Introduction
X-linked mental retardation (XLMR) syndrome is a
highly heterogeneous disorder characterized by cognitive impairment
and intellectual disability; this syndrome has an estimated
prevalence of 1/600 in male patients worldwide (1). XLMR can be classified into two types:
Non-syndromic XLMR and syndromic XLMR, according to the clinical
features (1–3). The XLMR type predominantly includes
Menkes syndrome, α-thalassemia with MR syndrome (4), Cabezas syndrome (5), fragile X syndrome (6), Holmes-Gang syndrome, Juberg-Marsidi
syndrome, Carpenter-Waziri syndrome, Chudley-Lowry syndrome and
Smith-Fineman-Myers syndrome (SFMS) (7), and its typical clinical symptoms
include short stature, hypogonadism, an abnormally large fontanel,
microcephaly, seizures and MR (7–9).
Smith et al (10) described two brothers in 1980 who
exhibited symptoms of MR, hypotonia, shortness of stature and
unusual facial appearance; however, these symptoms differed from
those of previously described syndromes. This syndrome was
designated as Smith-Fineman-Myers syndrome (SFMS; OMIM #309580)
(10), a rare form of XLMR. SFMS
is characterized by short stature, seizure, hypotonia, craniofacial
anomalies, psychomotor retardation with dysphasia, growth delay and
an intelligence level lower than that of boys of a similar age, in
addition to the aforementioned symptoms. SFMS is an uncommon
clinically heterogeneous disease with an X-linked recessive
inheritance trait in which hemizygous males are affected, although
heterozygous females appear normal. Among these clinical
manifestations, the craniofacial changes mainly comprise
microcephaly, macrostomia, hyperopia, micrognathia and a patulous
lower lip. Affected patients may also exhibit dolichocephaly, foot
deformities, hyperreflexia, hypogonadism and hypertelorism
(11,12).
The α-thalassemia/MR syndrome X-linked (ATRX;
OMIM #300032) gene is located on the X chromosome (Xq21.1), and is
also known as XH2 or XNP. This gene was named after
α-thalassemia with MR syndrome (OMIM #301040), which is
characterized by severe MR, genital abnormalities, characteristic
facial traits and α-thalassemia (6,13–18).
The ATRX gene is composed of 37 exons and spans 288,392 bp;
this is a disease-causing gene associated with XLMR (19). Being a member of the SWI/SNF2
superfamily of helicases/ATPases with a length of ≤2,492 amino
acids, the ATRX protein consists of several domains, including a
C2-C2 zinc-finger motif, plant homeodomain (PHD)-like motif and the
SWI/SNF2 ATPase/helicase-like motifs, which are involved in
regulation of transcription, DNA recombination and repair, and
meiosis and mitosis via effects on chromatin remodeling (4,15–17,19–24).
As well as SFMS (25), mutations
in ATRX can give rise to several other syndromes, including
Holmes-Gang syndrome (7),
Carpenter-Waziri syndrome (26),
Juberg-Marsidi syndrome (23) and
Chudley-Lowry syndrome (20).
In the present study, a novel missense mutation in
the ATRX gene was screened and identified by performing
whole exome sequencing (WES) in combination with Sanger sequencing
validation in a Chinese family with SFMS. The present study may
provide support to elucidate the complex genotype-phenotype
associations of SFMS and could enrich the ATRX pathogenic
variant spectrum, which may facilitate an improved early molecular
diagnosis and the timely treatment of children with SFMS.
Patients and methods
Patients
The subjects selected for the present study were
from a Chinese Han family based in Shandong province. A 2-year-old
male proband (IV1; Fig. 1) was
diagnosed with SFMS based on clinical manifestations; laboratory
investigations, including Bayley Scales of Infant Development of
China Revision (27); and the
results of molecular genetic detection at The Affiliated Hospital
of Qingdao University. Notably, all other family members were
identified to be asymptomatic. After having obtained written
informed consent from the family, including parents or guardians of
the minors, and 200 healthy control subjects (male:female ratio,
1:1.3, age range, 1–50 years), peripheral venous blood samples were
collected from the proband, other individuals in his family and
healthy controls. All of the clinical data were collected prior to
clinical treatment of the patient. The present study was approved
by the ethics committee of The Affiliated Hospital of Qingdao
University.
DNA extraction
Blood samples (400 µl) were collected from the
family (the proband, the mother, the grandmother and three maternal
granduncles) and the healthy controls, and DNA was extracted from
peripheral venous blood using a Qiagen DNA extraction kit (Qiagen,
Inc.), strictly according to the manufacturer's protocol.
Screening for mutations using WES
Genomic DNA was randomly fragmented using a Covaris
S220 sonicator (Covaris, Inc.) with 200 cycles per burst and 80 sec
processing time. T4 DNA polymerase and polynucleotide kinase (PNK;
Vazyme Biotech Co., Ltd.) were added to the DNA fragments for the
end-repair reaction, so as to obtain a 5′-blunt-end DNA short
fragment library containing phosphoric acid groups. Polymerase
chain reaction (PCR) amplification may be used to effectively
enrich DNA fragments with ligated adapter molecules at both ends.
To meet this purpose, an Agilent™ SureSelect Human All Exon V6 kit
(Agilent Technologies, Inc.) was used according to manufacturer's
protocol for library hybridization with an exome array to capture
the exons of the gene; subsequently, PCR using CAP-PCR Mastar Mix
and CAP Hotstar Enzyme (Kapa Biosystems; Roche Diagnostics) was
performed to amplify the exon DNA library as follows: An initial
denaturation of 98°C for 30 sec, 12 cycles of denaturation at 98°C
for 25 sec, annealing at 65°C for 30 sec, extension at 72°C for 30
sec, and a final extension of 72°C for 5 min. Finally, the
fragments from the purified DNA library were sequenced on Illumina
NextSeq 500 sequencer (Illumina, Inc) designed for paired-end 150
bp reads. All of the coding exon regions and exon-intron boundaries
in ATRX were sequenced by WES. The concentration of DNA used
was >10 ng/µl, the volume was >100 µl, and the purity aimed
for in terms of the DNA fragments was determined from measures of
absorbance (A260/280>1.8, and
A260/230>1.2).
Sanger sequencing validation
The ATRX variant of the proband discovered by
WES was validated by Sanger sequencing samples of the proband, the
mother, the grandmother, three maternal granduncles and 200 healthy
controls. A pair of fragments covering the mutation locus was
amplified by PCR in a total reaction volume of 20 µl, including 2
µl template DNA, 6 µl ddH2O, 10 µl Master Mix (Tsingke
Biological Technology Co., Ltd.), and 1 µl forward and reverse
primers respectively. The primer pair for exon 7 in ATRX was
designed using Primer Premier version 5.0 software (PREMIER
Biosoft) primer sequences: Forward, 5′-TGGAATTTGCCAAGGTTGTCA-3′ and
reverse, 5′-TCCGGTGTGCTCCCATAATC-3′). The reaction conditions were
as follows: Initial denaturation at 94°C for 5 min, followed by 35
cycles of denaturation at 94°C for 30 sec, annealing at 59°C for 30
sec and extension at 72°C for 30 sec, followed by final extension
at 72°C for 7 min. Amplified PCR products were analyzed by 1%
agarose gel electrophoresis stained with GoldViewI (Beijing Zoman
Biotechnology Co., Ltd.). If the target band was revealed to be
clearly separated and strongly fluoresced following
electrophoresis, the PCR product was purified and sequenced using
an ABI 3730XL automated sequencer (Applied Biosystems; Thermo
Fisher Scientific, Inc.). DNA sequences were analyzed using the
BioEdit program (V7.0.1; http://www.mbio.ncsu.edu/BioEdit/bioedit.html) and
compared with a reference sequence published on the National Center
Biotechnology Information (NCBI) website (https://www.ncbi.nlm.nih.gov/). The detected mutation
was predicted by bioinformatics software Polymorphism Phenotyping
v2 (PolyPhen-2; http://genetics.bwh.harvard.edu/pph2/) and was
identified in the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php),
1000 Genomes (http://browser.1000genomes.org/), dbSNP (https://www.ncbi.nlm.nih.gov/projects/SNP/index.html)
and ESP6500 (https://evs.gs.washington.edu/EVS/) databases.
Finally, the conservative analysis of the mutation site was
performed.
Results
Clinical phenotype
The general features and results of the clinical
examination of the proband (proband IV1; male; age, 2 years;
Fig. 1) are shown in Table I, including developmental delay,
characteristic facial appearance and hypogonadism. These physical
anomalies met the diagnostic criteria for SFMS. With the exception
of the proband, the phenotypes of the other family members were all
normal (data not shown).
 | Table I.Clinical findings in the patient with
Smith-Fineman-Myers syndrome. |
Table I.
Clinical findings in the patient with
Smith-Fineman-Myers syndrome.
| Clinical
phenotype | Patient (IV1) |
|---|
| Short stature | + |
| Psychomotor
retardation | + |
| Severe
dysphasia | + |
| Early
hypotonia | + |
| Restlessness | + |
| Undescended
testes | + |
| Microcephaly | + |
| Ocular
hypertelorism | + |
| Micrognathia | + |
Mutation analysis of ATRX
Following the screening of numerous mutations in the
entire exome using WES, candidate genes were detected by WES,
including ATRX, FCN3, TAS1R1, FOXD3, F5, TTN, CACNA2D3, NAPRT1,
VPS13A, HPS6 and RBM20. Notably, only mutated
ATRX, which matched the clinical phenotype of the proband,
was identified in the present study. In addition, ATRX is
associated with SFMS, Holmes-Gang syndrome, Carpenter-Waziri
syndrome, Juberg-Marsidi syndrome, Chudley-Lowry syndrome and
α-thalassemia with MR syndrome, while the clinical symptoms of the
proband were associated with SFMS and co-segregation analysis also
excluded dominant inherited α-thalassemia with MR syndrome after
comprehensive consideration.
ATRX mutational analysis of the proband
revealed a missense mutation, which was absent from the Exome
Aggregation Consortium (http://exac.broadinstitute.org/) and Genome
Aggregation Database (https://gnomad.broadinstitute.org/). The mutation
consisted of a substitution of thymine for cytosine at position
515, resulting in a change from threonine to isoleucine at codon
172 (c.515C>T or p.Thr172Ile) (Fig.
2). Polymorphism Phenotyping v2 (PolyPhen-2; http://genetics.bwh.harvard.edu/pph2/)
analysis predicted that this mutation would exert a significantly
damaging effect on the structure and function of ATRX due to a
prediction score of 1.000 for the effect of an amino acid
substitution on protein function. This specific mutation site was
subsequently screened for in the other family members and in 200
healthy control subjects using Sanger sequencing. The results
revealed that the boy's mother (III1; Fig. 1) and his maternal grandmother (II1;
Fig. 1) possessed the heterozygous
mutation, whereas the mutation was not possessed by three maternal
granduncles (II3, II4 and II5; Fig.
1); likewise, the mutation was absent from the 200 unrelated
normal individuals of identical ethnic origin (data not shown).
Therefore, the co-segregation analysis of this pedigree indicated
that this missense mutation was most likely to have been the
pathogenic variant responsible for the SFMS phenotype in this
family. Additional genetic studies of the family history revealed
that this mutated gene had been inherited from the proband's
grandmother. Based upon searches of the Human Gene Mutation
Database (http://www.hgmd.cf.ac.uk/ac/index.php), 1000 Genomes
(http://browser.1000genomes.org/), dbSNP
(https://www.ncbi.nlm.nih.gov/projects/SNP/index.html)
and ESP6500 (https://evs.gs.washington.edu/EVS/) databases, to the
best of our knowledge, this mutation has not previously been
detected, and is therefore considered a novel variant.
Bioinformatics analysis of the ATRX
mutation
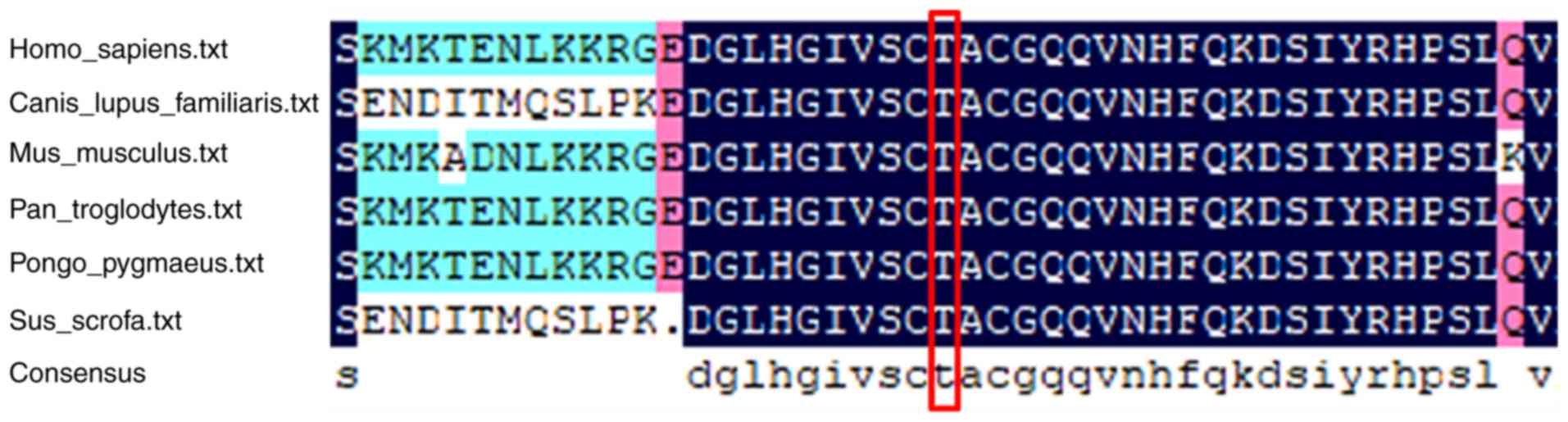
The ATRX sequences of various animal species,
including Homo sapiens, Bos taurus, Canis lupus familiaris, Mus
musculus, Pan troglodytes, Pongo pygmaeus and Sus
scrofa, were obtained from the NCBI (https://www.ncbi.nlm.nih.gov/) and UniProtKB
(https://www.uniprot.org/) websites, and multiple
sequence alignment was performed using DNAMAN software (V6.0.3.99;
Lynnon Corporation). The c.515C>T/p.Thr172Ile variant was
revealed to be located in a highly conserved region of the ATRX
protein (Fig. 3).
Discussion
In the present study, a Chinese pedigree affected by
SFMS was investigated, and a novel missense mutation in the
ATRX gene, c.515C>T/p.Thr172Ile, was subsequently
detected and confirmed using WES followed by Sanger sequencing
validation. This finding may expand our knowledge with regards to
the clinical spectrum and pathogenic mutants associated with SFMS.
That the mutation co-segregated with the syndrome in this family
was supported by the finding that this mutational variant was not
identified in 200 unrelated healthy controls of identical ethnic
origin. In addition, PolyPhen-2 analysis predicted that this
mutation, located in a highly conserved region of ATRX,
might result in a damaging impairment of ATRX protein function.
Bioinformatics analysis and co-segregation analysis revealed that
the c.515C>T mutation in ATRX is likely to give rise to
the disease-causing variant of SFMS. However, it was impossible to
draw definitive conclusions in this regard due to the absence of
functional experiments.
SFMS is an uncommon congenital mental deficiency
disorder that is governed by mutations in ATRX and is
transmitted in an X-linked recessive manner. Since SFMS was first
defined in 1980 by Smith et al (10), only a few cases have been reported.
Patients with SFMS have specific similar clinical symptoms,
including MR, characteristic facial appearance, short stature,
microcephaly, micrognathia, failure to grow, seizure, hypotonia,
genital abnormalities, abnormal speech, cortical atrophy and cleft
palate (19,28). Briefly, the assessment for SFMS,
like any other form of MR, is mainly based on an inspection of the
detailed family history, physical defects, clinical examinations
and genetic testing. Currently, a correct diagnosis for many
patients with SFMS, and those suffering from other forms of XLMR,
remains problematic due to clinical and genetic heterogeneity,
unknown mechanisms of pathogenesis, and the ever-growing number of
novel mutants of causative genes. Furthermore, it is even more
difficult to determine which gene is most likely to be associated
with the affected individuals based only on clinical
manifestations. Consequently, genetic detection and prenatal
genetic screening are vitally important in the diagnosis of
SFMS.
Mutations in the genes ATRX/XNP, AP1S2,
GDI1/OPHN2, UBE2A, OPHN1, MID1, MECP2 and ARX,
etc., have been reported to be associated with XLMR (1,6,19,25,29–31),
whereas patients with SFMS only possess mutations in ATRX
(NM-000489.4). ATRX was originally considered to be involved
in α-thalassemia with MR syndrome; this gene spans ~300 kb in
chromosome Xq21.1, is comprised of 37 exons and serves an important
role in sex differentiation (17).
Subsequently, it was identified that ATRX is associated with
other MR syndromes, including Holmes-Gang syndrome, Chudley-Lowry
syndrome, Carpenter-Waziri syndrome, MR with spastic paraplegia and
Juberg-Marsidi syndrome (4,7,19,20,23,25),
and with tumorigenesis, including pancreatic neuroendocrine tumors,
low-grade glioma, glioblastoma multiforme, osteosarcoma and
adrenocortical tumors (32).
ATRX is integral to the development of the brain, skeleton,
genital organs and facial morphogenesis (14), suggesting that different mutations
in ATRX may cause distinct symptoms, and varying severity
under different conditions. Although a growing number of mutations
have been reported in ATRX since it was revealed to be a
pathogenic gene of XLMR, only a few mutations have been identified
in SFMS (4,25).
The ATRX protein is a member of the SWI2/SNF2 family
of chromatin-remodeling proteins, with a length of 2,492 amino
acids (33). This protein has
several functional domains that exert differing influences on
associated proteins, a phenomenon which could account for the
clinical phenotypic heterogeneity of patients with SFMS (20). The N-terminus comprises a
cysteine-rich region considered to include a C2-C2 zinc finger and
a PHD-like zinc finger domain, whereas the ATPase/helicase-like
domain mainly controls transcriptional regulation, cell cycle
regulation and mitotic chromosome segregation (17). The central portion of ATRX includes
a predicted coiled-coil domain, a predicted nuclear localization
signal and a stretch of 21 glutamic acid residues. A conserved
region among different SNF2 proteins and a glutamine-rich region
are located in the C-terminal sequence of ATRX (17,34).
ATRX mutations can result in alterations in
the pattern of ribosomal DNA methylation, Y-specific repeats and
subtelomeric repeats (14,19,35),
when the mutational ‘hot-spots’ occur in the cysteine-rich region
and the ATPase/helicase-like motifs (36). However, patients with mutations in
the PHD-like zinc finger present with more severe phenotypes
compared with those that have mutations in the helicase domain
(37). In addition to the
expression of α-globin and an impaired intellectual level, mutated
ATRX has been reported to be associated with various degrees
of urogenital developmental disabilities (17).
With the expanding scope of genetic testing in
scientific research and clinical medicine, high-throughput
sequencing technology has undergone rapid development, and the
detection speed has been accelerated. At present, DNA sequencing
technology is not only advancing genetic research, but is also
being used for the detection of a variety of genetic diseases. The
traditional methods of genetic diagnosis often fail to lead to the
identification of the pathogenic gene, which means that patients do
not receive a definite diagnosis, let alone the corresponding
treatment or subsequent prenatal diagnosis. WES enables
high-throughput sequencing to be performed after DNA capture and
enrichment in the exon region of the genome using sequence capture
technology, and genetic variations associated with changes in
protein function can be directly detected. WES has the advantages
of high-sequencing depth, accurate mutation detection and a high
accuracy of data. Although exons account for only 1% of the genome,
>85% of Mendelian diseases are associated with this region
(38). Since the majority of
diseases are caused by mutations that may change the function of
encoded proteins, and diseases are mostly associated with rare
mutations, WES has currently established itself as the most
cost-effective and reliable method for studying Mendelian genetic
diseases.
In conclusion, recently, more cases associated with
mutations in ATRX have been identified on the basis of
severe MR with typical physical impairment (25,37).
In the present study, since the family was relatively small and
other family members did not exhibit similar phenotypes, it was
difficult to clearly diagnose and identify the pathogenic gene;
therefore, WES followed by Sanger sequencing validation was adopted
to detect the defective gene. Consequently, a novel missense
mutation was described that may have led to decreased ATRX activity
in a Chinese proband with SFMS, and which is available for the
accurate diagnosis of other patients, laying the foundation for
genetic counseling and prenatal diagnosis. Therefore, this study
may further broaden the phenotypic spectrum of SFMS associated with
ATRX mutations, thereby providing the basis for further
investigation of SFMS pathogenesis and gene therapy.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Key Research
and Development Program of China (grant no. 2016YFC1000306) and
Shandong Provincial Natural Science Foundation of China (grant no.
ZR2019PH072).
Availability of data and materials
The datasets used and analyzed during the study are
available from the corresponding authors on reasonable request.
Authors' contributions
LL performed the experiments and wrote the
manuscript. JY, XZ, MH and WL made important contributions to data
acquisition, analysis and interpretation. HL and SL designed the
present study and revised the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the ethics
committee of The Affiliated Hospital of Qingdao University. Written
informed consent was obtained for all patients, family members and
volunteers; for minors, consent was obtained from their parents or
guardians.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Al-Owain M, Kaya N, Al-Zaidan H, Al-Hashmi
N, Al-Bakheet A, Al-Muhaizea M, Chedrawi A, Basran RK and Milunsky
A: Novel intragenic deletion in OPHN1 in a family causing XLMR with
cerebellar hypoplasia and distinctive facial appearance. Clin
Genet. 79:363–370. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Borck G, Molla-Herman A, Boddaert N,
Encha-Razavi F, Philippe A, Robel L, Desguerre I, Brunelle F,
Benmerah A, Munnich A and Colleaux L: Clinical, cellular, and
neuropathological consequences of AP1S2 mutations: Further
delineation of a recognizable X-linked mental retardation syndrome.
Hum Mutat. 29:966–974. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ma QL, Yang F, Frautschy SA and Cole GM:
PAK in Alzheimer disease, Huntington disease and X-linked mental
retardation. Cell Logist. 2:117–125. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lossi AM, Millan JM, Villard L, Orellana
C, Cardoso C, Prieto F, Fontés M and Martínez F: Mutation of the
XNP/ATR-X gene in a family with severe mental retardation, spastic
paraplegia and skewed pattern of X inactivation: Demonstration that
the mutation is involved in the inactivation bias. Am J Hum Genet.
65:558–562. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Badura-Stronka M, Jamsheer A,
Materna-Kiryluk A, Sowińska A, Kiryluk K, Budny B and
Latos-Bieleńska A: A novel nonsense mutation in CUL4B gene in three
brothers with X-linked mental retardation syndrome. Clin Genet.
77:141–144. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
des Portes V: X-linked mental deficiency.
Handb Clin Neurol. 111:297–306. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Stevenson RE, Abidi F, Schwartz CE, Lubs
HA and Holmes LB: Holmes-Gang syndrome is allelic with
XLMR-hypotonic face syndrome. Am J Med Genet. 94:383–385. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chudley AE, Lowry RB and Hoar DI: Mental
retardation, distinct facial changes, short stature, obesity, and
hypogonadism: A new X-linked mental retardation syndrome. Am J Med
Genet. 31:741–751. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
du Souich C, Chou A, Yin J, Oh T, Nelson
TN, Hurlburt J, Arbour L, Friedlander R, McGillivray BC, Tyshchenko
N, et al: Characterization of a new X-linked mental retardation
syndrome with microcephaly, cortical malformation, and thin
habitus. Am J Med Genet A 149A. 2469–2478. 2009. View Article : Google Scholar
|
|
10
|
Smith RD, Fineman RM and Myers GG: Short
stature, psychomotor retardation, and unusual facial appearance in
two brothers. Am J Med Genet. 7:5–9. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Stephenson LD and Johnson JP:
Smith-Fineman-Myers syndrome: Report of a third case. Am J Med
Genet. 22:301–304. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wei J, Chen B, Jiang Y, Yang Y and Guo Y:
Smith-Fineman-Myers syndrome: Report on a large family. Am J Med
Genet. 47:307–311. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
McPherson EW, Clemens MM, Gibbons RJ and
Higgs DR: X-linked alpha-thalassemia/mental retardation (ATR-X)
syndrome: a new kindred with severe genital anomalies and mild
hematologic expression. Am J Med Genet. 55:302–306. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gibbons RJ and Higgs DR:
Molecular-clinical spectrum of the ATR-X syndrome. Am J Med Genet.
97:204–212. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gibbons RJ, Picketts DJ, Villard L and
Higgs DR: Mutations in a putative global transcriptional regulator
cause X-linked mental retardation with alpha-thalassemia (ATR-X
syndrome). Cell. 80:837–845. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schenkel LC, Kernohan KD, McBride A, Reina
D, Hodge A, Ainsworth PJ, Rodenhiser DI, Pare G, Bérubé NG, Skinner
C, et al: Identification of epigenetic signature associated with
alpha thalassemia/mental retardation X-linked syndrome. Epigenetics
Chromatin. 10:102017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tang P, Park DJ, Marshall Graves JA and
Harley VR: ATRX and sex differentiation. Trends Endocrinol Metab.
15:339–344. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Villard L and Fontes M:
Alpha-thalassemia/mental retardation syndrome, X-Linked (ATR-X, MIM
#301040, ATR-X/XNP/XH2 gene MIM #300032). Eur J Hum Genet.
10:223–225. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chiurazzi P, Tabolacci E and Neri G:
X-linked mental retardation (XLMR): From clinical conditions to
cloned genes. Crit Rev Clin Lab Sci. 41:117–158. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Abidi FE, Cardoso C, Lossi AM, Lowry RB,
Depetris D, Mattéi MG, Lubs HA, Stevenson RE, Fontes M, Chudley AE
and Schwartz CE: Mutation in the 5′alternatively spliced region of
the XNP/ATR-X gene causes Chudley-Lowry syndrome. Eur J Hum Genet.
13:176–183. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
De La Fuente R, Viveiros MM, Wigglesworth
K and Eppig JJ: ATRX, a member of the SNF2 family of
helicase/ATPases, is required for chromosome alignment and meiotic
spindle organization in metaphase II stage mouse oocytes. Dev Biol.
272:1–14. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Picketts DJ, Higgs DR, Bachoo S, Blake DJ,
Quarrell OW and Gibbons RJ: ATRX encodes a novel member of the SNF2
family of proteins: Mutations point to a common mechanism
underlying the ATR-X syndrome. Hum Mol Genet. 5:1899–1907. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Villard L, Gecz J, Mattéi JF, Fontés M,
Saugier-Veber P, Munnich A and Lyonnet S: XNP mutation in a large
family with Juberg-Marsidi syndrome. Nat Genet. 12:359–360. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wong LH, McGhie JD, Sim M, Anderson MA,
Ahn S, Hannan RD, George AJ, Morgan KA, Mann JR and Choo KH: ATRX
interacts with H3.3 in maintaining telomere structural integrity in
pluripotent embryonic stem cells. Genome Res. 20:351–360. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Villard L, Fontès M, Adès LC and Gecz J:
Identification of a mutation in the XNP/ATR-X gene in a family
reported as Smith-Fineman-Myers syndrome. Am J Med Genet. 91:83–85.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Abidi F, Schwartz CE, Carpenter NJ,
Villard L, Fontes M and Curtis M: Carpenter-Waziri syndrome results
from a mutation in XNP. Am J Med Genet. 85:249–251. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Qian X, Li J, Xu S, Wan Y, Li Y, Jiang Y,
Zhao H, Zhou Y, Liao J, Liu H, et al: Prenatal exposure to
phthalates and neurocognitive development in children at two years
of age. Environ Int. 131:1050232019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Guion-Almeida ML, Tabith A Jr,
Kokitsu-Nakata NM and Zechi RM: Smith-Fineman-Myers syndrome in
apparently monozygotic twins. Am J Med Genet. 79:205–208. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Budny B, Badura-Stronka M, Materna-Kiryluk
A, Tzschach A, Raynaud M, Latos-Bielenska A and Ropers HH: Novel
missense mutations in the ubiquitination-related gene UBE2A cause a
recognizable X-linked mental retardation syndrome. Clin Genet.
77:541–551. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Giannandrea M, Bianchi V, Mignogna ML,
Sirri A, Carrabino S, D'Elia E, Vecellio M, Russo S, Cogliati F,
Larizza L, et al: Mutations in the small GTPase gene RAB39B are
responsible for X-linked mental retardation associated with autism,
epilepsy, and macrocephaly. Am J Hum Genet. 86:185–195. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Huo L, Teng Z, Wang H and Liu X: A novel
splice site mutation in AP1S2 gene for X-linked mental retardation
in a Chinese pedigree and literature review. Brain Behav.
9:e012212019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Dyer MA, Qadeer ZA, Valle-Garcia D and
Bernstein E: ATRX and DAXX: Mechanisms and mutations. Cold Spring
Harb Perspect Med. 7(pii): a0265672017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Haase S, Garcia-Fabiani MB, Carney S,
Altshuler D, Núñez FJ, Méndez FM, Núñez F, Lowenstein PR and Castro
MG: Mutant ATRX: Uncovering a new therapeutic target for glioma.
Expert Opin Ther Targets. 22:599–613. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gibbons RJ, Bachoo S, Picketts DJ, Aftimos
S, Asenbauer B, Bergoffen J, Berry SA, Dahl N, Fryer A, Keppler K,
et al: Mutations in transcriptional regulator ATRX establish the
functional significance of a PHD-like domain. Nat Genet.
17:146–148. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nandakumar P, Mansouri A and Das S: The
role of ATRX in glioma biology. Front Oncol. 7:2362017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Juhasz S, Elbakry A, Mathes A and Löbrich
M: ATRX promotes DNA repair synthesis and sister chromatid exchange
during homologous recombination. Mol Cell. 71:11–24.e7. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Badens C, Lacoste C, Philip N, Martini N,
Courrier S, Giuliano F, Verloes A, Munnich A, Leheup B, Burglen L,
et al: Mutations in PHD-like domain of the ATRX gene correlate with
severe psychomotor impairment and severe urogenital abnormalities
in patients with ATRX syndrome. Clin Genet. 70:57–62. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ng SB, Buckingham KJ, Lee C, Bigham AW,
Tabor HK, Dent KM, Huff CD, Shannon PT, Jabs EW, Nickerson DA, et
al: Exome sequencing identifies the cause of a mendelian disorder.
Nat Genet. 42:30–35. 2009. View
Article : Google Scholar : PubMed/NCBI
|