Introduction
Wilson disease (WD; OMIM 27790) is a rare autosomal
recessive disorder affecting copper transport, characterized by
decreased biliary copper excretion and reduced copper incorporation
into ceruloplasmin (1). The
worldwide incidence is ~30 per million individuals, with a gene
frequency of 0.56% and a carrier frequency of 1/90 (2). The main clinical features are
hepatic, neurologic and psychiatric disorders and, Kayser-Fleischer
(KF) rings (3). Defects the ATPase
copper transporter 7B (ATP7B) gene, which encodes for a
membrane P-type ATPase, at 13q14.3 have been identified to be
responsible for WD (4). The gene
spans ~80 kb and consists of 21 exons. ATP7B encodes a
protein with 1,465 amino acid that consists of six copper binding
domains, eight transmembrane domains and one ATP loop that
transports copper into bile (5).
The present study identified a heterozygous missense mutation,
which, to the best of our knowledge, has not been previously
identified in the Chinese population. In addition, the present
analysis identified a novel heterozygous gross deletion mutation in
the ATP7B locus in a Chinese pediatric patient with WD.
Additionally, the sequence of the precise breakpoint junction was
validated. Analysis of the family members showed that the missense
mutation and the novel gross deletion mutation were inherited from
the mother and father, respectively.
Subjects and methods
Subjects
The proband was hospitalized at the department of
Pediatrics of the Affiliated Hospital of Guizhou Medical University
(Guiyang, China) due to anorexia in March 2015. The present study
was approved by the Ethics Committees of Affiliated Hospital of
Guizhou Medical University (approval no. 201011). Written informed
consent was obtained from the parents of the patient. Peripheral
venous blood samples (3 ml), were collected from each subject. In
addition, the following parameters of hospital routine examination
were analyzed: Alanine transaminase (ALT), aspartate transaminase
(AST), ceruloplasmin (CER), creatine kinase, myocardial band
isoenzyme of creatine kinase, lactate dehydrogenase, ammonia, total
bilirubin, lactic acid, α fetal protein, blood urea nitrogen, blood
routine, urine routine, antibody to hepatitis viruses A-E,
Epstein-Barr virus IgM, toxoplasmosis, rubella, cytomegalovirus and
herpes simplex virus screening, and autoimmune hepatitis antibodies
and antinuclear antibody spectrum. Brain MRI and abdominal CT were
performed on the patient. In total, three people were enrolled; a
female patient with WD (age, 2.7 years) and her father and mother
(aged 29 and 28 years, respectively).
Genetic analysis
Blood samples (3 ml) were collected from every
family member after informed consent was obtained, including from
the parents of the patient. Genomic DNA was then isolated from
peripheral blood lymphocytes using the Tianamp Genomic DNA kit
(DP304; Tiangen Biotech Co., Ltd.) according to the manufacturer's
recommendations.
Sanger-method sequencing
All of the coding exons and exon-intron boundaries
of ATP7B were amplified by PCR using the same primers as
previously described amplification conditions (6). The PCR products were sequenced using
ABI PRISM 3730XL DNA automated sequencer (Applied Biosystems;
Thermo Fisher Scientific, Inc.)
Multiplex ligation-dependent probe
amplification (MLPA)
The DNA in which two mutations in the ATP7B
gene could not be identified by sequencing was subsequently tested
for partial exon deletions/duplications using the ATP7B MLPA
assay (SALSA MLPA kit Wilson disease; MRC-Holland BV) including
primers and enzymes according to the manufacturer's protocol. The
thermocycling conditions included initial denaturation at 98°C for
5 min, pause at 25°C, hybridisation reaction at 95°C for 1 min,
60°C for 16 h, ligation reaction at 54°C for 15 min, 98°C for 5
min, pause at 20°C, followed by 35 cycles of 95°C for 30 sec, 60°C
for 30 sec, 72°C for 60 sec, and an extension at 72°C for 20
min.
Next-generation sequencing (NGS)
The DNA in which precise heterozygous deletions in
the ATP7B gene could not be identified by MLPA was
subsequently tested with NGS. The genomic DNA sample was sheared by
sonication. A xGen Exome Research Panel v1.0 kit (Integrated DNA
Technologies, Inc.) was used to capture exons; the full length of
all exons was of 35.58 Mb. The samples were then sequenced on an
Illumina Hiseq2500 System (Illumina, Inc.). The sequencing reads
were aligned to the human reference genome (GRCH37/hg19) using
Burrows-Wheeler Aligner (ver. 0.7.12, http://bio-bwa.sourceforge.net/). Single nucleotide
variations, insertions and deletions were analyzed with Samtools
and indel software (https://sourceforge.net/projects/samtools/files/).
Selected variants were checked in relevant variant frequency
databases ((Exome sequencing project (http://evs.gs.washington.edu/EVS/), dbSNP (http://www.ncbi.nlm.nih.gov/SNP/), 1000Genomes
(http://www.1000genomes.org/), ClinVar
(https://www.ncbi.nlm.nih.gov/clinvar/) and Human gene
mutation database (http://www.hgmd.cf.ac.uk/ac/index.php)). Pathogenicity
prediction tools (Polyphen 2 (http://genetics.bwh.harvard.edu/pph2/); SIFT
(http://sift.jcvi.org/); Mutation Taster
(http://www.mutationtaster.org/)) were
additionally used.
Junction sequence confirmed by
Sanger-method sequencing
The possible region deleted in ATP7B was
detected by NGS. The suspected breakpoint junction sequence was
amplified using PCR. Specific primers were designed on both sides
of the breakpoints. The following primers were used: F:
5′-GCCAGAGAAGCTGGGATGTT-3′ and R: 5′-GACTGACCCAGCCCTCTTTC-3′. DNA
polymerase was provided by the Tianamp Genomic DNA kit (ET105;
Tiangen Biotech Co., Ltd.).
The thermocycling conditions were: Initial
denaturation at 95°C for 5 min, followed by 30 cycles of 95°C for
30 sec, 62°C for 30 sec, 72°C for 30 sec, and a final extension at
72°C for 10 min. The amplification product of the gel extraction
purification was sequenced using ABI PRISM 3730XL DNA automated
sequencer (Applied Biosystems; Thermo Fisher Scientific, Inc.).
Bioinformatics analysis
Bioinformatics analysis of the 2,000 bp sequence
surrounding the two breakpoints using the RepeatMasker program
4.0.7 (http://www.repeatmasker.org/) showed
special elements. The secondary structures of nucleotides for the
1,000 bp sequence in the vicinity of proximal and distal
breakpoints were predicted using the UNAFold v3.8 suite (http://unafold.rna.albany.edu/?q=mfold/DNA-Folding-Form).
Results
The patient was a 2.7-year-old Chinese girl, born
(weight, 3 kg) to non-consanguineous parents as their only child
following a pregnancy of normal duration. The patient presented
with two-month history of anorexia, which was the only presenting
symptom. The physical examination of the patient showed that her
liver was palpable ~4 cm under the low processus xiphoideus, and ~3
cm under the right lowest rib. The texture was soft, the edge was
blunt and there was no tenderness. The spleen was impalpable. KF
rings were not observed. The patient had no neurological or
psychological symptoms.
The clinical data collected from the patient on
admission are presented in Table
I. The laboratory tests identified a low level of serum CER,
and high levels of ALT and AST. The MRI results of the brain were
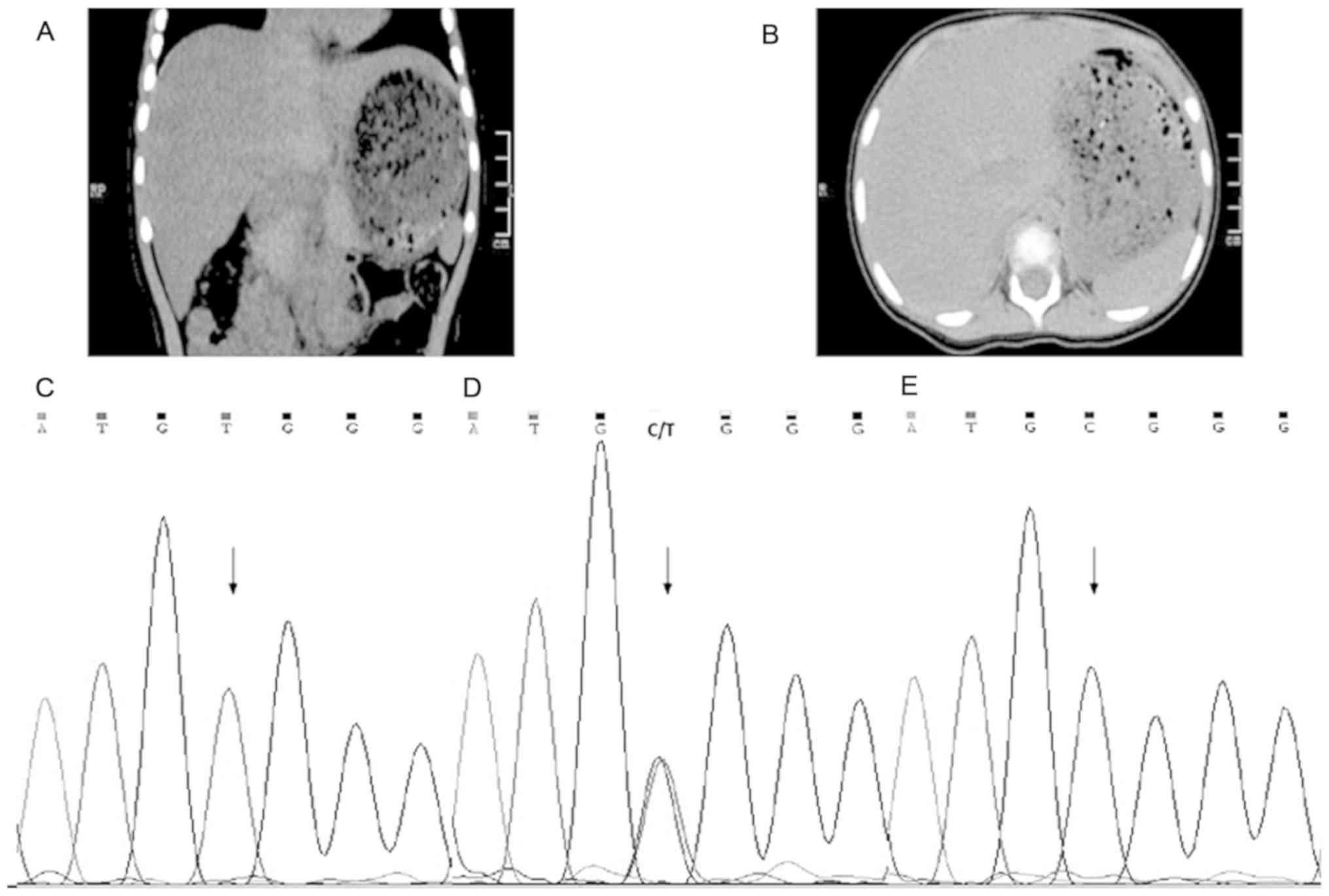
normal. The abdominal CT showed increased liver volume. The mean CT
value of liver was ~35 HU (Hounsfield units), with spleen
parenchymal CT values on the same plane of ~50 HU. The decreased
density of the liver had been proposed because the CT value of the
liver was less than the spleen on basis of the literature report
(7) (Fig. 1A and B). The parents of the patient
were asymptomatic and so no laboratory tests, CT scans or MRI
examinations were performed. Liver function of the patient was
normal after treatment with penicillamine and zinc sulfate.
 | Table I.Clinical data of the proband. |
Table I.
Clinical data of the proband.
| Clinical data | Patient | Normal range |
|---|
| ALT | 126.4 U/l | 7–40 U/l |
| AST | 86.5 U/l | 13–35 U/l |
| CER | 9.77 mg/dl | 22–58 mg/dl |
| CK | 179.2 U/l | 26–140 U/l |
| LDH | 301.1 U/l | 109–245 U/l |
| AMM | 80 µmol/l | 18–72 µmol/l |
| CKMB | 22.49 U/l | 0.0–25 U/l |
| TBIL | 5.96 µmol/l | 3.4–17 µmol/l |
| LACT | 1.93 mmol/l | 0.6–2.2 mmol/l |
| AFP | 3.1 ng/ml | 0.0–8.1 ng/ml |
| BUN | 3.61 mmol/l | 1.78–7.14
mmol/l |
| Blood routine | – |
|
| Urine routine | – |
|
| Antibody to
hepatitis viruses A-E | – |
|
| Epstein-Barr virus
IgM | – |
|
| TORCH | – |
|
| Autoimmune
hepatitis antibodies | – |
|
| ANA antibody
spectrum | Negative |
|
| Brain MRI | Normal | Normal |
| Abdominal CT | Abnormal | Normal |
Mutation analysis of ATP7B showed that the
proband was compound heterozygous for c.3121C>T (p. Arg1041Trp)
in exon 14, as assessed by Sanger sequencing (Fig. 1C) and had a large deletion of exons
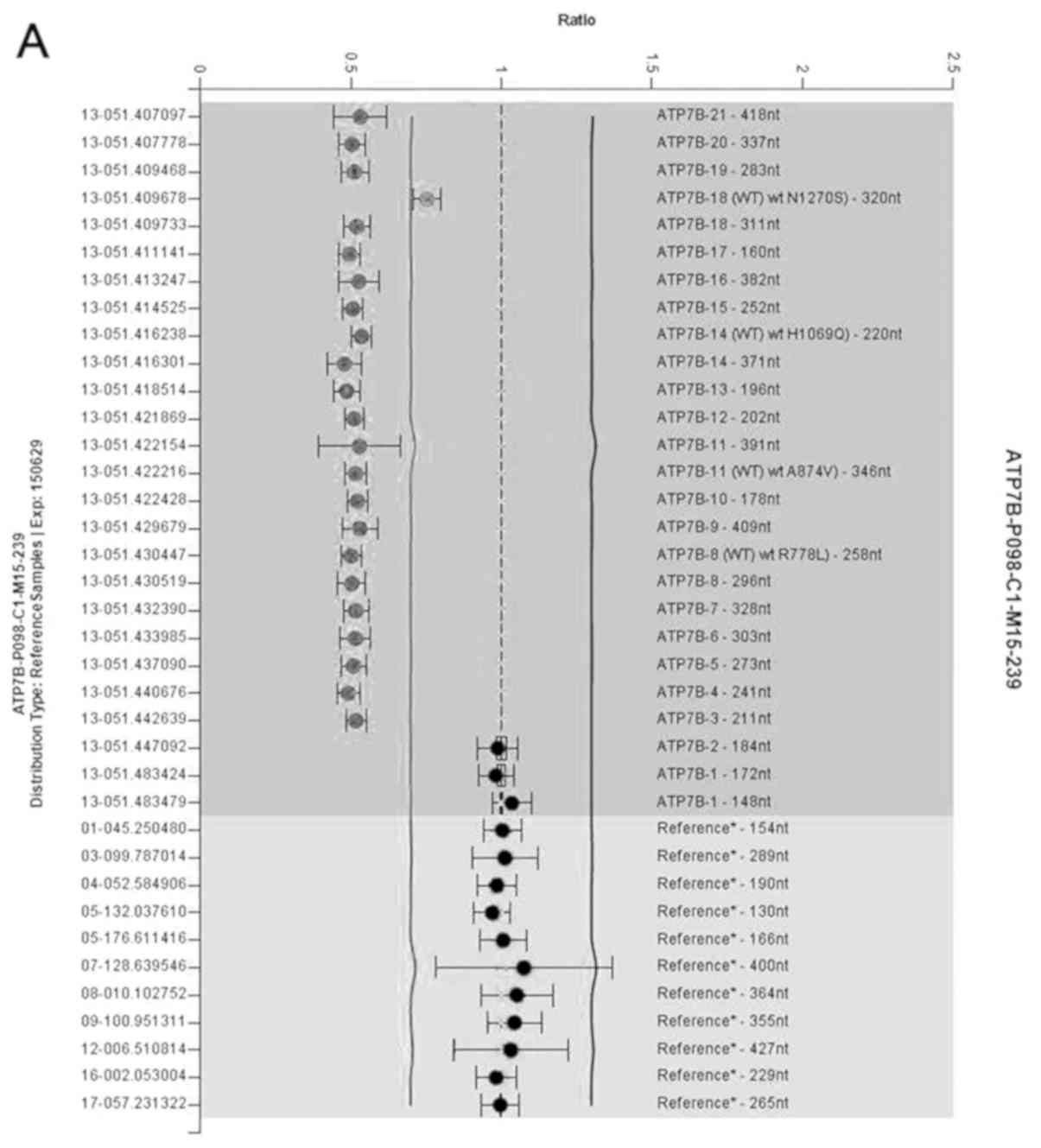
3–21 detected via MLPA (Fig. 2A).
The confirmed known missense mutation c.3121C>T in exon 14
causes a substitution from arginine to tryptophan at position 1041,
and occurs in the ATP loop of the copper transporting P type ATPase
(8). The mutation was inherited
from the mother, who did not present any signs of WD (Fig. 1D). For the mutant site, her mother
was heterozygous, whereas her father presented a wild-type allele
(Fig. 1E). Suggesting that the
patient presented a heterozygous and not a homozygous mutation.
ATP7B deletion/duplication detection using MLPA was
therefore performed, and MLPA results showed that the proband and
her father have a heterozygous gross deletion (Fig. 2). In order to accurately identify
the deletions, NGS analysis was performed. A heterozygous deletion
of exons 2–21 in the proband was found by NGS. This mutation was
identified as a novel gross deletion mutation in the ATP7B
gene sequence, which has never been previously reported. The large
deletion was inherited from her father who was asymptomatic. There
is a single copy exons 2–21 of ATP7B in the patient. The
gross deletion mutation is predicted to cause the loss of almost
all functional domains, including copper binding domains,
transmembrane domains and the ATP loop (5). The present study found that the
pathogenic mutations p.Arg1,041Trp and the gross deletion were
compound heterozygous on two chromosomes in the proband, which were
inherited separately from her mother and father. The sequencing
analysis of the 868 bp band of the amplified breakpoint junction
showed a deletion of a 57,771 bp fragment (chr13:
52490972-52548742)(GRCh37) from partial exons 2–21 to external
ATP7B sequence (15.833 bp), without including additional
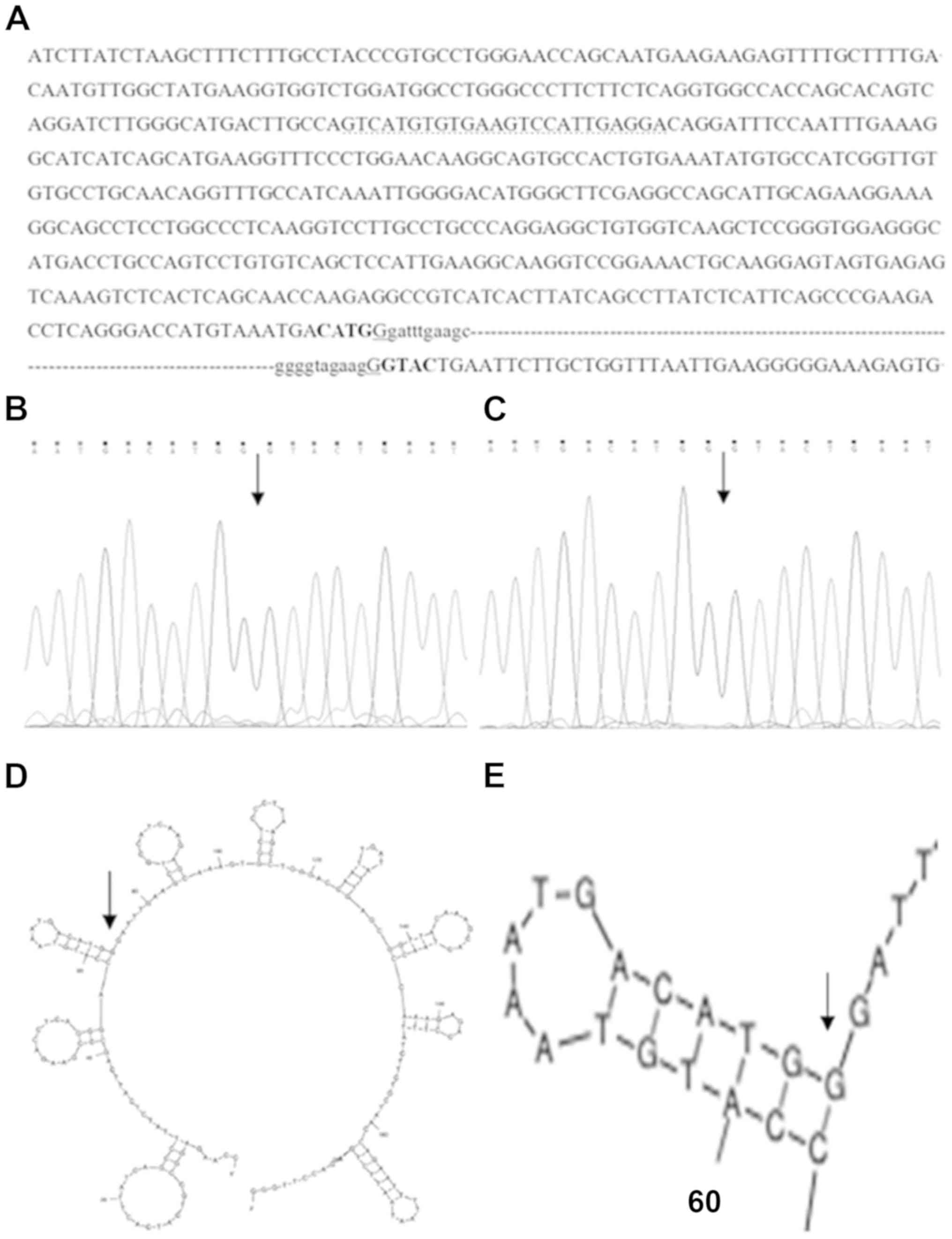
genes (Fig. 3). The sequence of
the breakpoint junction was confirmed in the proband and her father
(Fig. 4A-C). The probe sequence
(5′-GTCATGTGTGAAGTCCATTGAGGA-3′) of MLPA for exon 2 of ATP7B
binds within the remaining 562 bp of exon 2 (Fig. 4A), and for this reason MLPA failed
to detect the deletion of exon 2 in the ATP7B gene.
The sequence analysis of both sides of the
breakpoints identified microhomology of one base (G) next to
inverted repeat sequences CATG-GTAC (Fig. 4A).
Bioinformatics analysis of the 2,000 bp sequence
surrounding the two breakpoints showed no consequential elements.
The secondary structures of nucleotides for the 1,000 bp sequence
in the vicinity of proximal and distal breakpoints were predicted
that the proximal breakpoint was found in the proximity of a
hairpin structure (Fig. 4D and
E).
The deletion may be caused by the inverted repeat
sequences CATG-GTAC, which may lead to genomic instability, and by
the microhomology sequence of one base (G) at the proximal
breakpoint, which may interact with the base C on the complementary
strand in the proximity of the distal breakpoint, mediating
non-homologous end joining repair. The same heterozygous deletion
and breakpoint junction sequence were also present in the father of
the proband (Fig. 4C), suggesting
that the deletion event occurred in the family of the father of the
proband.
Discussion
Wilson disease (WD) is a rare autosomal recessive
genetic disorder that causes abnormal copper metabolism, which may
result in a pathological accumulation of copper in the liver, brain
and other organs (9). Diagnostic
criteria are based on symptoms, biochemical testing and the
presence of KF rings (10).
Patients with cirrhosis, neurological manifestations and KF rings
are diagnosed with classic WD. The type of clinical symptoms can be
highly variable in pediatric patients with WD, as hepatic symptoms
usually appear in late childhood and vary from asymptomatic,
exhibiting only biochemical abnormalities, to acute, presenting
liver failure and neurological or psychiatric manifestations that
generally occur in the second or third decade of life (11–13).
Laboratory diagnosis is based on low serum CER, increased 24-h
urine copper excretion and elevated liver copper levels (10). The presence of KF rings reflects
copper deposition in the brain (11). KF rings are usually absent in
pediatric patients with WD with liver disease in the earlier period
of the disease, and are not found in all patients with WD. KF rings
are not specific to WD, and have also been seen in other forms of
chronic liver diseases (14). When
symptoms are mild in the early stages of WD, genetic investigation
of ATP7B mutations can be valuable in confirming a diagnosis
of WD (11). Early diagnosis and
treatment can effectively prevent excessive copper accumulation and
tissue damage (15).
In the present study, a pediatric patient presented
with early-onset hepatic disease. The patient did not present
neurological or psychiatric manifestations, or KF rings. Laboratory
tests showed a low level of serum CER, and high levels of ALT and
AST. Biochemical investigations, including 24-h urine copper
excretion and hepatic biopsy, were not carried out due to technical
limitations. The early diagnosis of the patient with WD was made by
testing for ATP7B mutations. The early diagnosis of WD
allowed a timely treatment.
Previous molecular genetic analyses identified
>1,000 distinct mutations associated with WD worldwide, the most
common mutations are the point mutations, and other type of
mutations are rare (http://www.wilsondisease.med.ualberta.ca/database.asp).
The distribution of mutations in the ATP7B gene for WD are
associated with ethnicity or geographic origin (11). The most prevalent mutation of the
ATP7B among Europeans and US Caucasian is p.H1069Q in exon
14 (16,17), whereas the mutation p.R778L in exon
8 is most common in East Asia including China, South Korea and
Japan (18–20).
The present study identified a previously described
heterozygous mutation (p.Arg1,041Trp) and a heterozygous gross
deletion including exons 2–21. The Arg1,041Trp mutation was first
reported in an Italian patient in 1998 (8), and has never been reported previously
in China, whereas, according to our knowledge, the heterozygous
gross deletion including exons 2–21 of the ATP7B gene has
not been previously reported. In the present study, the mutations
p.Arg1,041Trp and the gross deletion including exons 2–21 could
alter the conformation of the ATP loop domain and all functional
domains, respectively (5); thus
resulting in a severe impairment of P-type ATPase function. The
gross deletion mutation in the patient may be classified as a
severe mutation, causing early hepatic disease onset. In the
present study, the pediatric patient lacked the typical symptoms of
WD and presented indeterminate biochemical results, which made
early diagnosis more difficult. Therefore, molecular analysis of
ATP7B mutations may facilitate an early diagnosis of WD. A
missense mutation was found in the homozygous state, due to the
large heterozygous gene deletion on its corresponding allele, but
no homologous mutations were inherited from the parents of the
patient. Molecular diagnosis of WD in the patient was carried out
using a combination of direct sequencing, MLPA and NGS. MLPA and
NGS were used for the detection of exonic deletions/duplications of
genomic DNA sequences. This analysis is of particular importance
when ≤1 mutation is detected in a patient with clinical symptoms of
WD, as the limitations of traditional Sanger sequencing mean large
deletions/duplications cannot be detected. MLPA can only detect
exon deletions/duplications because of restricted location of
probes. However, in the present study, NGS in combination with
Sanger sequencing was more effective than MLPA in detecting precise
deletions.
Microhomology sequences that are closely associated
with deletion mutations in the vicinity of deletion breakpoints
have previously been reported in the ATP7B gene, including
c.51 + 384_1708-953del (21),
c.1870-45_2355 + 189del (22),
c.4021 + 87_4125-2del (23),
c.3556 + 281_4001del, c.3134_3556 + 689del (24), c.532_574del (25), c.52-9329_1286-11del, c.2447 +
1612_2730 + 31delins12bp and c.3700-592_*409del (26). Alu repeat elements related to
deletion events have also been discovered in the ATP7B gene
[c.3134_3556 + 689del (24) and
c.52-2671_368del (27)]. In
addition, sequence analysis surrounding the breakpoints of the
deletion identified microhomology and inverted repeat sequences in
the patient enrolled in the present study, which may also mediate
deletion mutations in the ATP7B gene.
The present study reported the clinical and
molecular characterization of one Chinese pediatric patient with
early onset WD, with liver disease as the presenting feature. In
the patient, a heterozygous mutation (p. Arg1041Trp) and a
heterozygous gross deletion of a 57,771 bp fragment (chr13:
52490972-52548742) from partial exons 2–21 to external ATP7B
sequence (15.833bp) were identified through direct sequencing and
NGS. Microhomology and inverted repeat sequences were found in the
proband, which warrants further investigation of DNA break and
recombination mechanisms. To the best of our knowledge, the
Arg1041Trp mutation has not been previously reported in Chinese
patients, and the novel heterozygous gross deletion mutation has
not previously been identified, expanding the mutational spectrum
of the ATP7B gene. The present results suggested that the
NGS assay in combination with Sanger sequencing of the breakpoint
junctions may be more efficient than MLPA in detecting precise
deletions.
Acknowledgements
The authors would like to thank Dr Haijian-Cheng
(Beijing Kangso Medical Laboratory Co., Ltd., Beijing, China) for
genetic data analysis.
Funding
The present work was supported by the Scientific
Research Projects of Guizhou Health Commission in China (grant no.
gzwjkj2018-1-048).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
WLL, FL and LL carried out the genetic studies, the
data analysis and wrote the manuscript. ZXH performed the genetic
studies, the data analysis and manuscript revision. WC, HG and RA
contributed to the clinical diagnosis and the clinical analysis.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The study was approved by the Ethics Committees of
Affiliated Hospital of Guizhou Medical University (approval no.
201011). Written informed consent was obtained from the parents of
the patient.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Danks DM: Disorders of copper transport.
The metabolic basis of inherited diseases. Scriver CR, Beaudet AL,
Sly WS and Valle D: 1. 6th. McGraw-Hill; New York, NY: pp.
1416–1422. 1989
|
|
2
|
Scheinberg IH and Sternlieb I: Wilson
disease. Major Problems in Internal Medicine. Lloyd H and Smith J:
Saunders; Philadelphia, PA: 1984
|
|
3
|
Sternlieb I: Perspectives on Wilson's
disease. Hepatology. 12:1234–1239. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Petrukhin K, Fischer SG, Pirastu M, Tanzi
RE, Chernov I, Devoto M, Brzustowicz LM, Cayanis E, Vitale E and
Russo JJ: Mapping, cloning and genetic characterization of the
region containing the Wilson disease gene. Nat Genet. 5:338–343.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Terada K, Schilsky ML, Miura N and
Sugiyama T: ATP7B (WND) protein. Int J Biochem Cell Biol.
30:1063–1067. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Thomas GR, Forbes JR, Roberts EA, Walshe
JM and Cox DW: The Wilson disease gene: Spectrum of mutations and
their consequences. Nat. Genet. 9:210–217. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Piekarski J, Goldberg HI, Royal SA, Axel L
and Moss AA: Difference between liver and spleen CT numbers in the
normal adult: Its usefulness in predicting the presence of diffuse
liver disease. Radiology. 137:727–729. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Loudianos G, Dessì V, Lovicu M, Angius A,
Nurchi A, Sturniolo GC, Marcellini M, Zancan L, Bragetti P, Akar N,
et al: Further delineation of the molecular pathology of wilson
disease in the mediterranean population. Hum Mutat. 12:89–94. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Subramanian I, Vanek ZF and Bronstein JM:
Diagnosis and treatment of Wilson's disease. Curr Neurol Neurosci
Rep. 2:317–323. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Medici V, Rossaro L and Sturniolo GC:
Wilson disease-A practical approach to diagnosis, treatment and
follow-up. Dig Liver Dis. 39:601–609. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Das SK and Ray K: Wilson's disease: An
update. Nat Clin Pract Neurol. 2:482–493. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Brewer GJ and Askari FK: Wilson's disease:
Clinical management and therapy. J Hepatol. 42 (Suppl):S13–S21.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hedera P: Update on the clinical
management of Wilson's disease. Appl Clin Genet. 10:9–19. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mak CM and Lam CW: Diagnosis of Wilson's
disease: A comprehensive review. Crit Rev Clin Lab Sci. 45:263–290.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dalvi A and Padmanaban M: Wilson's
disease: Etiology, diagnosis, and treatment. Dis Mon. 60:450–459.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ferenci P: Regional distribution of
mutations of the ATP7B gene in patients with Wilson disease: Impact
on genetic testing. Hum Genet. 120:151–159. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shah AB, Chernov I, Zhang HT, Ross BM, Das
K, Lutsenko S, Parano E, Pavone L, Evgrafov O, Ivanova-Smolenskaya
IA, et al: Identification and analysis of mutations in the Wilson
disease gene (ATP7B): Population frequencies, genotype-phenotype
correlation, and functional analyses. Am J Hum Genet. 61:317–328.
1997. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu XQ, Zhang YF, Liu TT, Hsiao KJ, Zhang
JM, Gu XF, Bao KR, Yu LH and Wang MX: Correlation of ATP7B genotype
with phenotype in Chinese patients with Wilson disease. World J
Gastroenterol. 10:590–593. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim EK, Yoo OJ, Song KY, Yoo HW, Choi SY,
Cho SW and Hahn SH: Identification of three novel mutations and a
high frequency of the Arg778Leu mutation in Korean patients with
Wilson disease. Hum Mutat. 11:275–278. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Okada T, Shiono Y, Hayashi H, Satoh H,
Sawada T, Suzuki A, Takeda Y, Yano M, Michitaka K, Onji M and
Mabuchi H: Mutational analysis of ATP7B and genotype-phenotype
correlation in Japanese with Wilson's disease. Hum Mutat.
15:454–462. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Incollu S, Lepori MB, Zappu A, Dessì V,
Noli MC, Mameli E, Iorio R, Rannuci G, Cao A and Loudianos G: DNA
and RNA studies for molecular characterization of a gross deletion
detected in homozygosity intheNH2-terminal region of the ATP7B gene
in a Wilson disease patient. Mol Cell Probes. 25:195–198. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tatsumi Y, Shinohara T, Imoto M, Wakusawa
S, Yano M, Hayashi K, Hattori A, Hayashi H, Shimizu A, Ichiki T, et
al: Potential of the international scoring system for the diagnosis
of Wilson disease to differentiate Japanese patients who need
anti-copper treatment. Hepatol Res. 41:887–896. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Møller LB, Ott P, Lund C and Horn N:
Homozygosity for a gross partial gene deletion of the C-terminal
end of ATP7B in a Wilson patient with hepatic and no neurological
manifestations. Am J Med Genet A. 138:340–343. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Todorov T, Balakrishnan P, Savov A, Socha
P and Schmidt HH: Intragenic deletions in ATP7B as an unusual
molecular genetics mechanism of Wilson's disease pathogenesis. PLoS
One. 11:e01683722016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu G, Ma D, Cheng J, Zhang J, Luo C, Sun
Y, Hu P, Wang Y, Jiang T and Xu Z: Identification and
characterization of a novel 43-bp deletion mutation of the ATP7B
gene in a Chinese patient with Wilson's disease: A case report. BMC
Med Genet. 19:612018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen YC, Yu H, Wang RM, Xie JJ, Ni W,
Zhang Y, Dong Y and Wu ZY: Contribution of intragenic deletions to
mutation spectrum in Chinese patients with Wilson's disease and
possible mechanism underlying ATP7B gross deletions. Parkinsonism
Relat Disord. 62:128–133. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mameli E, Lepori MB, Chiappe F, Ranucci G,
Di Dato F, Iorio R and Loudianos G: Wilson's disease caused by
alternative splicing and Alu exonization due to a homozygous
3039-bp deletion spanning from intron 1 to exon 2 of the ATP7B
gene. Gene. 569:276–279. 2015. View Article : Google Scholar : PubMed/NCBI
|