100
Bioanalyzer (Agilent Technologies, Inc.) and quantified by RT-qPCR.
Library sequencing was performed on an Illumina HiSeq 4000
(Illumina, Inc.), using NaOH base denaturing single-strand
generation, Illumina flow cell in situ amplification and 150
double-end cycle sequencing. Image processing and base recognition
was performed using Solexa Pipeline software version 1.8. The
FastQC software (version 0.11.5; Babraham Bioinformatics; Babraham
Institute) was used to assess the sequencing quality of the reads
and remove joints. The library was compared to the reference genome
using Hisat2 software (version 2.1.; http://ccb.jhu.edu/software/hisat2/faq.shtml).
StringTie software (version v1.3.3; http://ccb.jhu.edu/software/stringtie/) was used to
estimate the transcript abundance. R software Ballgown was used to
determine fragments per kilobase of exon per million fragments
mapped at the gene level and at the transcript level and to screen
the DEGs between samples. DEGs with |FC| ≥1.5 were highlighted;
where FC is fold change.
Bioinformatics analysis
Gene Ontology (GO) clustering and Kyoto Encyclopedia
of Genes and Genomes (KEGG; http://www.kegg.jp) pathway analysis were performed on
all DEGs identified following RNA sequencing. GO enrichment
analysis of DEGs was retrieved using the DAVID database (http://david.abcc.ncifcrf.gov) (14,15).
KEGG pathway enrichment analysis and results were visualized using
Cytoscape (http://cytoscape.org).
Protein-protein interaction (PPI)
network and downstream effects of uc.412
To determine the downstream effects of uc.412 on the
proliferation of MC, the STRING database (http://www.string-db.org) was used to analyze the PPI
networks of DEGs. The cut-off criterion was set as a combined PPI
score of >0.4.
Statistical analysis
SPSS 17.0 software (SPSS, Inc.) was used to conduct
the statistical analyses. Student's t-test was used to compare two
groups. Results were presented as the mean ± the SD. Each
experiment was repeated at least three times. P<0.05 was
considered to indicate a statistically significant difference.
Results
MC proliferation is significantly
increased following uc.412 overexpression
RT-qPCR results demonstrated that uc.412 was
upregulated in TGF-β1-treated MCs (P<0.05; Fig. 1A). Furthermore, uc.412 expression
level was significantly increased in oeuc.412 MCs compared with
Control oeVec MCs (P<0.05; Fig.
1B). In addition, the proliferation rate of MCs overexpressing
uc.412 was significantly increased after 48 and 72 h compared with
oeVec-transfected MCs (P<0.05; Fig.
1C).
DEG analysis
Following oeuc.412 transfection, a total of 1,305
DEGs were identified from RNA sequencing, of which 462 were
upregulated and 843 were downregulated. The scatter plot of the
expression level of the DEGs between oeuc.412-transfected group and
oeVec-transfected control group is presented in Fig. 2A, and the differences among the top
50 DEGs in each group are highlighted in the heatmaps in Fig. 2B. The full lists of up- and
downregulated genes are presented in Tables SII and SIII, respectively.
 | Figure 2.Identification of DEGs between
oeuc.412 and oeVec MCs. (A) Scatter plot used to determine gene
expression variations between uc.412-overexpressing MCs and control
MCs. Expression levels were detected and normalized as tag counts
per million of TPM. In the scatter-plot, values on × and y axes
represent the averaged TPM values of each group (log2
scaled). Red dots represent upregulated genes, and green dots
represent downregulated genes, identified as changes of >1.5
fold change between the two comparison groups; gray dots indicate
genes that were not differentially expressed. (B) DEGs between
oeuc.412 MCs and oeVEC negative control. The top 50 upregulated
genes and 50 downregulated genes with the highest difference in
expression levels. In the left figure, red represent relatively
high expression, and white represents relatively low expression. In
the right figure, blue represents relatively high expression and
white represent relatively low expression. DEGS, differentially
expressed genes; MCs, mesangial cells; oe, overexpressing; uc.412,
long non-coding RNA uc.412; vec, vector; TPM, total aligned tRNA
reads. |
To verify the DEGs detected by RNA sequencing, nine
genes (16) were randomly selected
and their mRNA expression levels were examined by RT-qPCR in
oeuc.412- and oeVec-transfected MCs; these included five
upregulated genes [phospholipase A2 group IIA (PLA2G2A), integrin
subunit a6 (ITGA6), fms related tyrosine kinase 1 (FLT1), serpin
family E member 1 (SERPINE1) and cd55], and four downregulated
genes [kelch-like family member 13 (KLHL13), argonaute RISC
component 1 (AGO1), matrix metallopeptidase 3 (MMP3) and tumor
necrosis factor receptor superfamily member 1B (TNFRS1B)]. The
results demonstrated that the expression levels of PLA2G2A, ITGA6,
FLT1, SERPINE1 and CD55 were significantly increased in MCs
overexpressing uc.412 compared with control cells (Fig. 3A), whereas KLHL13, AGO1, MMP3 and
TNFRS1B expression levels were significantly decreased in oeuc.412
compared with oeVEC MCs (Fig. 3B),
which was consistent with the RNA sequencing results. This data
identified an interlaced transcript network associated with
uc.412.
 | Figure 3.Reverse transcription-quantitative PCR
confirmation of the mRNA expression levels of the identified
differentially expressed genes. The data indicated that (A)
PLA2G2A, ITGA6, FLT1, SERPINE1 and CD55 expression levels were
significantly increased, and (B) KLHL13, AGO1, MMP3 and TNFRS1B
expression levels were significantly decreased in
oeuc.412-transfected MCs compared with oeVec-transfected MCs. Each
experiment was performed three times and with five replicates;
*P<0.05. AGO1, argonaute RISC component 1; FLT1, fms related
tyrosine kinase 1; ITGA6, integrin subunit alpha 6; KLHL13,
kelch-like family member 13; MMP3, matrix metallopeptidase 3; oe,
overexpressing; PLA2G2A, phospholipase A2 group IIA; SERPINE1,
serpin family E member 1; TNFRS1B, tumor necrosis factor receptor
superfamily member 1B; uc.412, long non-coding RNA uc.412; vec,
vector. |
GO clustering of DEGs
GO clustering was performed on the 1,305 DEGs and
included the biological processes, the cellular components and the
molecular function categories (Fig.
4). The top nine biological processes associated with the
identified DEGs that were identified included ‘extracellular matrix
organization’, ‘positive regulation of cell proliferation’,
‘epithelial to mesenchymal transition’, ‘negative regulation of
protein kinase activity’ and ‘angiogenesis’ (Fig. 4A). The cellular components
identified included ‘cytosol’, ‘cytoplasm’, ‘extracellular matrix’,
‘cytoskeleton’ and ‘proteinaceous extracellular matrix’ (Fig. 4B). The molecular functions
identified included ‘protein binding’, ‘sequence-specific DNA
binding’, ‘extracellular matrix binding’, ‘protein kinase inhibitor
activity’ and ‘transcriptional activator activity’ (Fig. 4C).
KEGG pathway analysis of DEGs
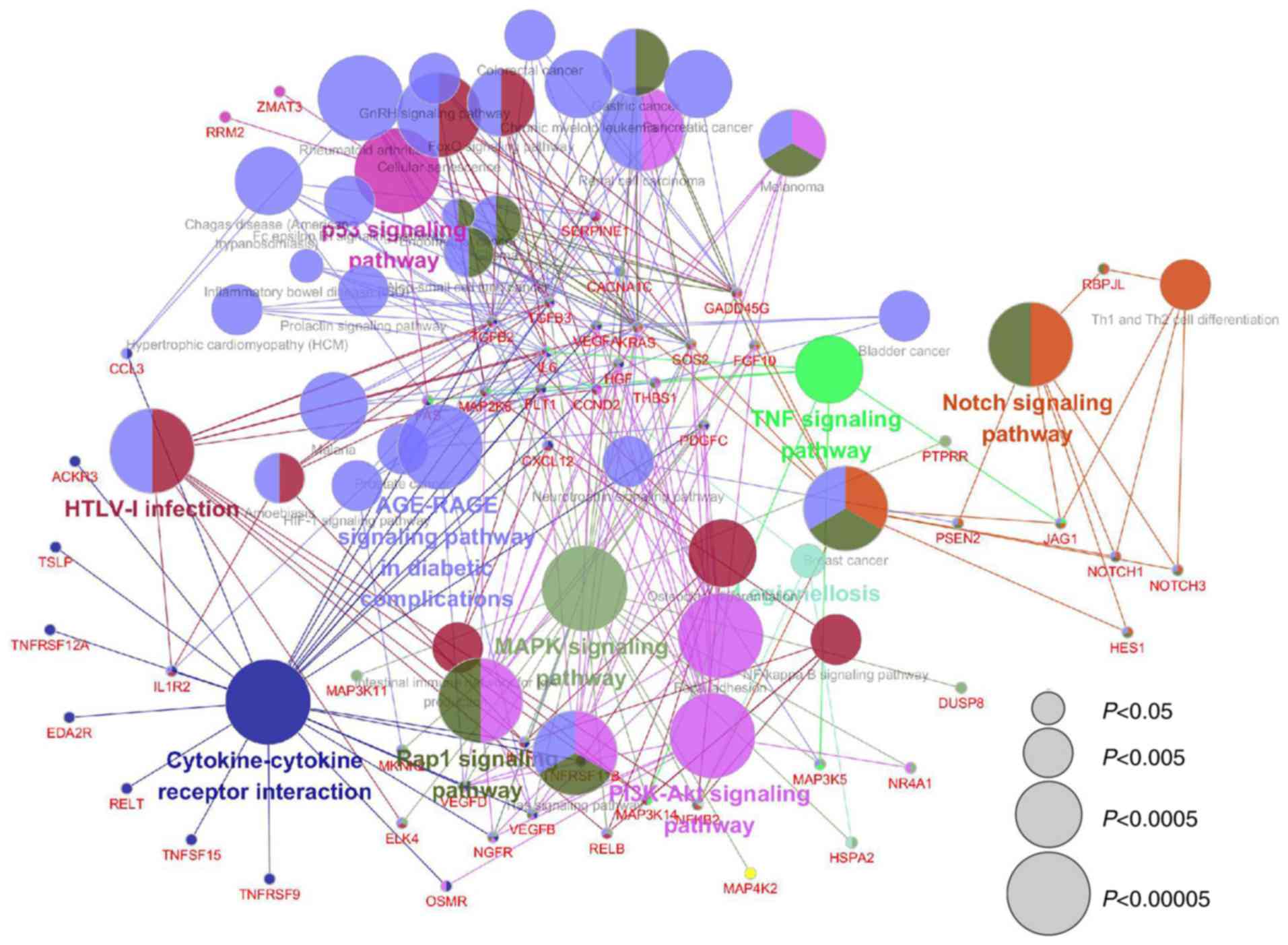
KEGG pathway analysis was performed on the
identified DEGs. The pathways identified included ‘p53 signaling
pathway’, ‘Notch signaling pathway’, ‘MAPK signaling pathway’,
‘PI3K-Akt singling pathway’, ‘Cytokine-cytokine receptor
interaction’, ‘HTLV-1 infection’, ‘AGE-RAGE signaling pathway in
diabetic complications’ and ‘TNF signaling pathway’ (Fig. 5). The results indicated that the
DEGs associated with uc.412 may be associated with several
signaling pathways and make up a complex network.
PPI network and downstream effects of
uc.412
The STRING database was used to predict the putative
downstream functional genes of uc.412. The results demonstrated
that SERPINE1 interacted with connective tissue growth factor
(CTGF), hepatocyte growth factor (HGF), FLT1, interleukin 6 (IL6),
prostaglandin G/H synthase 2 (PTGS2), tissue-type plasminogen
activator (PLAT) and insulin-like growth factor-binding protein 5
(IGFBP5) in the network of the upregulated genes (Fig. S1A), and, therefore, may be a hub
protein. MMP3 interacts with TNFRS1B, ETS domain-containing protein
ELK4 (ELK4), integrin subunit β-like 1 (ITGBL1), endothelin-1
receptor (EDNRA) and cysteine-rich secretory protein 1 (CRISP1)
(Fig. S1B).
Discussion
TGF-β1 serves a crucial role in promoting fibrosis
in tissue (4). Since antifibrotic
therapy consisting in targeting TGF-β1 to treat renal fibrosis can
lead to dysregulation of other normal physiological processes
(4), it is necessary to understand
the specific downstream mechanisms of TGF-β1-induced fibrosis to
identify additional precise targets. It has been demonstrated that
lncRNAs can directly regulate some biological processes in a
disease- and tissue-specific manner (6). For example, the lncRNA SARCC binds to
and destabilizes the androgen receptor post-transcriptionally to
regulate its expression, which can contribute to tumor progression
in renal cell carcinoma (17).
Targeting lncRNAs may therefore provide a novel therapeutic
approach in various types of cancer. Results from the present study
demonstrated that uc.412 was upregulated in MCs following TGF-β1
treatment and that uc.412 overexpression significantly increased MC
proliferation. It is therefore crucial to understand the underlying
mechanism of uc.412 in the regulation of MC proliferation and to
determine whether uc.412 may be considered as a therapeutic target
in mesangial proliferative kidney diseases.
To determine the potential biological functions of
uc.412 in MCs, DEGs were identified between MCs that overexpressed
uc.412 and negative control MCs using high-throughput RNA
sequencing. The results from this analysis identified 1,305 DEGs,
including 462 up- and 843 downregulated genes.
The number of samples analyzed in the present study
was limited, criteria for selecting DEGs was set as |FC| ≥1.5. In
order to verify the RNA-sequencing results, expression levels of
five upregulated and 4 downregulated DEGs were subsequently
verified by RT-qPCR. Serum secretory phospholipase A2-IIa, which is
encoded by PLA2G2A, has been demonstrated to be associated with
cell signaling, apoptosis, remodeling of cell membranes and
inflammatory responses (16). The
β-catenin-dependent Wnt signaling pathway may regulate PLA2G2A
expression through the β-catenin-dependent Wnt signaling pathway;
the Wnt pathway is involved in numerous kidney diseases, including
glomerular diseases, ischemic kidney injury, interstitial fibrosis,
diabetic nephropathy and cystic kidney diseases (18). Deregulation of the Wnt pathway has
been associated with fibrotic diseases in kidney (19). Numerous molecules have been
demonstrated to be associated with the establishment of the
fibrotic condition, including angiotensin II, TGF-β1 and CTGF
(19). Furthermore, a crosstalk
between the Wnt pathway and these signaling molecules has been
identified (19). uc.412 may
therefore regulate the expression of certain genes involved in
proliferation and fibrosis of MCs and may serve a role in the
pathways that contribute to the development of kidney diseases.
The results from GO analysis indicated that the
identified DEGs may be involved in certain biological process,
which may serve crucial roles in the incidence and development of
certain kidney diseases. For example, podocyte injury may lead to
glomerulosclerosis in IgA nephropathy (IgAN) (19). Various factors contributing to
epithelial-to-mesenchymal transition (EMT) have been demonstrated
to be responsible for podocyte damage. For example, hyperglycemia
has been demonstrated to induce podocyte EMT through TGF-β/Smad
classic pathway, the Wnt/β-catenin signaling pathway, the
integrin/integrin-linked kinase signaling pathway, the
mitogen-activated protein kinase signaling pathway, the
Jagged/Notch signaling pathway and the nuclear factor-κB signaling
pathway and contribute to podocyte damage (20). However, it was previously reported
that the PI3K/AKT-signaling pathway may be involved in EMT in
podocytes following treatment with mesangial medium from cells
isolated from IgAN patients (19).
In addition, previous studies reported that directly modulating
cell cycle progress could regulate RMC proliferation (21,22).
Furthermore, immunosuppressive agents could affect the cell cycle
progression, apoptosis and proliferation of MCs (23). Some kidney diseases can be
ameliorated through cell cycle-dependent mechanisms (23); for example, MC cell cycle can be
regulated using rapamycin to induce an inhibition of MC
proliferation, a reduction of IgA deposition and a slow IgAN
progression (24). Results from
the present study indicated that uc.412 downstream genes may
regulate cell cycle, which indicated that uc.412 may be involved in
MC proliferation.
The results from KEGG clustering analysis,
identified several key pathways and a complex regulatory network.
It has been reported that p53 is upregulated in a mouse model of
Habu nephritis, which suggested that p53 can modulate MC
proliferation and apoptosis (25).
Furthermore, Notch signaling pathway could activate TGF-β to
accelerate the development of diabetic nephropathy (26). The dysregulation of Notch signaling
molecules has been reported to serve a vital role in fibrosis
models, renal injuries and diabetic kidney biopsies (26). For example, Jagged1, which is the
canonical Notch ligand, is upregulated in a TGF-β dependent manner
during chronic kidney diseases (26). The present study therefore
hypothesized that uc.412 may serve a crucial role in the
development of mesangial proliferative glomerulonephritis.
STRING was used to construct PPI networks from RNA
sequencing data. The results demonstrated that SERPINE1 interacted
with a number of proteins in the upregulated genes PPI network. For
example, SERPINE1 may interact with CTGF, which has been
demonstrated to serve a crucial role in the progression of kidney
fibrosis, as it participates in cell migration, proliferation and
inflammation (27). CTGF can also
stimulate MCs to synthesize more fibronectin and collagen type I
(28). MMP3 was also revealed to
interact with a number of proteins in the downregulated genes PPI
network. In addition, a series of genes associated with cell
proliferation were involved in the interaction networks. For
example, Notch3 interacted with cyclin D2, dynamin 3 and EPH
receptor A7 (29). Downregulating
cellular expression of Notch3 may therefore improve cell
proliferation (29). In
conclusion, the results from the present study exhibited an
interlaced transcript network associated with uc.412, which may
have important effects on MC proliferation.
Identifying the role of uc.412 in MC proliferation
is crucial to understanding certain glomerular diseases. The
present study identified the downstream genes of uc.412 and a
complicated mesangial cell network. These results may aid in
developing novel therapeutic strategies to limit the pathological
process of glomerular diseases associated with uncontrolled MC
proliferation. However, the underlying mechanism and biological
function of uc.412 in the regulation of MC proliferation require
further investigation.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by The National
Natural Science Foundation of China (grant no. 81670650), The
Natural Science Foundation of Jiangsu Province (grant no.
BK20161071), The Project of Nanjing Medical Youth Talent (grant no.
QRX17106), The Project of Jiangsu Provincial Maternal and Child
Talent (grant no. FRC201737), The Research Project of Jiangsu
Provincial Health Commission (grant no. LGY2018071) and The Science
and Technology Development Foundation of Nanjing Medical University
(grant no. 2016NJMU036).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
SL, AZ, BW and WG conceived and designed the study.
MY, ZG and XW performed the experiments. HS, GQ, XL, QF and XZ
interpreted the data. MY wrote the manuscript. AZ critically
reviewed and revised the manuscript. SL and QF revised the
manuscript. All authors have read and approved the final version of
the manuscript.
Ethics approval and consent to
participate
The present study was approved by the Medical Ethics
Committee of the Second Affiliated Hospital of Nanjing Medical
University (Nanjing, China; approval number: 2016KY008-01).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Cove-Smith A and Hendry BM: The regulation
of mesangial cell proliferation. Nephron Exp Nephrol. 108:e74–e79.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kurogi Y: Mesangial cell proliferation
inhibitors for the treatment of proliferative glomerular disease.
Med Res Rev. 23:15–31. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang A, Han Y, Wang B, Li S and Gan W:
Beyond gap Junction channel function: The expression of Cx43
contributes to aldosterone-induced mesangial cell proliferation via
the ERK1/2 and PKC pathways. Cell Physiol Biochem. 36:1210–1222.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Loeffler I and Wolf G: Transforming growth
factor-β and the progression of renal disease. Nephrol Dial
Transplant. 29 (Suppl 1):i37–i45. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rodrigues-Diez R, Rayego-Mateos S, Orejudo
M, Aroeira LS, Selgas R, Ortiz A, Egido J and Ruiz-Ortega M:
TGF-beta blockade increases renal inflammation caused by the
C-terminal module of the CCN2. Mediators Inflamm. 2015:5060412015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kopp F and Mendell JT: Functional
classification and experimental dissection of long noncoding RNAs.
Cell. 172:393–407. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tang PM, Tang PC, Chung JY and Lan HY:
TGF-β1 signaling in kidney disease: From Smads to long non-coding
RNAs. Noncoding RNA Res. 2:68–73. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tang W, Zhang D and Ma X: RNA-sequencing
reveals genome-wide long non-coding RNAs profiling associated with
early development of diabetic nephropathy. Oncotarget.
8:105832–105847. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bai X, Geng J, Li X, Wan J, Liu J, Zhou Z
and Liu X: Long noncoding RNA LINC01619 regulates
MicroRNA-27a/Forkhead box protein O1 and endoplasmic reticulum
stress-mediated podocyte injury in diabetic nephropathy. Antioxid
Redox Signal. 29:355–376. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Qu L, Ding J, Chen C, Wu ZJ, Liu B, Gao Y,
Chen W, Liu F, Sun W, Li XF, et al: Exosome-transmitted lncARSR
promotes sunitinib resistance in renal cancer by acting as a
competing endogenous RNA. Cancer Cell. 29:653–668. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang A, He Y, Wang B, Shi H and Gan W:
Analysis of the differential expression of long noncoding RNAs in
experimental mesangial cells proliferation induced by TGF-β. Chin J
Nephrol. 31:774–779. 2015.
|
|
12
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ma F, Fuqua BK, Hasin Y, Yukhtman C, Vulpe
CD, Lusis AJ and Pellegrini M: A comparison between whole
transcript and 3′RNA sequencing methods using Kapa and Lexogen
library preparation methods. BMC Genomics. 20:92019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Huang da W, Sherman BT and Lempicki RA:
Bioinformatics enrichment tools: Paths toward the comprehensive
functional analysis of large gene lists. Nucleic Acids Res.
37:1–13. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Huang da W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2008. View Article : Google Scholar
|
|
16
|
Cui Y, Huang Y, Wu X, Zheng M, Xia Y, Fu
Z, Ge H, Wang S and Xie H: Hypoxia-induced tRNA-derived fragments,
novel regulatory factor for doxorubicin resistance in
triple-negative breast cancer. J Cell Physiol. 234:8740–8751. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhai W, Sun Y, Jiang M, Wang M, Gasiewicz
TA, Zheng J and Chang C: Differential regulation of LncRNA-SARCC
suppresses VHL-mutant RCC cell proliferation yet promotes
VHL-normal RCC cell proliferation via modulating androgen
receptor/HIF-2α/C-MYC axis under hypoxia. Oncogene. 35:4866–4880.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dong T, Peng Y, Zhong N, Liu F, Zhang H,
Xu M, Liu R, Han M, Tian X, Jia J, et al: Perfluorodecanoic acid
(PFDA) promotes gastric cell proliferation via sPLA2-IIA.
Oncotarget. 8:50911–50920. 2017.PubMed/NCBI
|
|
19
|
Kawakami T, Ren S and Duffield JS: Wnt
signalling in kidney diseases: Dual roles in renal injury and
repair. J Pathol. 229:221–231. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ying Q and Wu G: Molecular mechanisms
involved in podocyte EMT and concomitant diabetic kidney diseases:
An update. Ren Fail. 39:474–483. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lan T, Wu T, Chen C, Chen X, Hao J, Huang
J, Wang L and Huang H: Berberine attenuates high glucose-induced
proliferation and extracellular matrix accumulation in mesangial
cells: Involvement of suppression of cell cycle progression and
NF-κB/AP-1 pathways. Mol Cell Endocrinol. 384:109–116. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen D, Li Y, Mei Y, Geng W, Yang J, Hong
Q, Feng Z, Cai G, Zhu H, Shi S, et al: miR-34a regulates mesangial
cell proliferation via the PDGFR-β/Ras-MAPK signaling pathway. Cell
Mol Life Sci. 71:4027–4042. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhou X, Workeneh B, Hu Z and Li R: Effect
of immunosuppression on the human mesangial cell cycle. Mol Med
Rep. 11:910–916. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tian J, Wang Y, Liu X, Zhou X and Li R:
Rapamycin ameliorates IgA nephropathy via cell cycle-dependent
mechanisms. Exp Biol Med (Maywood). 240:936–945. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lu Y, Wen J, Chen D, Wu L, Li Q, Xie Y, Wu
D, Liu X and Chen X: Modulation of cyclins and p53 in mesangial
cell proliferation and apoptosis during Habu nephritis. Clin Exp
Nephrol. 20:178–186. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu L, Gao C, Chen G, Li X, Li J, Wan Q
and Xu Y: Notch signaling molecules activate TGF-β in rat mesangial
cells under high glucose conditions. J Diabetes Res.
2013:9797022013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Toda N, Mukoyama M, Yanagita M and Yokoi
H: CTGF in kidney fibrosis and glomerulonephritis. Inflamm Regen.
38:142018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Riser BL, Denichilo M, Cortes P, Baker C,
Grondin JM, Yee J and Narins RG: Regulation of connective tissue
growth factor activity in cultured rat mesangial cells and its
expression in experimental diabetic glomerulosclerosis. J Am Soc
Nephrol. 11:25–38. 2000.PubMed/NCBI
|
|
29
|
Zhao WX, Zhuang X, Huang TT, Feng R and
Lin JH: Effects of Notch2 and Notch3 on cell proliferation and
apoptosis of trophoblast cell lines. Int J Med Sci. 12:867–874.
2015. View Article : Google Scholar : PubMed/NCBI
|