Introduction
Sudden cardiac arrest (CA) is a major public health
challenge and the leading cause of death worldwide, imposing a
heavy burden on patients and society (1). Only 17–49% of CA victims are able to
regain self-circulation. Meanwhile, 80% of CA survivors present
with a certain degree of coma, and very few fully recover brain
function. Brain damage after continuous hypoxia remains the leading
cause of death in CA (2).
Postresuscitation syndrome is characterized by cerebral injury,
cardiovascular injury, ischemia/reperfusion (I/R) injury and
systemic inflammatory reaction after hypoxia (3), which may aggravate ischemic
encephalopathy (4). Effective
intervention for brain injury after CA has important clinical
significance and social benefits. How to reduce brain function
damage after CA has become a research ‘hot’ topic. Unfortunately,
there is no specific pharmacological treatment for I/R injury after
restoration of spontaneous circulation (ROSC) (3,4).
Nogo-A is widely found in neurons of the central
nervous system (CNS) and oligodendrocytes, and is considered an
inhibitor of neurite outgrowth and axon regeneration after CNS
injury. Nogo-A expression increases after focal cerebral ischemia
reperfusion injury (5) and stroke
(6). Increased Nogo-A can affect
the plasticity of the CNS and prevent the improvement of neural
function. Anti-Nogo-A treatment promotes axonal sprouting and
neuro-structural plasticity to recover neural function after
ischemic stroke and injury (7).
Nogo-A plays an important role in nerve regeneration in the CNS;
therefore, regulating Nogo-A is of great significance for nerve
function recovery (5). These
results were derived from a study of focal cerebral ischemia, but
there are few reports assessing total cerebral ischemia, especially
Nogo-A expression in the CA/CPR (cardiac arrest/cardiopulmonary
resuscitation) model. Meanwhile, there is no report concerning the
effect of Nogo-A antibody on brain function after
intracerebroventricular injection in rats after CA/CPR. Studies
found that anti-Nogo-A treatment affects neurogenesis after stoke.
Research assessing Nogo-A focuses on regeneration inhibition in the
chronic period (8), and few
studies have evaluated Nogo-A changes in the CA/CPR model in the
acute phase. In recent years, studies evaluating Nogo-A have shown
that intervention with myocardial Nogo-A could reduce apoptosis in
cardiomyocytes (9). We speculated
that Nogo-A may be associated with apoptosis in the brain.
Ephedrine with or without hyperbaric oxygen inhibits caspase-3 and
Nogo-A expression, and reduces the degree of brain damage caused by
ischemia (10) in the neonatal
brain injury model. We speculated that Nogo-A may be associated
with apoptosis after CA/CPR. Nogo-A contains double lysine motifs
and is mainly found in the endoplasmic reticulum (ER), where it
aids in the formation of ER tubules (11) and maintenance of normal ER shape
(12). Based on the above facts,
we hypothesized that Nogo-A may be associated with ER
stress-related apoptosis in neuron cells during global brain I/R
injury in the CA/CPR model. It is well-known that C/EBP homologous
protein (CHOP) and glucose regulated protein 78 (GRP78) are ER
stress markers, and cysteinyl aspartate specific proteinase-12
(casapse-12) is a specific marker of ER stress-related apoptosis
(13). To investigate the effect
of Nogo-A antibody on brain tissue structure and function in rats
with CA/CPR and to explore the possible mechanism, providing an
experimental basis for brain protection in the CNS, the effects of
Nogo-A antibody on neuron cell morphology and caspase-3, Nogo-A,
GPR78, CHOP, caspase-12, Bcl-2 and Bax expression levels were
assessed in the CA/CPR rat model at different time-points after
ROSC.
Materials and methods
Animal preparation
A total of 115 Wistar rats (male, age 3-months old,
weight 300–400 g) were purchased from the Sichuan University
Laboratory Animal Center. They were housed in standard rodent
housing with soft wood bedding, maintained at room temperature (23
± 2°C, humidity 50±5%), on a 12:12-h light:dark cycle with ad
libitum access to water and food. The West China Hospital's
Experimental Animal Ethics Committee approved the study protocol.
Rats were anesthetized by intraperitoneal injection of 45 mg/kg
1.5% sodium pentobarbital. Animal tracheas were intubated (14G
artery puncture needle cannula; BD Bioscience) and mechanically
ventilated (HX-100E Ventilator, Chengdu Taimeng Technology Co.,
Ltd.; 60 breaths per min). A 22G artery puncture needle cannula was
inserted into the right femoral artery and vein. A pressure
transducer (PT-100 blood pressure transducer; Chengdu Taimeng
Technology Co. Ltd.) was used to monitor arterial blood pressure
online. All catheters were blushed intermittently with saline
containing 2.5 IU/ml crystalline bovine heparin. Subcutaneous
needle electrodes were used to monitor electrocardiographic
recordings continuously. Data were recorded using a BL-420F
biological signal acquisition and procession system (BL-420F;
Chengdu Taimeng Technology Co. Ltd.).
Modeling cardiac arrest
Experimental procedures in the rat studies were
executed based on Utstein-Style Guideline for Uniform Reporting of
Laboratory CPR Research (14). A
5F pacing catheter with ring electrodes (Medtronic, Inc.) was
placed in the esophagus for transesophageal induction of
ventricular fibrillation (15).
The distance between the electrode and incisor was 7 cm. A heating
plate and light were used to maintain the temperature between 36.5
and 37.5°C. The ECG and hemodynamic data were monitored for 15 min.
Ventricular fibrillation was induced using 20 V/30 Hz with a 10
msec wave width alternating current via an esophageal electrode
(Medtronic Inc.). CA criteria: i) ECG showed ventricular
fibrillation, pulseless electricity activity, or asystole. ii) mean
arterial pressure (MAP) below 25 mmHg. Artificial ventilation was
stopped when inducing CA. After 5 min of CA, CPR was started by
artificial ventilation (80 breaths/min, tidal volume of 6 ml/kg,
with pure oxygen) and thoracic compression with 2 fingers over the
sternum at a rate of 200 compressions/min paced by a metronome with
the depth of 1/3 of anteroposterior diameter of chest and adjusting
according to maintain aortic diastolic pressure over 20 mmHg.
Advanced cardiac life support, including epinephrine (Shanghai
Hefeng Pharmaceutical Co., Ltd.; 20 µg/kg body weight, i.v.)
administration, external defibrillation (5J, HeartSmart XL
defibrillator; Philips Medical Systems B.V.), was initiated. If
first defibrillation did not result in ROSC, CPR was sustained and
defibrillation was attempted every 2 min. Resuscitation procedures
were sustained until restoration of spontaneous circulation (ROSC).
ROSC criteria: i) ECG showed supraventricular rhythm (sinus,
atrial, borderline rhythm); ii) mean arterial pressure (MAP) over
60 mmHg maintained for more than 10 min. If ROSC did not occur
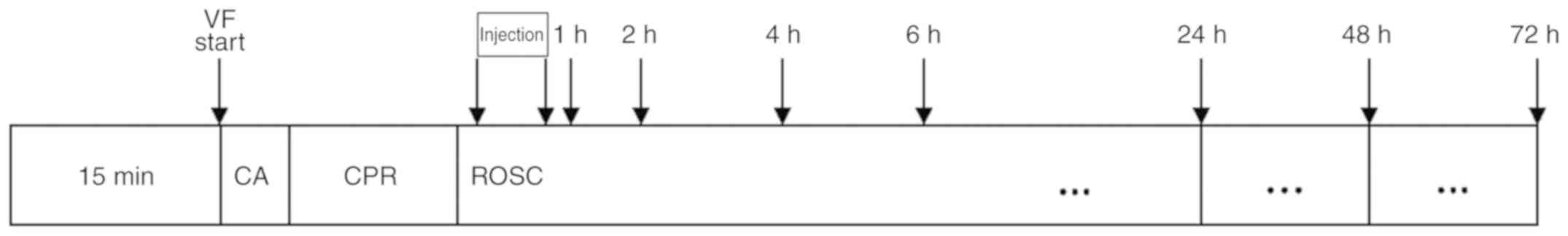
within 10 min of CPR, resuscitation was terminated (Fig. 1).
Intracerebroventricular administration
of Nogo-A antibody
The rats were randomized into three groups after
ROSC. The Treatment group received Nogo-A antibody (n=50) while the
Model group (n=50) received saline. In addition, the Sham-operated
rats without CA served as non-ischemic controls (n=15). Ten minutes
after ROSC, rats were placed into a stereotaxic apparatus (RWD
Desktop digital brain stereotaxic instrument 68025; Shenzhen Reward
Life Technology Co. Ltd.). A stainless cannula was inserted into
the right lateral ventricle (bregma coordinates: 1.5 mm lateral and
0.8 mm posterior to the bregma, at a depth of 3.5 mm) (16) and connected to an osmotic minipump
(Microsyringe pump ALC_IP 600Gb; Shanghai Alcott Biotechnology Co.,
Ltd.). Continuous intracerebroventricular application of Nogo-A
antibody (17) (200 µg/ml, 1.0
µl/min pump rate, 20 µl) or placebo saline (1.0 µl/min pump rate,
20 µl) was carried out for 20 min (18). After 1, 2, 4, 6, 24, 48 and 72 h,
respectively, rats were anesthetized and decapitated. The brains
were removed and cryo-fixed overnight before embedding and
sectioning. At the hippocampal level (approximately at the bregma
−3.5 mm), coronal brain sections (10-µm) were cut and placed on
glass slides. Then, hippocampal samples were collected for western
blot and RT-PCR analysis.
Assessment of neurological deficit
score (NDS)
NDSs were measured to evaluate the neurologic state
in the Sham, Model and Treatment groups at 1, 2 and 3 days,
respectively, after CA/CPR as described previously (19). NDS assessment included seven
variables such as general behavior, brainstem function, exercise
assessment, sensory function, motor behavior, behaviors and
convulsions. An NDS of 80 was assigned to reflect normal brain
function, whereas an NDS of 0 indicated brain death. All
assessments were examined and confirmed by two separate
investigators blinded to the treatment (20).
Reverse transcription-quantitative
(RT-q) PCR
Total RNA was obtained from dissected hippocampal
samples using TRIzol reagent as directed by the manufacturer
(Invitrogen; Thermo Fisher Scientific, Inc.). First-strand cDNA was
synthesized with Revert Aid™ First Strand cDNA Synthesis Kit from
Fermentas (Thermo Fisher Scientific, Inc.). Amplification was
performed on an FTC2000 real-time fluorescent quantitative gene
amplification instrument (Funglyn). Each reaction contained 1 µl of
TaqMan probe, 15.34 µl of PCR water, 5 µl of cDNA and 1 µl of each
primer (final concentration 10 µM). Thermal cycling included an
amplification cycle of denaturation at 94°C for 2 min, followed by
40 cycles of 94°C for 20 sec and 54°C for 20 sec (β-actin) and 52°C
for 20 sec (caspase-3) (21).
Primers included: β-actin (111 bp), forward
5′-GAAGATCAAGATCATTGCTCCT-3′ and reverse
5′-TACTCCTGCTTGCTGATCCACA-3′; caspase-3 (156 bp), forward
5′-CCGAAACTCTTCATCATTCA-3′ and reverse
5′-CCAGGAATAGTAACCGGGT-3′.
Relative mRNA expression was assessed by the
comparative Cq (2−ΔΔCq) method (22,23).
Transcript levels of caspase-3 were determined by RT-PCR and
normalized to β-actin as an endogenous control.
Western blot analysis
Hippocampal tissue samples were homogenized in
ice-cold suspension buffer (Bioteke Corp.), and centrifuged (20
min, 4°C, 12000 × g), and supernatants were transferred into a
20-µl tube. After protein quantitation using the BCA protein
concentration kit (Beyotime Biotechnology), protein extracts (30 µg
each) were separated by 10% SDS-PAGE (sodium dodecyl
sulfate-polyacrylamide gel electrophoresis) and transferred onto
polyvinylidene fluoride (PVDF) membranes (Millipore Corp.). The
membranes were then blocked with 5% non-fat dried milk in PBS.
After electroblotting, the membranes were incubated overnight with
anti-Nogo-A (cat. no. SC25660; dilution 1:500; Santa Cruz
Biotechnology, Inc.), anti-caspase-3 (cat. no. 9662S; dilution
1:1,000; Cell Signaling Technology, Inc.), anti-β-actin (cat. no.
bsm-33036M; dilution 1:1,000; BIOSS), anti-GRP78 BiP (cat. no.
ab108613; dilution 1:1,000 Abcam), anti-DDIT3 (cat. no. ab179823;
dilution 1:1,000; Abcam), anti-caspase-12 (cat. no. ab62484;
dilution 1:1,000; Abcam), anti-Bcl-2 (cat. no. ab59348; dilution
1:1,000; Abcam) and anti-Bax (cat. no. ab32503; dilution 1:1,000;
Abcam) primary antibodies, respectively, followed by a 1-h
incubation with horseradish peroxidase-conjugated goat-anti-rabbit
antibody (dilution 1:1,000; ZB-2301; OriGene Technologies, Inc.).
The film was photographed and scanned, and Quantity One Analysis
Software 4.6 (Bio-Rad Laboratories, Inc.) was used for quantifying
protein gray values. Relative expression of the target protein was
assessed as its gray-scale value divided by that of the internal
control β-actin.
Nissl staining
The animals were anesthetized by 1.5% sodium
pentobarbital and sacrificed at various time-points. Rats were
transcardially perfused with 250 ml saline at 4°C (24). Brains were removed and fixed for
one week at 4°C. A series of 70, 80, 90 and 95% alcohol solutions
and xylene were used to dehydrate the brain samples. The prepared
paraffin-embedded blocks were cut coronally and mounted onto
slides. Five fields in each slide were randomly assessed under an
Olympus BX41 optical microscope (Olympus Corporation) with a
magnification of ×400, photographed by an attached camera (Olympus
DP71, Olympus Corporation) and quantified with Image Pro Plus
software version 6.0 (Media Cybernetics, Inc.). Average optical
density (AOD) values were analyzed according to a previous report
(25).
Electron microscopy
Rats were deeply anesthetized with 1.5% sodium
pentobarbital and transcardially perfused with 200 ml 4%
paraformaldehyde at 4°C. The brain tissue was quickly removed,
placed on ice, cut at 0.5×0.5×0.5 cm with a sharp blade, and placed
in a pre-cooled 2.5% glutaraldehyde solution for at least 2 h at
4°C. The, the tissues were rinsed 3 times with a 0.2 M phosphate
buffer saline solution (pH=7.4) for 15 min. Next, they were placed
in 1% osmium solution for at least 1 h and rinsed again as
described previously (16). The
samples were soaked in gradient ethanol series for 15 min,
respectively, for dehydration. The samples were embedded in epoxy
resin, and 70-nm-thick sections were prepared. The slices were
stained with saturated acetate 30 min and treated with lead for 5–8
min (24). After staining, slices
were rinsed with distilled water and assessed under a transmission
electron microscope (Hitachi H-7500 transmission electron
microscopy; Hitachi Corp., Japan). The changes in neurons,
mitochondria and ER were observed.
Terminal deoxynucleotidyl transferase
(TdT)-mediated dUTP nick-end labelling (TUNEL) assay
Histological analysis by TUNEL assay is
characterized by the incorporation of deoxyuridine triphosphate
fluorescein-12 (12-d-UTP) at the DNA 30-OH ends, whose signal is
amplified by the reaction involving the enzyme terminal
deoxynucleotidyl transferase (rTdT); the fragmented DNA labeled
with 12-dUTP fluorescein becomes visible under a fluorescence
microscope. TUNEL staining was applied at 72 h after ROSC for the
detection of neuronal apoptosis. For in situ staining of DNA
fragmentation and apoptotic bodies, the TUNEL assay was used as
described previously (21). An
in situ cell death detection kit, the DeadEnd™ Fluorometric
TUNEL system (Promega, USA), was used for coronal sections mounted
on 12-µm slides (n=3 per group). The slides were washed with 0.05 M
PBS and incubated for 2 min in 1% sodium citrate solution in 0.05 M
PBS at 4°C. After additional washes in 0.1 M PBS, 50 µl of TUNEL
reaction mixture was pipetted onto each slide. The slides were
incubated for 60 min at 37°C in the dark and washed again with 0. M
PBS three times. Then, slides were incubated in DAPI solution
(1:65,000) for 5 min in the dark, washed with 0.1 M PBS three
times, allowed to dry and mounted with glycerol (26). The images were acquired under a
DM4000B-M fluorescence microscope (magnification, ×400) equipped
with a color digital camera and diagnostic instruments (Leica Co.,
Germany). Image analysis adopted the double-blind method. Five film
sections were randomly selected from each group of rats for
analysis. TUNEL fluorescent signals located in the nucleus appeared
as bright green dots, representing apoptotic cells; DAPI was used
for nuclear staining. Apoptosis index (AI)=green-stained
cells/blue-stained cells.
Statistical analysis
The IBM-SPSS19 statistical software (SPSS Inc.,
Chicago, IL, USA) was used for statistical analysis. All western
blot and PCR data are represented as relative expression based on
the sham group. Continuous variables with normal distribution were
presented as mean ± standard deviation (mean ± SD). Multiple group
comparisons were performed by one way analysis of variance (ANOVA),
with the least-significant difference (LSD) test for group pair
comparisons. Pearson correlation analysis was used to examine the
association of Nogo-A with caspase-3 in the hippocampus. P<0.05
was considered as indicative of statistical significance.
Results
Rats after ROSC were randomized to a Model group and
a Treatment group with n=6 for 1, 2, 4, 6, 48 and 72 h time-points
and n=9 for 24 h time-point. Five rats were dead within 72 h in
each group. There were no significant differences in weights, heart
rates, mean arterial pressures among groups at baseline. There were
no significant differences in the number of defibrillations
required and CPR duration between the Model and Treatment group.
The survival rates at 72 h after ROSC in the Model group was 6 of 7
rats (85.7%); the survival rates in the Treatment group was 6 of 7
rats (85.7%). The survival rates in the Sham group was 6 of 6 rats
(100%).
Nissl staining
The cells in the Sham operation group (Fig. 2A) under light microscope were
large, with Nissl bodies clearly visible. Reduced amounts of cells
were observed in the Model group (Fig.
2B) compared with the Sham group. In the Treatment group
(Fig. 2C), there were more cells
in an arranged order; some Nissl bodies were clearly visible.
Average optical density (AOD) values in the Model
and Treatment groups were significantly decreased compared with
that of the Sham group. Meanwhile, AOD in the Treatment group was
significantly higher than that of the Model group (Fig. 2D). There were significant
differences in AOD among the three groups (P<0.001). These
results indicated that Nogo-A antibody could preserve the Nissl
bodies.
Ultrastructure of the brain
samples
As shown in Fig.
3A, neuronal structure was clearly visible in the Sham group,
with abundant cytoplasm; the rough ER was large and linear. The
mitochondria were elliptical, rich and complete; the mitochondrial
membrane was smooth, with visible double-layer structure. The
mitochondrial ridge was clear and orderly. Ribosomes and glycogen
granules were abundant. The nuclear membrane was clear and the
chromatin dispersed uniformly in the nucleus. As shown in Fig. 3B, the rough ER was expanded
obviously in the Model group; the mitochondria were swollen, and
the mitochondrial crest decreased and disappeared. There was cell
edema and vacuolar degeneration. Perinuclear plasmids, ribosomes
and glycogen granules were sparse. As shown in Fig. 3C, ER expansion was less pronounced
in the Treatment group than that in the Model group. The
mitochondrial structure was well preserved, and the mitochondria
were mildly swollen. The membrane was slightly fuzzy. The
mitochondrial ridge was partially visible, and cavitation was
reduced. Sub-organelle structure was preserved as assessed by
electron microscopy in the Treatment group.
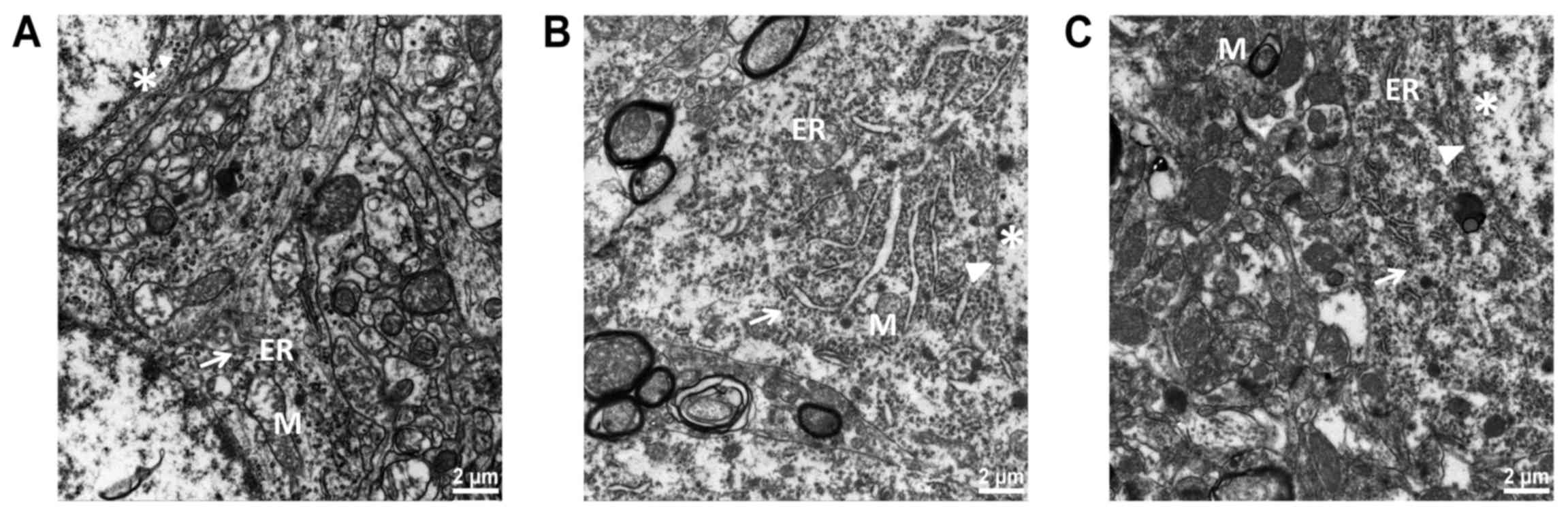 | Figure 3.The hippocampal endoplasmic reticulum
(ER) and mitochondrial ultrastructure examined by transmission
electron microscope (TEM) (magnification, ×30,000) (n=3). (A) the
membranes of normal ER were continuous and integrated. Many
ribosomes attached to the ER. (B) In the Model group, the structure
of the ER was irregular, faulted and expanded irregularly. (C) In
the Treatment group, the ER had a more complete structure. M,
mitochondria; ER, endoplasmic reticulum. Arrows indicate ribosomes;
arrowheads indicate nuclear membrane; asterisk indicates chromatin.
Groups: Treatment, received Nogo-A antibody (n=50); Model, received
saline (n=50); Sham, Sham-operated rats without cardiac arrest (CA)
serving as non-ischemic controls (n=15). |
Apoptosis quantitated by TUNEL
staining
Only a few cells in the Sham group underwent
apoptosis, with an apoptotic index of 3±1.58%; apoptotic index
values were 46.6±12.95 and 26.2±9.85% in the CPR Model and
Treatment groups at 72 h, respectively. Differences among the three
groups were statistically significant (F=26.686, P<0.001). The
apoptosis index of the Treatment group was significantly lower than
that of the Model group (P<0.05) (Fig. 4). These results revealed that the
Nogo-A antibody overtly reduced the number of TUNEL-positive
cells.
Changes in neurological function
In the Sham group, the neurological deficit scores
(NDSs) showed no significant differences at the three time-points
(F=0.830, P=0.447). NDSs in both the Model and Treatment groups
increased with time. Indeed, NDSs varied at different time-points
in the Model (F=31.249, P<0.001) and Treatment (F=33.924,
P<0.001) groups. The difference between the 48 and 72 h
time-points was not significant in the Treatment group (P=0.055).
NDSs in the Treatment and Model groups were lower than those of the
Sham group, and differences were statistically significant
(P<0.05). NDSs at all time-points in the Treatment group were
higher than those of the Model group, and differences were
statistically significant (P<0.05) (Fig. 5). These findings demonstrated that
Nogo-A antibody could improve neurological function in rats after
ROSC.
Caspase-3 mRNA levels in the cerebral
hippocampus
Hippocampal caspase-3 mRNA levels in the Model group
were increased after CPR and decreased at 4 h, and then increased
at 6 h. The Treatment group showed decreased levels until 4 h,
followed by an increase. Hippocampal caspase-3 mRNA amounts in the
treatment group were lower than those of the Model group, and the
difference was statistically significant (P<0.05). Hippocampal
caspase-3 mRNA amounts in the Treatment group and in the Model
group were higher than those of the Sham group, and the difference
was statistically significant (P<0.05; Fig. 6). Nogo-A antibody injection reduced
caspase-3 mRNA expression (P<0.05).
Nogo-A and caspase-3 protein levels in
the hippocampus
In the Model group, hippocampal Nogo-A protein
levels increased gradually reaching a peak at 24 h, and then
decreased. The Treatment group showed decreased amounts at 4 h,
followed by an increase. Hippocampal Nogo-A protein amounts in the
Model group were higher than those of the Sham group at all
time-points (P<0.05), and hippocampal Nogo-A protein amounts in
the Treatment group were lower than those of the Model group within
24 h (P<0.05), and the decrease was most obvious at 4 h
(Fig. 7A-C). Hippocampal caspase-3
protein levels in the Model group increased over time. In the
Treatment group, hippocampal caspase-3 protein increased over time
as well. But hippocampal caspase-3 protein levels in the Treatment
group were significantly lower than those of the Model group
(P<0.05), but higher than those of the Sham group (P<0.05).
(Fig. 7A, B and D).
 | Figure 7.Expression of Nogo-A and caspase-3
protein in the hippocampus (n=3). (A and B) Western blotting for
Nogo-A and caspase-3 expression in the hippocampus. (C) Relative
levels of Nogo-A. (D) Relative levels of caspase-3, Data are
presented as mean ± SD. Sham group (white), Model group (black),
Treatment group (gray). 1, 2, 4, 6, 24, 48 and 72 h: Model group at
1, 2, 4, 6, 24, 48 and 72 h. T1, T2, T4, T6, T24, T48 and T72 h:
Treatment group at 1, 2, 4, 6, 24, 48 and 72 h. *P<0.05,
**P<0.01 and ***P<0.001, comparison between the Model and
Treatment group; #P<0.05, comparison with the Sham group;
one-way ANOVA followed by LSD test. Groups: Treatment, received
Nogo-A antibody (n=50); Model, received saline (n=50); Sham,
Sham-operated rats without cardiac arrest (CA) serving as
non-ischemic controls (n=15). |
Pearson's correlation showed that Nogo-A protein in
the hippocampus was significantly positively correlated with
caspase-3 (r=0.790, P<0.001). These findings indicated that
Nogo-A antibody injection could reduce Nogo-A and caspase-3 protein
amounts (P<0.05).
GRP78, CHOP, caspase-12, Bcl- and Bax
protein levels in the hippocampus
The expression of GRP78 gradually increased in the
hippocampus and decreased after peaking at 2 h in the Model group.
However, GRP78 expression gradually increased and peaked at 4 h,
then increased again at 24 h in the Treatment group. GRP78 protein
levels were higher in the Treatment group compared with the Model
group at 4, 6 and 24 h (P<0.01). Hippocampal GRP78 protein
levels in the Treatment and Model group were higher than those of
the Sham group (P<0.05) (Fig.
8A).
 | Figure 8.Expression of GRP78, CHOP,
caspase-12, Bcl-2 and Bax protein in the hippocampus (n=3).
Relative levels of (A) GRP78 expression, (B) CHOP expression, (C)
caspase-12 expression, (D) Bcl-2 expression and (E) Bax expression
in the hippocampus. (F) Western blotting for GRP78, CHOP,
caspase-12, Bcl-2, Bax expression in the hippocampus, GRP78,
glucose regulated protein 78; CHOP, C/EBP homologous protein;
caspase-12, cysteinyl aspartate specific proteinase-12. Data are
presented as mean ± SD, Sham group (white), Model group (black),
Treatment group (gray). 2, 4, 6 and 24 h: Model group at 2, 4, 6
and 24 h. T2, T4, T6 and T24 h: Treatment group at 2, 4, 6 and 24
h. *P<0.05, **P<0.01 and ***P<0.001, comparison between
the Model and Treatment group; #P<0.05, comparison with the Sham
group; one-way ANOVA followed by LSD test. Groups: Treatment,
received Nogo-A antibody (n=50); Model, received saline (n=50);
Sham, Sham-operated rats without cardiac arrest (CA) serving as
non-ischemic controls (n=15). |
CHOP protein expression varied significantly at
different time-points. CHOP protein amounts in the Model group
increased with time and peaked at 6 h. The Treatment group had a
similar profile. CHOP protein levels were lower in the Treatment
group than levels in the Model group at 2, 4, 6 and 24 h
(P<0.01). Hippocampal CHOP protein levels in the Model group
were higher than those of the Sham group (P<0.05). Hippocampal
CHOP protein levels in the Model group were higher compared with
the Sham group at 4, 6 and 24 h (P<0.05) (Fig. 8B).
Caspase-12 expression increased at 2 h, and peaked
at 24 h in the Model group. Hippocampal caspase-12 levels in the
Treatment group showed a trend of decrease with time, and no
significant peak was observed. Caspase-12 protein levels were lower
in the Treatment group compared with the Model group at 2, 6 and 24
h (P<0.05). Hippocampal caspase-12 protein levels in the Model
group were significantly higher than those of the Sham group at 4,
6 and 24 h (P<0.05). Hippocampal caspase-12 protein levels in
the Treatment group were lower than those of the Sham group at 2, 6
and 24 h (P<0.05) (Fig.
8C).
Bcl-2 protein levels were decreased at 2 h, and
increased to a peak at 6 h, then decreased in the Model group.
Bcl-2 protein amounts in the Treatment group peaked at 2 h and
decreased with time. Bcl-2 protein levels were significantly higher
in the Treatment group than the Model group at 2 h (P<0.001).
Hippocampal Bcl-2 protein levels in the Model group were
significantly higher than those of the Sham group at 6 h and
significantly lower than those of the Sham group at 2 and 24 h
(P<0.05). Hippocampal Bcl-2 protein levels in the Treatment
group were significantly higher than those of the Sham group at 2
and 6 h and significantly lower than those of the Sham group at 24
h (P<0.05; Fig. 8D).
Hippocampal Bax protein levels increased gradually
with time in the Model group, and decreased after peaking at 6 h.
In the Treatment group, they were slightly increased at 2 h,
significantly decreased at 4 h, and further increased after 6 h.
Hippocampal Bax protein amounts in the Treatment group were
significantly lower than those of the Model group at all
time-points (P<0.05). There were differences in Bax protein
levels between the Sham group and the Model and Treatment groups
(P<0.05). Hippocampal Bax protein levels in the Model group were
significantly higher than those of the Sham group at 2, 4, 6 and 24
h (P<0.05). Hippocampal Bax protein levels in the Treatment
group were higher than those of the Sham group at 2, 6 and 24 h and
lower than those of the Sham group at 4 h (P<0.05; Fig. 8E).
These findings demonstrated that Nogo-A antibody
injection could upregulate GRP78 and Bcl-2 at the protein level,
and reduce CHOP, caspase-12 and Bax protein amounts within 24 h
(P<0.05).
Discussion
The present study assessed the expression changes in
Nogo-A after cardiac arrest/cardiopulmonary resuscitation (CA/CPR)
in rats. The results showed that Nogo-A protein levels increased
over time in the hippocampus of rats after CA/CPR, consistent with
findings by Zhao et al (27) for brain injury and similar with rat
studies of ischemia and hypoxia (5,6,28,29).
The present study showed that Nogo-A protein levels in the
hippocampus were lower in the Treatment group than the Model group,
corroborating Weinmann et al (30) and Zhou et al (17). Zhou (17) found that Nogo-A protein levels in
brain tissue were significantly reduced by injection with the
Nogo-A antibody into the cerebral ventricle of newborn rats with
hypoxic ischemic brain injury. He believed that the reason for the
decrease in Nogo-A protein was the formation of antigen-antibody
complex of Nogo-A. Weinmann et al (30) also found that the total Nogo-A
protein amounts in tissues were significantly reduced after Nogo-A
antibody injection into the lateral ventricle or subdural cavity of
adult rats and rhesus monkeys. Moreover, his study discovered that
the internalized Nogo-A and endogenous Nogo-A co-localized in
organelles similar to lysosomes or lysosomal progenitors. He also
found that antibodies bound to the cell surface and were
internalized as antibody ligand complexes. Weinmann et al
speculated that the decrease in Nogo-A was due to the reduction of
intracellular storage caused by the degradation induced by enhanced
antibodies, but whether the synthesis of Nogo-A is also affected
still needs to be further studied.
The present study demonstrated that Nogo-A protein
levels were significantly positively correlated with caspase-3
expression in the rat hippocampus (r=0.79, P<0.001). Previous
studies on ischemic hypoxic brain damage in a newborn SD rat model
found that caspase-3 expression was significantly increased along
with Nogo-A expression. Application of ephedrine and hyperbaric
oxygen treatment could inhibit Nogo-A and caspase-3 expression,
alleviating the degree of brain injury caused by ischemia hypoxia
(10). Another study assessing the
optic nerve also showed increased Nogo-A and caspase-3 amounts
after acute injury (31), and
found that glucocorticoids reduce Nogo-A and caspase-3 levels.
However, the above study failed to assess whether Nogo-A and
caspase-3 are concomitant or causal. The present research revealed
that inhibition of Nogo-A expression could reduce caspase-3
expression, suppressing apoptosis. As shown above, caspase-3 mRNA
and protein expression levels, and Nogo-A protein amounts were
increased after restoration of spontaneous circulation (ROSC), but
decreased after intraventricular injection of the Nogo-A antibody
in rats. The expression trend of the Nogo-A protein after
intraventricular injection was consistent with that of caspase-3.
Specific intervention of Nogo-A expression could reduce caspase-3
expression. Inhibition of Nogo-A expression could downregulate
caspase-3 and inhibit apoptosis. It was also demonstrated that
caspase-3 was associated with Nogo-A, indicating that Nogo-A is
involved in the process of apoptosis, which can be reduced by
intraventricular injection of Nogo-A antibody. In cultured
cardiomyocytes, a study found that knockdown of Nogo-A reduced
caspase-3 cleavage and apoptosis in hypoxia/reoxygenation
cardiomyocytes (9), corroborating
the present findings. Therefore, downregulation of Nogo-A reduces
caspase-3 expression, decreases apoptosis and protects neural
function.
The present study found that after ROSC, GRP78
levels in the hippocampal tissue were increased compared with those
of the Model group, and rapidly increased at 2 h. This is
consistent with the GRP78 results for focal ischemic rat brain
tissue (32). GRP78, a member of
the heat shock protein family, is a classic marker of endoplasmic
reticulum (ER) stress and promotes the correct assembly, folding
and modification of proteins. Increased expression of GRP78 plays a
protective role in cells under stress. After CA/CPR, the brain
tissue experiences total cerebral ischemia and reperfusion, which
causes ER stress and activates the unfolded protein response (UPR)
process. When unfolded proteins accumulate in the ER, GRP78 binds
to them, dissociating with inositol-requiring enzyme1 (IRE1),
activating transcription factor 6 (ATF6) and double-stranded RNA
dependent protein kinase-like ER kinase (PERK), and causing their
activation (33). GRP78 activates
these three pathways, which regulate GRP78 after activation, and
GRP78 subtly modulates the activation of the pathways. Through
these mechanisms, GRP78 expression is increased, and the protein
binds to misfolded and unfolded proteins in the ER lumen to reduce
the ER burden and restore ER function. However, when ER stress is
extremely strong, this self-stabilizing effect is weakened,
resulting in dysfunctional ER and decreased GRP78 expression
(34). This is why GRP78 protein
amounts were significantly increased at 4, 6 and 24 h in the
Treatment group compared with the Model group, with a delayed peak
(P<0.01) in this study. The peak time of the GRP78 protein in
the Treatment group was 4 h, which was consistent with the low ebb
time for the Nogo-A protein, and subsequent Nogo-A decrease was
consistent with subsequent elevation of GRP78, indicating that
reduced Nogo-A protein results in increased expression of the GRP78
protein, alleviates ER stress, and exerts a protective effect on
cell survival.
CHOP, also known as growth arrest and
DNA-damage-inducible gene 153 (GADD153), is also a classical marker
of ER stress. Increased expression of CHOP initiates the apoptotic
pathway (35). The present study
found that CHOP in the hippocampal tissue gradually increased to
peak at 6 h after CA/CPR, and then decreased. CHOP levels were
lower in the Treatment group compared with the Model group, and
differences between the Model and Treatment groups and Sham group
were statistically significant at 2, 4, 6 and 24 h. CHOP is
normally widely expressed at a very low level, but highly expressed
in cells under stress. After ER stress, the UPR occurs and three
pathways are activated. Since CHOP expression (at the gene and
protein levels) occurs upon activation of the three pathways of
unfolded reactions by GRP78, CHOP can be induced by the above three
pathways simultaneously, and its protein expression gradually
increases. At the early stage of ER stress, GRP78 is highly
expressed and inhibits CHOP expression through the three pathways
in order to restore homeostasis. At the later stage, GRP78
expression decreases and CHOP expression is rapidly upregulated.
Therefore, the peak of CHOP occurs later than that of GRP78. CHOP
is a transcription factor that regulates genes involved in cell
survival or death. When cell adaptation through the UPR is
unsuccessful due to prolonged or unresolved ER stress, new signals
transmitted from the ER induce cell death. UPR's PERK pathway
initially mediates the pro-survival response and promotes GRP78
expression; however, in case of severe or prolonged ER stress, a
pro-apoptotic response takes place instead (36). CHOP can induce apoptosis in many
ways, possibly through inhibition of Bcl-2 transcription.
Overexpression of CHOP leads to decreased Bcl-2 protein levels. In
the present study, Bcl-2 levels increased at 2 h in the Treatment
group, which may be related to decreased CHOP protein expression at
this time-point. We found that CHOP protein expression levels in
the Treatment group were lower at 2, 4, 6 and 24 h compared with
those of the Model group. The CHOP protein decrease in the
Treatment group was highest at 4 h, which was consistent with the
most obvious decrease in Nogo-A protein levels, indicating that a
decrease in Nogo-A protein downregulates CHOP at the protein level,
reduces ER stress, alleviates CHOP protein-induced apoptosis, and
has a protective effect on cell survival (37). The present study demonstrated that
during CPR, CHOP, GRP78, caspase-12 and Bax in hippocampal tissues
were all increased compared with the values of the Sham operation
group, Bcl-2 expression was decreased, and the differences were
statistically significant. GRP78 peaked at 2 h and CHOP at 6 h. The
variation pattern of CHOP in our model was similar to previous
findings (38). GRP78 and CHOP are
both classical markers of ER stress. Increased expression of GRP78
plays a protective role in cells under stress, and increased
expression of CHOP is a pathway that initiates apoptosis. Both of
them reflect ER stress occurrence.
Caspase-12 is located at the cytoplasmic side of the
ER, which hosts caspases. It represents a specific molecule that
mediates ER stress and induces apoptosis, and is not activated in
the death receptor and mitochondrial pathways, the other two axes
of apoptosis (39). Caspase-12
mediates the apoptosis pathway specific to the ER. It directly
enters the cytoplasm and activates caspase-9 (3) without relying on the mitochondrial
cytochrome C/Apaf-1 pathway, and activates caspase-3 to induce
apoptosis (40).
The present study showed that caspase-12 in the
hippocampal tissue was gradually increased after CA/CPR compared
with the Sham group, and significantly increased at 6 h, peaking at
24 h, and differences were statistically significant. This is
consistent with findings by Osada et al (37). In the latter study, the hippocampal
tissue of mice with transient anterior cerebral ischemia was
assessed, and caspase-12 was shown to be increased significantly at
24 h. Caspase-12 also increased in the ischemic core 24 h after
ischemia in the permanent middle cerebral artery occlusion (MCAO)
model (41). GRP78, ATF4 and CHOP
levels were also shown to be significantly increased in the
ischemia reperfusion model of MCAO (38). The increase of ER stress markers
was consistent with caspase-12 upregulation in the core area of
ischemia at 24 h. In the mouse MCAO model, caspase-12 after 1-h
ischemia/reperfusion was found to be associated with GRP78
upregulation and cell apoptosis. CHOP and caspase-12 levels were
shown to be increased in the same model at 6–12 h after reperfusion
(42). Meanwhile, caspase-12
expression was increased in the hippocampus of rats with traumatic
brain injury, which was found to be associated with neuronal
injury. In the present study, the caspase-12 level increased after
CA/CPR indicating apoptotic induction by ER stress. As shown above,
CHOP peaked at 6 h after CA/CPR, and caspase-12 also increased
significantly at that time-point. While CHOP levels decreased in
the Treatment group, caspase-12 amounts did not increase
significantly, and no significant peak was observed. Meanwhile,
caspase-12 protein expression in the Treatment group was lower than
that of the Model group at 2, 6 and 24 h, respectively, with the
most significant reductions at 6 and 24 h, and no significant peak
formation. We also found that a decrease in Nogo-A protein could
inhibit the specific caspase-12 apoptosis pathway induced by ER
stress and eventually lead to caspase-3 downregulation, thereby
reducing apoptosis.
In addition to its role in the mitochondrial
apoptotic pathway, the Bcl-2 protein is also located in the ER and
reduces the level of calcium ions in a stable state through IP3Rs.
The apoptotic protein Bax is also located in the ER and antagonizes
the activity of Bcl-2 in regulating the concentration of calcium
ions (43). Meanwhile, Bax binds
to IRE1α after ER stress, leading to pathological activation
(44), which in turn neutralizes
the anti-apoptotic activity of Bcl-2. Bax may play an executor role
in ER stress-induced apoptosis. The present study found that there
were no significant differences in hippocampal Bcl-2 protein levels
between the Treatment and Model groups except at 2 h, which may be
associated with the insignificant increase in CHOP protein levels
observed at this time-point, as overexpression of CHOP leads to
decreased Bcl-2 protein expression. Hippocampal Bax protein levels
in the Treatment group were lower than those of the Model group at
2, 4, 6 and 24 h, respectively, since CHOP overexpression leads to
increased Bax expression. The expression of CHOP was inhibited in
the Treatment group, thus Bax expression was decreased.
The limitations of this research should be
mentioned. Firstly, the observation time after ROSC was relatively
short (72 h), and further studies should prolong the observation
period to at least 1 to 3 months after ROSC to observe long-term
effects. Secondly, a single drug dose was assessed; optimal timing
and dosage should be comprehensively tested in further
investigation. Thirdly, only hippocampal injury was evaluated after
ROSC, and it remains unclear whether other parts of the brain such
as the corpus striatum were equally affected. Fourthly, this study
adopted a bolus injection for 20 min. We found that the effects
would not exceed 24 h, thus further studies should deliver
antibodies over several days or weeks to maintain the drug effects
for a longer time. Meanwhile, further studies are required in
posttreatment settings.
In conclusion, the present study found that after
intracerebroventricular injection of Nogo-A antibody to rats after
ROSC, cell apoptosis was reduced, the morphological structure and
ultrastructure were preserved, and neurological function was
improved. These results suggest that the Nogo-A antibody has
certain protective effects on brain structure and function in rats
after ROSC. The protective mechanism possibly includes GRP78 and
Bcl-2 upregulation, and CHOP, Bax and caspase-12 downregulation, ER
stress inhibition, and finally suppression of caspase-3-associated
apoptosis. The anti-Nogo-A antibody could protect brain structure
and function in rats after ROSC by reducing ER stress-induced
apoptosis.
Acknowledgements
Not applicable.
Funding
This study was supported by the Sichuan Science and
Technology Program (grant nos. 2013SZ0079 and 2017RZ0046) and the
Youth Teacher Research Startup Fund of Sichuan University (grant
no. 2016SCU11016).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
All authors designed the experiments. QW and HZ
carried out the experiments and associated data analysis. QW
drafted the manuscript. ZZ and HN interpreted the data and revised
the manuscript. All authors read and approved the final version of
the manuscript.
Ethics approval and consent to
participate
The West China Hospital's Experimental Animal Ethics
Committee approved the study protocol.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Madder RD and Reynolds JC:
Multidisciplinary management of the post-cardiac arrest patient.
Cardiol Clin. 36:85–101. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Neumar RW, Nolan JP, Adrie C, Aibiki M,
Berg RA, Böttiger BW, Callaway C, Clark RS, Geocadin RG, Jauch EC,
et al: Post-cardiac arrest syndrome: Epidemiology, pathophysiology,
treatment, and prognostication. A consensus statement from the
International Liaison Committee on Resuscitation (American Heart
Association, Australian and New Zealand Council on Resuscitation,
European Resuscitation Council, Heart and Stroke Foundation of
Canada, InterAmerican Heart Foundation, Resuscitation Council of
Asia, and the Resuscitation Council of Southern Africa); the
American Heart Association Emergency Cardiovascular Care Committee;
the Council on Cardiovascular Surgery and Anesthesia; the Council
on Cardiopulmonary, Perioperative, and Critical Care; the Council
on Clinical Cardiology; and the Stroke Council. Circulation.
118:2452–2483. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Taccone FS, Crippa IA, Dell'Anna AM and
Scolletta S: Neuroprotective strategies and neuroprognostication
after cardiac arrest. Best Pract Res Clin Anaesthesiol. 29:451–464.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cherry BH, Sumien N and Mallet RT:
Neuronal injury from cardiac arrest: Aging years in minutes. Age
(Dordr). 36:96802014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang H, Yao Y, Jiang X, Chen D, Xiong Y
and Mu D: Expression of Nogo-A and NgR in the developing rat brain
after hypoxia-ischemia. Brain Res. 1114:212–220. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhou CM, Li Y, Nanda A and Zhang JH: HBO
suppresses Nogo-A, Ng-R, or RhoA expression in the cerebral cortex
after global ischemia. Biochem Biophys Res Commun. 309:368–376.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ulndreaj A, Badner A and Fehlings MG:
Promising neuroprotective strategies for traumatic spinal cord
injury with a focus on the differential effects among anatomical
levels of injury. F1000Res. 6:19072017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang ZW, Jiang JJ, Luan MC, Ma ZJ, Gao F
and Yu SJ: Nogo-A antibody treatment enhances neuron recovery after
sciatic nerve transection in rats. Eur Rev Med Pharmacol Sci.
21:1780–1786. 2017.PubMed/NCBI
|
|
9
|
Sarkey JP, Chu M, McShane M, Bovo E, Ait
Mou Y, Zima AV, de Tombe PP, Kartje GL and Martin JL: Nogo-A
knockdown inhibits hypoxia/reoxygenation-induced activation of
mitochondrial-dependent apoptosis in cardiomyocytes. J Mol Cell
Cardiol. 50:1044–1055. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen S, Xiao N and Zhang X: Effect of
combined therapy with ephedrine and hyperbaric oxygen on neonatal
hypoxic-ischemic brain injury. Neurosci Lett. 465:171–176. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen S, Novick P and Ferro-Novick S: ER
structure and function. Curr Opin Cell Biol. 25:428–433. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rämö O, Kumar D, Gucciardo E, Joensuu M,
Saarekas M, Vihinen H, Belevich I, Smolander OP, Qian K, Auvinen P
and Jokitalo E: NOGO-A/RTN4A and NOGO-B/RTN4B are simultaneously
expressed in epithelial, fibroblast and neuronal cells and maintain
ER morphology. Sci Rep. 6:359692016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu D, Zhang M and Yin H: Signaling
pathways involved in endoplasmic reticulum stress-induced neuronal
apoptosis. Int J Neurosci. 123:155–162. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Idris AH, Becker LB, Ornato JP, Hedges JR,
Bircher NG, Chandra NC, Cummins RO, Dick W, Ebmeyer U, Halperin HR,
et al: Utstein-style guidelines for uniform reporting of laboratory
CPR research. A statement for healthcare professionals from a task
force of the American Heart Association, the American College of
Emergency Physicians, the American College of Cardiology, the
European Resuscitation Council, the Heart and Stroke Foundation of
Canada, the Institute of Critical Care Medicine, the Safar Center
for Resuscitation Research, and the Society for Academic Emergency
Medicine. Writing Group. Circulation. 94:2324–2336. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen MH, Liu TW, Xie L, Song FQ, He T,
Zeng ZY and Mo SR: Ventricular fibrillation induced by
transoesophageal cardiac pacing: A new model of cardiac arrest in
rats. Resuscitation. 74:546–551. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Labak M, Foniok T, Kirk D, Rushforth D,
Tomanek B, Jasiński A and Grieb P: Metabolic changes in rat brain
following intracerebroventricular injections of streptozotocin: A
model of sporadic Alzheimer's disease. Acta Neurochir Suppl.
106:177–181. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhou XG, Liu RH and Xiong AH: Effect of
ventricle injection of Nogo-A antibody on neuronal regeneration
following hypoxic-ischemic brain damage in the neonatal rat.
Zhongguo Dang Dai Er Ke Za Zhi. 9:301–304. 2007.(In Chinese).
PubMed/NCBI
|
|
18
|
Ineichen BV, Schnell L, Gullo M, Kaiser J,
Schneider MP, Mosberger AC, Good N, Linnebank M and Schwab ME:
Direct, long-term intrathecal application of therapeutics to the
rodent CNS. Nat Protoc. 12:104–131. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Geocadin RG, Ghodadra R, Kimura T, Lei H,
Sherman DL, Hanley DF and Thakor NV: A novel quantitative EEG
injury measure of global cerebral ischemia. Clin Neurophysiol.
111:1779–1787. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ye S, Weng Y, Sun S, Chen W, Wu X, Li Z,
Weil MH and Tang W: Comparison of the durations of mild therapeutic
hypothermia on outcome after cardiopulmonary resuscitation in the
rat. Circulation. 125:123–129. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Birnie M, Morrison R, Camara R and Strauss
KI: Temporal changes of cytochrome P450 (Cyp) and
eicosanoid-related gene expression in the rat brain after traumatic
brain injury. BMC Genomics. 14:3032013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mavridis K, Stravodimos K and Scorilas A:
Downregulation and prognostic performance of microRNA 224
expression in prostate cancer. Clin Chem. 59:261–269. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Eslamizade MJ, Madjd Z, Rasoolijazi H,
Saffarzadeh F, Pirhajati V, Aligholi H, Janahmadi M and Mehdizadeh
M: Impaired memory and evidence of histopathology in CA1 pyramidal
neurons through injection of Aβ1–42 peptides into the frontal
cortices of rat. Basic Clin Neurosci. 7:31–41. 2016.PubMed/NCBI
|
|
25
|
Zhao BB, Long QH, Wang CY, Chen LL, Xie
GJ, Bo WJ, Xu B, Li ZF, Li HM and Wang P: Protective effects of Liu
Wei Di Huang Wan on the liver, orbitofrontal cortex nissl bodies,
and neurites in MSG+PH-induced liver regeneration rat model. Evid
Based Complement Alternat Med. 2018:90901282018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
França MS, Moron AF, Araujo Júnior E,
Avedissian M, Pares DB, Nardozza LM, Jaqueta CB and Mello LE:
Neonatal neuronal apoptosis after betamethasone administration in
pregnant Wistar rats. J Matern Fetal Neonatal Med. 29:1089–1093.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhao X, Song JN, Xi L, Sui L, Wang WB and
Liu XB: Expression changes of Nogo-A and its significance in rat
brain with diffusive axonal injury. J Xi'an Jiaotong Univ (Medical
Sciences). 36:80–84. 2015.
|
|
28
|
Marklund N, Fulp CT, Shimizu S, Puri R,
McMillan A, Strittmatter SM and McIntosh TK: Selective temporal and
regional alterations of Nogo-A and small proline-rich repeat
protein 1A (SPRR1A) but not Nogo-66 receptor (NgR) occur following
traumatic brain injury in the rat. Exp Neurol. 197:70–83. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jiang W, Xia F, Han J and Wang J: Patterns
of Nogo-A, NgR, and RhoA expression in the brain tissues of rats
with focal cerebral infarction. Transl Res. 154:40–48. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Weinmann O, Schnell L, Ghosh A, Montani L,
Wiessner C, Wannier T, Rouiller E, Mir A and Schwab ME:
Intrathecally infused antibodies against Nogo-A penetrate the CNS
and downregulate the endogenous neurite growth inhibitor Nogo-A.
Mol Cell Neurosci. 32:161–173. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yu J, Lin L, Luan X, Jing C and Maierab:
Impacts of Rho kinase inhibitor Fasudil on Rho/ROCK signaling
pathway in rabbits with optic nerve injury. Int J Clin Exp Pathol.
8:14717–14724. 2015.PubMed/NCBI
|
|
32
|
Aoki M, Tamatani M, Taniguchi M, Yamaguchi
A, Bando Y, Kasai K, Miyoshi Y, Nakamura Y, Vitek MP, Tohyama M, et
al: Hypothermic treatment restores glucose regulated protein 78
(GRP78) expression in ischemic brain. Brain Res Mol Brain Res.
95:117–128. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang S and Kaufman RJ: The impact of the
unfolded protein response on human disease. J Cell Biol.
197:857–867. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xiaoyan S, Zhao YB, Zhou XL, Wu YC and Liu
WW: Changes in the expression of endoplasmic reticulum
stress-related factors after cerebral ischemia reperfusion in rats.
Chin J Nerv Ment Dis. 33:624–626. 2007.
|
|
35
|
Zhao J, Xiang X, Zhang H, Jiang D, Liang
Y, Qing W, Liu L, Zhao Q and He Z: CHOP induces apoptosis by
affecting brain iron metabolism in rats with subarachnoid
hemorrhage. Exp Neurol. 302:22–33. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gardner BM, Pincus D, Gotthardt K,
Gallagher CM and Walter P: Endoplasmic reticulum stress sensing in
the unfolded protein response. Cold Spring Harb Perspect Biol.
5:a0131692013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Osada N, Kosuge Y, Ishige K and Ito Y:
Characterization of neuronal and astroglial responses to ER stress
in the hippocampal CA1 area in mice following transient forebrain
ischemia. Neurochem Int. 57:1–7. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nakka VP, Gusain A and Raghubir R:
Endoplasmic reticulum stress plays critical role in brain damage
after cerebral ischemia/reperfusion in rats. Neurotox Res.
17:189–202. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Nan L, Yan L and Niu ZH: Effects of
ischemic postconditioning on endoplasmic reticulum stress pathways
in rats with cerebral ischemia/reperfusion injury. J Stroke Neurol
Dis. 31:203–206. 2014.
|
|
40
|
Weston RT and Puthalakath H: Endoplasmic
reticulum stress and BCL-2 family members. Adv Exp Med Biol.
687:65–77. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Mouw G, Zechel JL, Gamboa J, Lust WD,
Selman WR and Ratcheson RA: Activation of caspase-12, an
endoplasmic reticulum resident caspase, after permanent focal
ischemia in rat. Neuroreport. 14:183–186. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhu H, Zhu H, Xiao S, Sun H, Xie C and Ma
Y: Activation and crosstalk between the endoplasmic reticulum road
and JNK pathway in ischemia-reperfusion brain injury. Acta
Neurochir (Wien). 154:1197–1203. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Byrd AE, Aragon IV and Brewer JW:
MicroRNA-30c-2* limits expression of proadaptive factor XBP1 in the
unfolded protein response. J Cell Biol. 196:689–698. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hetz C, Bernasconi P, Fisher J, Lee AH,
Bassik MC, Antonsson B, Brandt GS, Iwakoshi NN, Schinzel A,
Glimcher LH and Korsmeyer SJ: Proapoptotic BAX and BAK modulate the
unfolded protein response by a direct interaction with IRE1alpha.
Science. 312:572–576. 2006. View Article : Google Scholar : PubMed/NCBI
|