Introduction
Endothelial-mesenchymal transition (EndMT) is a
similar process to epithelial-mesenchymal transition, which
involves a phenotypic conversion from endothelial cells to
mesenchymal cells. During this process, endothelial cells lose
their specific endothelial markers, including CD31, vascular
endothelial (VE)-cadherin and von Willebrand Factor (vWF), and
begin to express specific mesenchymal markers, including vimentin,
fibronectin, collagens and α smooth muscle actin (α-SMA) (1). EndMT has been confirmed to
participate in several cardiovascular diseases, including cardiac
fibrosis, atherosclerosis, atrial fibrillation and pulmonary
hypertension (2,3). For example, Kato et al
(4) reported the occurrence of
EndMT in the atria of patients with atrial fibrillation.
Circulating endothelial cells (CECs) represent
desquamated mature cells sloughed off from vessel walls in response
to endothelial injury. Previous studies have demonstrated that the
number of CECs may be a biomarker for several vascular disorders,
such as inflammatory vasculitis, cardiovascular diseases and
metabolic pathologies (5–7). CECs also have important roles in
tumor progression, and are involved in endothelial homeostasis and
angiogenesis (8). It has been
reported that increased counts of viable CECs are a marker of
progressive disease in patients with cancer (9). Freestone et al (10) suggested that the numbers of CECs
were increased in patients with atrial fibrillation and acute
vascular complications. Alongside vascular endothelial cells, CECs
may also undergo EndMT in response to injury (11). However, little is known about the
molecular mechanism of CECs undergoing EndMT.
Several signaling molecules contribute to the
process of EndMT, including transforming growth factor (TGF)-β,
epidermal growth factor, Wnt, Notch and bone morphogenetic proteins
(BMPs) (12). Among these, TGF-β1
has been identified as a potent inducer of EndMT in several
diseases (13,14). TGF-β indirectly phosphorylates
Smad2 and Smad3 through binding to type II TGF-β receptor, which
phosphorylates the type I receptor. The phosphorylated Smad2 and
Smad3 then interact with Smad4 and the complex containing Smad2,
Smad3 and Smad4 was translocate into the nucleus, regulating the
transcription of target genes. It has been demonstrated that
TGF-β1-induced EndMT can be inhibited by BMP-7, which belongs to
the TGF-β superfamily (15). BMP7
can inhibit Smad2/3 phosphorylation through phosphorylating Smad1,
5 and 8 (16). In the present
study, the effects of recombinant human BMP-7 (rhBMP-7) on
TGF-β1-induced EndMT in CECs were assessed. Additionally, the role
of Smad5 in the EndMT process of CECs regulated by rhBMP-7 was
further investigated.
Materials and methods
Cell isolation, culture and
treatments
Peripheral blood samples (100 ml) were collected
from 10 healthy volunteers (6 male and 4 female, age range 20–40
years) at Taizhou Hospital (Taizhou, China) between October 2017
and March 2018, once informed consent was obtained. The present
study was approved by the ethics committee of Taizhou Hospital. CEC
isolation was performed according to previous studies (9,17).
Briefly, after discarding the first 3–5 ml peripheral blood drawn
through venipuncture, the remaining blood was incubated with
magnetic beads conjugated to a monoclonal antibody against CD146
(also known as Sendo-I, cat. no. 361036, BioLegend, Inc.). PBS-BSA
(0.1%, Shanghai Fanke Biotechnology Co., Ltd.) was used to rinse
the bead-bound cell fraction. The viable endothelial cells within
the isolate were quantified using a fluorescence microscope after
staining with CalceinAM (Sigma-Aldrich; Merck KGaA). The selected
bead-bound CECs were isolated using cytospin (5 min at 100 × g) on
glass slides at 37°C. The primary antibodies against CD31 (1:20;
cat. no. ab28364; Abcam), von Willebrand factor (vWF) (1:400; cat.
no. ab6994; Abcam) and vascular endothelial growth factor
(VEGF)-receptor 2 (VEGF-R2) (1:50; cat. no. ab2349; Abcam) were
used for the phenotypic analysis of CECs by immunofluorescence.
CECs were co-cultured with an endothelial feeder
layer as described in a previous study (9). Human umbilical vein endothelial cells
(HUVECs; Shanghai Hongshun Biological Technology Co., Ltd.;
7×104/well) were used as the feeder layer.
Carboxyfluorescein diacetate succinimidyl ester-labeled CECs (500
cells; Bio-Rad Laboratories, Inc.), obtained by incubating
Carboxyfluorescein diacetate succinimidyl ester with CECs for 30
min at 37°C, were co-cultured with HUVECs (7×104/well)
in EGM-2 endothelial growth medium (Beijing Fubo Biotechnology Co.,
Ltd.) in a 6-well plate, which was pre-coated with fibronectin. For
the treatments, CECs were divided into various groups: Control,
TGF-β1 (10 ng/ml rhTGF-β1; 24 h; 37°C; cat. no. GF346;
Sigma-Aldrich; Merck KGaA) treatment group, TGF-β1 (10 ng/ml) +
rhBMP (1 or 10 or 100 ng/ml; 24 h; 37°C; cat. no. 354-BP-010;
R&D Systems, Inc.) treatment group, and TGF-β1 (10 ng/ml) +
rhBMP (100 ng/ml) + Jun activation domain-binding protein 1 (JAB1;
20 ng/ml; 24 h; 37°C; cat. no. H00010987-P01; Abnova) treatment
group.
Reverse transcription quantitative
(RT-q)PCR
Total RNA was extracted from CECs using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) following the manufacturer's instructions. Following
quantification by NanoDrop (Thermo Fisher Scientific, Inc.), RNA
was used as a template to synthesize cDNA using qRT SuperMix
(Vazyme). The temperature protocol was as follows: 70°C for 3 min,
42°C for 60 min and 70°C for 15 min. Subsequently, RT-qPCR was
carried out using SYBR Green kits (Takara Biotechnology Co., Ltd.,
cat. no. RR820Q) and an Applied Biosystems 7500 Real-Time PCR
system (Applied Biosystems; Thermo Fisher Scientific, Inc.). The
reaction steps were: 95°C for 30 sec; 40 cycles of 95°C for 5 sec
and 60°C for 40 sec. Primer sequences used in RT-qPCR are presented
in Table I. Each experiment was
performed three times. The 2−ΔΔCq method was used to
calculate relative gene expression (18).
 | Table I.Primer sequences used for reverse
transcription-quantitative PCR. |
Table I.
Primer sequences used for reverse
transcription-quantitative PCR.
| Gene | Forward primer
(5′-3′) | Reverse primer
(5′-3′) |
|---|
| vWF |
TGCAACACTTGTGTCTGTCG |
CGAAAGGTCCCAGGGTTACT |
| E-selectin |
AAAGAGAGTGGAGCCTGGTC |
CCTACCCAGACCCACACATT |
| VE-cadherin |
TACCAGGACGCTTTCACCAT |
AAAGGCTGCTGGAAAATGGG |
| Vimentin |
GAGTCCACTGAGTACCGGAG |
ACGAGCCATTTCCTCCTTCA |
| Fibronectin |
GTATACGAGGGCCAGCTCAT |
CCCAGGAGACCACAAAGCTA |
| α-SMA |
ACCCAGCACCATGAAGATCA |
TTTGCGGTGGACAATGGAAG |
| GAPDH |
CCATCTTCCAGGAGCGAGAT |
TGCTGATGATCTTGAGGCTG |
Western blot analysis
Total proteins in CECs were extracted using ice-cold
cell extraction buffer (Invitrogen; Thermo Fisher Scientific,
Inc.). Protein concentration was quantified using the BCA Protein
Assay kit (Takara Biotechnology Co., Ltd.). Subsequently, equal
amounts of protein (20 µg) were separated by 10% SDS-PAGE and
transferred onto polyvinylidene difluoride membranes (EMD
Millipore). The membranes were blocked in 5% skimmed milk for 2 h
at 37°C and then incubated with primary antibodies, including
anti-vWF (1:2,000; cat. no. ab218333; Abcam), anti-E-selectin
(1:1,000; cat. no. ab18981; Abcam), anti-VE-cadherin (1:2,000; cat.
no. ab33168; Abcam), anti-vimentin (1:1,000; cat. no. ab45939;
Abcam), anti-fibronectin (1:2,000; cat. no. ab2413 Abcam),
anti-α-SMA (1:2,000; cat. no. ab5694; Abcam), anti-Smad2 (1:2,000;
cat. no. ab40855; Abcam), anti-Smad3 (1:1,000; cat. no. ab40854;
Abcam) and anti-β-actin (1:2,000; cat. no. ab8227; Abcam) overnight
at 4°C. The secondary antibody used was a species appropriate
horseradish peroxidase (HRP)-conjugated secondary antibody
(1:2,000; cat. no. ab7090) Abcam). Immunoreactive bands were
visualized with the ECL detection system (EMD Millipore) and
analyzed by ImageJ v1.8 (National Institutes of Health).
Immunofluorescence staining
Following exposure to 4% paraformaldehyde for 30 min
at 37°C, the CECs were co-incubated with antibodies against
VE-cadherin (1:1,000; cat. no. ab33168; Abcam) or vimentin
(1:1,000; cat. no. ab45939; Abcam), or CD31 (1:20; cat. no.
ab28364; Abcam), von Willebrand factor (vWF) (1:400; cat. no.
ab6994; Abcam) and vascular endothelial growth factor
(VEGF)-receptor 2 (VEGF-R2) (1:50; cat. no. ab2349; Abcam) at 4°C
overnight. The samples were washed with PBS three times and
incubated with the secondary HRP-conjugated immunoglobulin G
antibody (1:1,000; cat. no. ab7090; Abcam) at room temperature for
2 h, then incubated with 100 ng/ml DAPI (Sigma-Aldrich; Merck KGaA)
for 10 min at 37°C to stain nuclei. For VE-cadherin and vimentin
expression, fluorescence microscopy (Nikon Corporation,) and
Image-Pro Plus v6.0 (Media Cybernetics, Inc.) were used.
ELISA of type I collagen content
CECs were cultured in six-well plates for 24 h and
were then subjected to different treatments. The supernatants were
collected to measure type I collagen content using a COL-I ELISA
kit (Shanghai Walan Biotech Co., Ltd., cat. no. ABE10204) according
to the manufacturer's protocol. Absorbance at 450 nm was determined
using a microplate reader. Type I collagen concentration was
calculated according to a standard curve.
Statistical analysis
Data are expressed as the mean ± standard deviation.
Statistical analysis was performed using SPSS v16.0 (SPSS Inc.).
The differences between two groups were evaluated by Student's
t-test or one-way ANOVA followed by Tukey's post hoc test was used
for analyzing the differences of multiple groups. P<0.05 was
considered statistically significant.
Results
Isolation and identification of
CECs
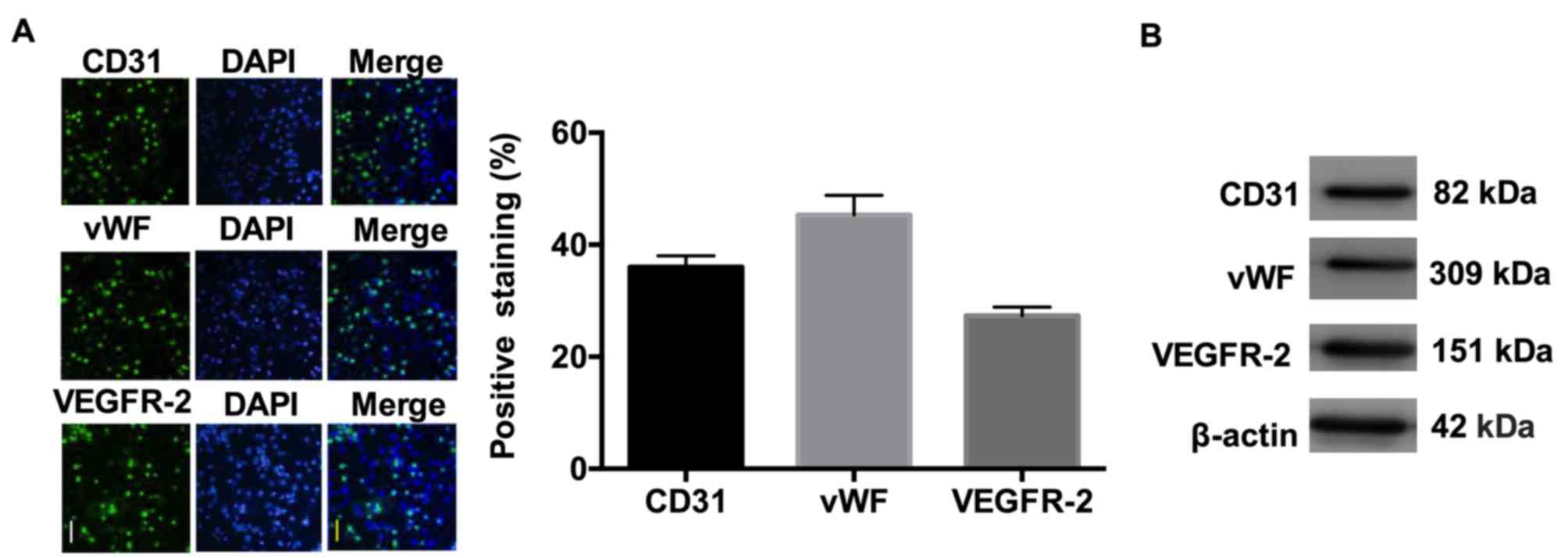
CECs were isolated by anti-CD146-coupled magnetic
beads and were identified by immunofluorescence using antibodies
against CD31, vWF and VEGF-R2. As demonstrated in Fig. 1, the isolated CECs were positive
for expression of CD31, vWF and VEGF-R2.
rhBMP-7 attenuates TGF-β1-induced
endothelial cell injury
rhBMP-7 shares a similar signal transduction
mechanism with TGF-β1, and can inhibit EndMT and collagen synthesis
(19). Endothelial cells were
treated with TGF-β1 or TGF-β1 + rhBMP-7 to elucidate the effect of
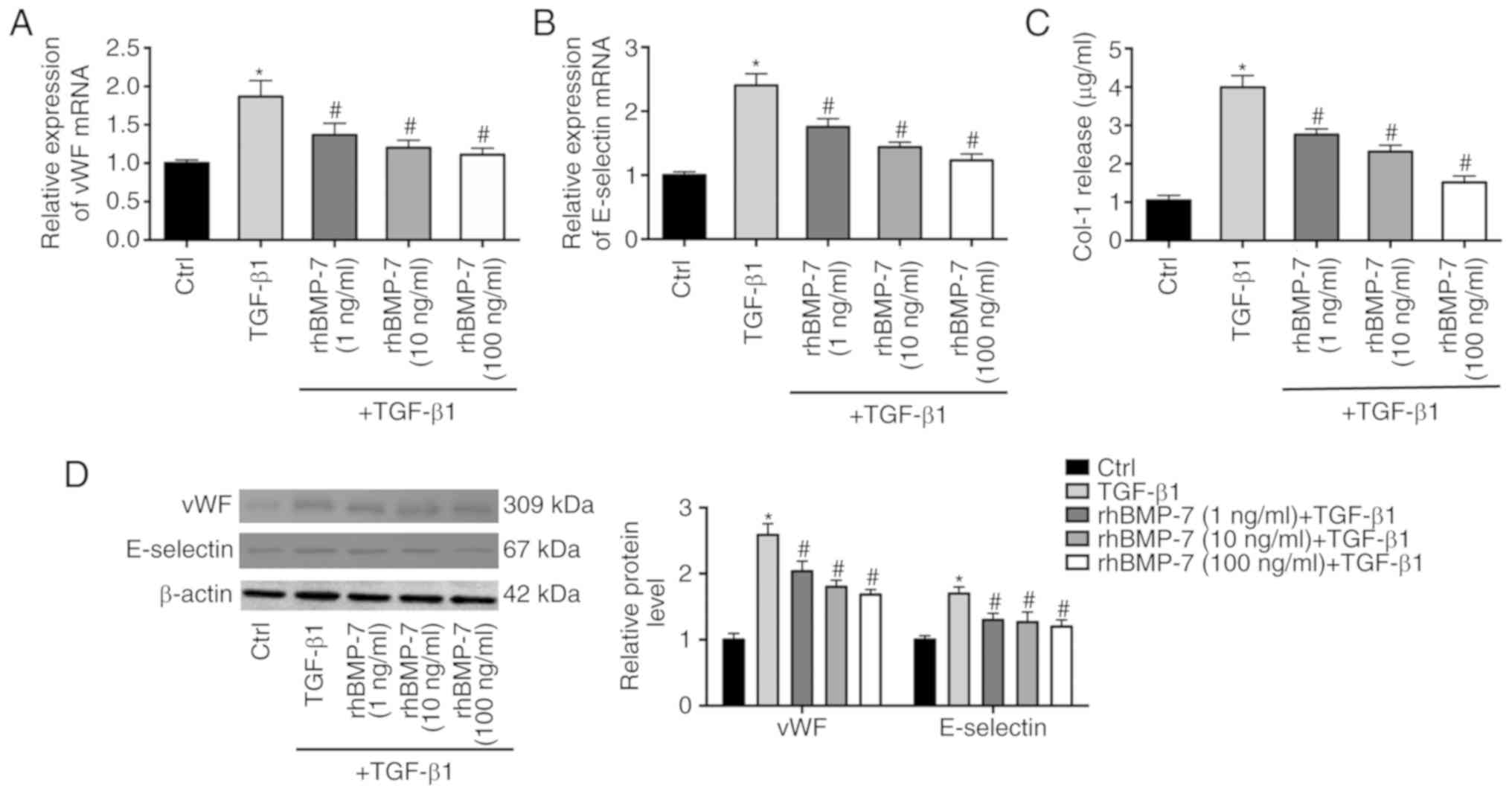
rhBMP-7 on TGF-β1-induced endothelial cell injury. vWF and
E-selectin are known biomarkers of endothelial cell injury. As
demonstrated in Fig. 2A and B,
TGF-β1 increased the mRNA expression levels of vWF and E-selectin
in endothelial cells, which was decreased by rhBMP-7 in a
dose-dependent manner. Western blotting further demonstrated the
effects of TGF-β1 and rhBMP-7 on the protein levels of vWF and
E-selectin (Fig. 2D). In addition,
the expression of type I collagen was upregulated by TGF-β1
treatment, whereas rhBMP-7 reduced its expression (Fig. 2C).
rhBMP-7 inhibits TGF-β1-induced
EndMT
To detect TGF-β1-induced EndMT, an
immunofluorescence assay for endothelial marker VE-cadherin and
mesenchymal marker vimentin expression was performed. As
demonstrated in Fig. 3A,
VE-cadherin and vimentin were co-expressed in TGF-β1-treated
endothelial cells, indicating the occurrence of EndMT induced by
TGF-β1. RT-qPCR results demonstrated that TGF-β1 greatly reduced
the mRNA expression levels of VE-cadherin and induced the mRNA
expression levels of vimentin, fibronectin and α-SMA (Fig. 3B-E). rhBMP-7 reversed the effects
of TGF-β1 on the expression of these genes, indicating that rhBMP-7
inhibited TGF-β1-induced EndMT. These results were further
validated by western blot analysis (Fig. 3F).
 | Figure 3.rhBMP-7 inhibits TGF-β1-induced
endothelial to mesenchymal transition. (A) Immunofluorescence assay
for VE-cadherin and vimentin (magnification, ×100). Endothelial
cells were treated with TGF-β1 or TGF-β1 + rhBMP-7. The mRNA
expression levels of (B) VE-cadherin, (C) vimentin, (D) fibronectin
and (E) α-SMA were detected in these cells. (F) Protein levels of
VE-cadherin, vimentin, fibronectin and α-SMA were detected in these
cells. n=3; *P<0.05 vs. Ctrl group; #P<0.05 vs.
TGF-β1 group. α-SMA, α smooth muscle actin; Ctrl, control; rhBMP-7,
recombinant human bone morphogenetic protein 7; TGF, transforming
growth factor; VE, vascular endothelial. |
Smad5 antagonist reverses the effect
of rhBMP-7 on TGF-β1-induced EndMT
To elucidate the mechanism underlying the effects of
Smad5 on rhBMP-7 inhibiting TGF-β1-induced EndMT, the Smad5
antagonist Jab-1 was used to treat endothelial cells in the TGF-β1
+ rhBMP-7 group. VE-cadherin expression was reduced by Jab-1, which
conversely increased the expression of vimentin, fibronectin and
α-SMA (Fig. 4). These results
indicated that the Smad5 antagonist reversed the effect of rhBMP-7
on TGF-β1-induced EndMT.
Smad5 antagonist upregulates
rhBMP-7-inhibited Smad2/3 expression
The present study further analyzed whether Smad2 and
Smad3 expression was mediated by rhBMP-7 and Smad5. As demonstrated
in Fig. 5, rhBMP-7 mitigated
TGF-β1-induced Smad2 and Smad3 expression. Conversely, Jab-1
increased the expression of Smad2 and Smad3 inhibited by rhBMP-7.
These data suggested that the Smad5 antagonist reversed
rhBMP-7-induced inhibition of Smad2/3 expression.
Discussion
CECs are identified by morphological features and
the presence of endothelial markers, including vWF, CD146 and CD31.
Mounting evidence has demonstrated that high levels of CECs might
indicate considerable damage to the endothelial cell layer
(20,21). Additionally, endothelial
dysfunction has been demonstrated to be involved in diverse
cardiovascular diseases (22).
Increased numbers of CECs have been observed in several
cardiovascular diseases, including hypertension, heart failure,
peripheral vascular disease and atrial fibrillation (10,23,24).
Wang et al (25) identified
that CEC numbers were higher in patients with acute myocardial
infarction compared with healthy controls. Patients with venous
thromboembolism also demonstrate higher numbers of CECs, as well as
vWF and vascular cell adhesion molecule 1 (26). Woywodt et al (27) demonstrated that CECs can serve as
markers of endothelial damage or repair in stroke. CECs are also
related to the pathogenesis of other diseases. Lombardo et
al (7) suggested that type 2
diabetes mellitus increases the count of CECs in peripheral blood.
A high CEC count is also correlated with shorter overall survival
and progression-free survival in patients with non-small cell lung
cancer (28).
Recently, Agarwal et al (11) identified that CECs undergo EndMT
following migration to the wound site of musculoskeletal injury,
which suggests the potential of CECs as a target to prevent
EndMT-related pathologies. However, there are few studies on the
molecular mechanism of CECs undergoing EndMT. EndMT is a phenomenon
occurring under several pathological conditions, including cardiac
fibrosis (29). EndMT may be an
important source of mesenchymal cells, which exhibit a high
migratory potential and increased extracellular matrix production.
In addition, EndMT can cause endothelial dysfunction during
inflammatory conditions (5) It has
been demonstrated that TGF-β and the BMP family of growth factors
are the best-studied mediators of EndMT via Smad-dependent and
Smad-independent pathways (12).
The present study identified that TGF-β1 induced
endothelial cell injury and EndMT in CECs. TGF-β1 significantly
reduced VE-cadherin expression, and induced the expression of
vimentin, fibronectin and α-SMA. To further investigate the
mechanism underlying TGF-β1-induced EndMT in CECs, CECs were
further exposed to rhBMP-7. The present study demonstrated that
rhBMP-7 inhibited TGF-β1-induced EndMT in CECs. Consistent with
these results, several studies have demonstrated that BMP-7
suppresses EndMT in vivo and in vitro (19,30).
Zhang et al (19) reported
that BMP-7 inhibits hypoxia-induced EndMT in pulmonary artery
endothelial cells and in experimental models of pulmonary artery
hypertension. BMP-7 treatment has also been reported to have a
positive impact on the severity of liver disease by attenuating
EndMT (31). Furthermore,
supplementation of exogenous rhBMP-7 effectively ameliorated EndMT
and experimental endocardial fibroelastosis in rats (30).
Mechanistically, BMP-7 is capable of suppressing
Smad2/3 phosphorylation through phosphorylating Smad1, 5 and 8.
BMP-7-induced inhibition of mesenchymal markers requires Smad5 in
mesangial cells (32). In the
present study, the Smad5 antagonist Jab-1 was used to treat
endothelial cells in the TGF-β1 + rhBMP-7 group. It was identified
that the Smad5 antagonist reversed the effects of rhBMP-7 on
TGF-β1-induced EndMT. Furthermore, the Smad5 antagonist reversed
the inhibitory effects of rhBMP-7 on Smad2/3 expression. These data
suggested that rhBMP-7 may suppress TGF-β1-induced EndMT in CECs
through regulating Smad5.
In summary, this study revealed that TGF-β could
induce EndMT in CECs, and rhBMP-7 could suppress this process by
regulating Smad5. These data suggested a therapeutic target
associated with the inhibition of EndMT in CECs for cardiovascular
diseases.
Acknowledgements
Not applicable.
Funding
The present study was supported by Zhejiang Medical
and Health Science and Technology Plan (grant no. 2017KY164) and
Taizhou Science and Technology Plan Class A (grant no.
1601KY75).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
WG and JJ designed and performed the research. YM,
SX, TL and YL performed the cell experiments. WG analyzed the data
and wrote the paper.
Ethics approval and consent to
participate
The present study was approved by the ethics
committee of Taizhou Hospital. Informed consent was obtained from
all individuals that participated in this study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Jiang Y, Zhou X, Hu R and Dai A:
TGF-β1-induced SMAD2/3/4 activation promotes RELM-β transcription
to modulate the endothelium-mesenchymal transition in human
endothelial cells. Int J Biochem Cell Biol. 105:52–60. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jackson AO, Zhang J, Jiang Z and Yin K:
Endothelial-to-mesenchymal transition: A novel therapeutic target
for cardiovascular diseases. Trends Cardiovasc Med. 27:383–393.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sato Y and Nakanuma Y: Role of
endothelial-mesenchymal transition in idiopathic portal
hypertension. Histol Histopathol. 28:145–154. 2013.PubMed/NCBI
|
|
4
|
Kato T, Sekiguchi A, Sagara K, Tanabe H,
Takamura M, Kaneko S, Aizawa T, Fu LT and Yamashita T:
Endothelial-mesenchymal transition in human atrial fibrillation. J
Cardiol. 69:706–711. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kluz J, Kopeć W, Jakobsche-Policht U and
Adamiec R: Circulating endothelial cells, endothelial apoptosis and
soluble markers of endothelial dysfunction in patients with
systemic lupus erythematosus-related vasculitis. Int Angiol.
28:192–201. 2009.PubMed/NCBI
|
|
6
|
Rakic M, Persic V, Kehler T, Bastiancic
AL, Rosovic I, Laskarin G and Sotosek Tokmadzic V: Possible role of
circulating endothelial cells in patients after acute myocardial
infarction. Med Hypotheses. 117:42–46. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lombardo MF, Iacopino P, Cuzzola M,
Spiniello E, Garreffa C, Ferrelli F, Coppola A, Saccardi R,
Piaggesi A, Piro R, et al: Type 2 diabetes mellitus impairs the
maturation of endothelial progenitor cells and increases the number
of circulating endothelial cells in peripheral blood. Cytometry A.
81:856–864. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
O'Reilly M, Holmgren L, Shing Y, Chen C,
Rosenthal RA, Cao Y, Moses M, Lane WS, Sage EH and Folkman J:
Angiostatin: A circulating endothelial cell inhibitor that
suppresses angiogenesis and tumor growth. Cold Spring Harb Symp
Quant Biol. 59:471–482. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Beerepoot LV, Mehra N, Vermaat JS,
Zonnenberg BA, Gebbink MF and Voest EE: Increased levels of viable
circulating endothelial cells are an indicator of progressive
disease in cancer patients. Ann Oncol. 15:139–145. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Freestone B, LiP GH, Chong A, Nadar S, Lee
KW and Blann AD: Circulating endothelial cells in atrial
fibrillation with and without acute cardiovascular disease. Thromb
Haemost. 94:702–706. 2005.PubMed/NCBI
|
|
11
|
Agarwal S, Loder S, Cholok D, Peterson J,
Li J, Fireman D, Breuler C, Hsieh HS, Ranganathan K, Hwang C, et
al: Local and circulating endothelial cells undergo endothelial to
mesenchymal transition (EndMT) in response to musculoskeletal
injury. Sci Rep. 6:325142016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu JU, Dong F, Jeong J, Masuda T and Lobe
CG: Constitutively active Notch1 signaling promotes
endothelial-mesenchymal transition in a conditional transgenic
mouse model. Int J Mol Med. 34:669–676. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang Z, Han Z, Tao J, Wang J, Liu X, Zhou
W, Xu Z, Zhao C, Wang Z, Tan R and Gu M: Role of
endothelial-to-mesenchymal transition induced by TGF-β1 in
transplant kidney interstitial fibrosis. J Cell Mol Med.
21:2359–2369. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yang W, Li X, Qi S, Li X, Zhou K, Qing S,
Zhang Y and Gao MQ: lncRNA H19 is involved in TGF-β1-induced
epithelial to mesenchymal transition in bovine epithelial cells
through PI3K/AKT signaling pathway. PeerJ. 5:e39502017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xu Y, Wan J, Jiang D and Wu X: BMP-7
counteracts TGF-beta1-induced epithelial-to-mesenchymal transition
in human renal proximal tubular epithelial cells. J Nephrol.
22:403–410. 2009.PubMed/NCBI
|
|
16
|
Manson SR, Austin PF, Guo Q and Moore KH:
BMP-7 signaling and its critical roles in kidney development, the
responses to renal injury, and chronic kidney disease. Vitam Horm.
99:91–144. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Beerepoot LV, Mehra N, Linschoten F, Jorna
AS, Lisman T, Verheul HM and Voest EE: Circulating endothelial
cells in cancer patients do not express tissue factor. Cancer Lett.
213:241–248. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang H, Liu Y, Yan L, Du W, Zhang X,
Zhang M, Chen H, Zhang Y, Zhou J, Sun H and Zhu D: Bone
morphogenetic protein-7 inhibits endothelial-mesenchymal transition
in pulmonary artery endothelial cell under hypoxia. J Cell Physiol.
233:4077–4090. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gendron N and Smadja DM: Circulating
endothelial cells: A new biomarker of endothelial dysfunction in
hematological diseases. Ann Biol Clin (Paris). 74:395–404.
2016.PubMed/NCBI
|
|
21
|
Erdbruegger U, Dhaygude A, Haubitz M and
Woywodt A: Circulating endothelial cells: Markers and mediators of
vascular damage. Curr Stem Cell Res Ther. 5:294–302. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sampson UK, Engelgau MM, Peprah EK and
Mensah GA: Endothelial dysfunction: A unifying hypothesis for the
burden of cardiovascular diseases in sub-Saharan Africa. Cardiovasc
J Afr. 26 (2 Suppl 1):S56–S60. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Blann AD, Seigneur M, Steiner M, Boisseau
MR and McCollum CN: Circulating endothelial cell markers in
peripheral vascular disease: RelationshiPto the location and extent
of atherosclerotic disease. Eur J Clin Invest. 27:916–921. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Martínez-Sales V, Sánchez-Lázaro I, Vila
V, Almenar L, Contreras T and Reganon E: Circulating endothelial
cells in patients with heart failure and left ventricular
dysfunction. Dis Markers. 31:75–82. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang C, Li H, Fu P, Zhang S and Xiu R:
Serum C-reactive protein and circulating endothelial cells in
patients with acute myocardial infarction. Clin Hemorheol
Microcirc. 32:287–296. 2005.PubMed/NCBI
|
|
26
|
Torres C, Matos R, Morais S, Campos M and
Lima M: Soluble endothelial cell molecules and circulating
endothelial cells in patients with venous thromboembolism. Blood
Coagul Fibrinolysis. 28:589–595. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Woywodt A, Gerdes S, Ahl B, Erdbruegger U,
Haubitz M and Weissenborn K: Circulating endothelial cells and
stroke: Influence of stroke subtypes and changes during the course
of disease. J Stroke Cerebrovasc Dis. 21:452–458. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ilie M, Long E, Hofman V, Selva E,
Bonnetaud C, Boyer J, Vénissac N, Sanfiorenzo C, Ferrua B,
Marquette CH, et al: Clinical value of circulating endothelial
cells and of soluble CD146 levels in patients undergoing surgery
for non-small cell lung cancer. Br J Cancer. 110:1236–1243. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Geng H and Guan J: MiR-18a-5p inhibits
endothelial-mesenchymal transition and cardiac fibrosis through the
Notch2 pathway. Biochem Biophys Res Commun. 491:329–336. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xu X, Friehs I, Zhong Hu T, Melnychenko I,
Tampe B, Alnour F, Iascone M, Kalluri R, Zeisberg M, Del Nido PJ
and Zeisberg EM: Endocardial fibroelastosis is caused by aberrant
endothelial to mesenchymal transition. Circ Res. 116:857–866. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ribera J, Pauta M, Melgar-Lesmes P,
Córdoba B, Bosch A, Calvo M, Rodrigo-Torres D, Sancho-Bru P, Mira
A, Jiménez W and Morales-Ruiz M: A small population of liver
endothelial cells undergoes endothelial-to-mesenchymal transition
in response to chronic liver injury. Am J Physiol Gastrointest
Liver Physiol. 313:G492–G504. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang S and Hirschberg R: Bone
morphogenetic protein-7 signals opposing transforming growth factor
beta in mesangial cells. J Biol Chem. 279:23200–23206. 2004.
View Article : Google Scholar : PubMed/NCBI
|