Introduction
Chromium is the sixth most abundant element in the
earth's crust. Trivalent Cr(III) and hexavalent Cr(VI) chromium are
the most stable oxidative states occurring in nature. Cr(VI) is
more cytotoxic than Cr(III) due to its stronger oxidizing capacity
and rapid absorption by the cells through non-specific anion
carriers (1,2). Toxic Cr(VI) can be released into the
environment through soil, sea water, and fresh water. As a result,
individuals may be at a risk of exposure to Cr(VI) when consuming
contaminated drinking water (3,4). The
development of an optimal treatment scheme against acute or chronic
Cr(VI) exposure remains a critical issue.
When Cr(VI) enters the cells, it can be rapidly
reduced into Cr(III), resulting in the production of several
reactive chromium intermediates and reactive oxygen species (ROS),
all of which are responsible for altering normal cellular function
and promoting apoptosis (5,6).
Potassium dichromate (K2Cr2O7) is
a toxic compound of Cr(VI) that has been demonstrated to induce
nephrotoxicity in humans and animals (7). In a clinical setting, acute and
chronic Cr(VI) exposure can cause severe damage to proximal renal
tubular cells and result in significant renal function
deterioration, thereby requiring hemodialysis (8–11).
Cr(VI) toxicity causes acute tubular necrosis, whereby Cr(VI)
directly damages the tubular epithelium (12). Less frequently, Cr(VI) also causes
interstitial renal injury (13,14).
Furthermore, Cr exposure can lead to hepatorenal syndrome, severe
coagulopathy, and intravascular hemolysis that may indirectly
contribute to the aggravation of renal dysfunction (13).
Cr(VI) induces free radical production by
Fenton-type or Haber-Weiss reaction or by reacting directly with
cellular molecules, triggering multiple possible apoptotic
signaling pathways in various cell types (15,16).
Several studies have suggested that Cr(VI) toxicity induced cell
apoptosis mainly via intrinsic mitochondrial pathways in several
types of cells, including human tumor cell lines, anterior
pituitary cells, hepatocytic cells, and also in a rat model
(5,17–19).
Although ROS is also linked to the extrinsic apoptotic pathway
(20), it is not well understood
whether the extrinsic apoptotic pathway is induced in renal cells
post-Cr(VI) exposure. The aim of the present study was to
investigate the activation of the extrinsic apoptotic pathway in
Cr(VI)-exposed HK-2 human immortalized proximal tubular epithelial
cell line HK-2.
Materials and methods
HK-2 cell culture
HK-2 cells (ATCC CRL-2190), an immortalized proximal
tubular epithelial cell line derived from normal adult human
kidneys, were purchased from the American Type Culture Collection.
Culture conditions have been described in our previous study
(21).
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT)
assay for cell viability
To determine the toxicity of chromium on kidney
cells, HK-2 cells (1×104 cells) were seeded in each well
of a 96-well culture plate (Falcon; BD Biosciences) in triplicates.
The cells were exposed to potassium dichromate
(K2Cr2O7; Sigma-Aldrich; Merck
KGaA), which was used as the source of chromium (VI), at
concentrations of 0.1, 0.3, 1, 10, 30, and 100 µM for 24, 48, and
72 h. Cell viability was directly examined by an Eclipse Ti-U
inverted microscope (Nikon Corporation) at a magnification of ×400
and indirectly assayed by an MTT assay (Sigma-Aldrich; Merck KGaA)
according to the manufacturer's instructions. The absorbance at 570
nm was determined using a microplate reader (Multiskan EX;
LabSystems).
Oxidative stress assay
HK-2 cells (1×106) were incubated in
10-cm dishes with 5 µM 2′7′-dichlorofluorescein diacetate
(Sigma-Aldrich; Merck KGaA) at 37°C for 30 min. After
centrifugation (1,000 × g) and washing in phosphate-buffered
saline, the HK-2 cells were exposed to 10 µM
K2Cr2O7 each in triplicates. After
30 min of incubation, the fluorescence intensity, which is
correlated to the concentration of hydroxyl radicals, was detected
by Partec CyFlow (Sysmex Partec GmbH). The data were analyzed by
FCS Express 4 Flow Cytometry (De Novo Software). All procedures
were performed in the dark on ice.
Western blotting
Protein lysates were extracted using
radioimmunoprecipitation lysis buffer (Amresco, LLC) containing 1%
proteinase inhibitor on ice for 20 min. Proteins were quantified
using the protein assay kit (based on Bradford method, Bio-Rad
Laboratories, Inc.) and 30 µg protein per lane was loaded and
separated by 10% sodium dodecyl sulfate-polyacrylamide
electrophoresis at 100 V for 1 h. The separated proteins were
transferred onto Hybond-P polyvinylidene difluoride membranes
(Amersham Biosciences; GE Healthcare) and blocked with 3% bovine
serum albumin in Tris-buffered saline containing Tween-20 for 1 h.
The membranes were incubated overnight at 4°C with commercial
primary monoclonal antibodies against poly (ADP-ribose) polymerase
(PARP; catalogue number 9542S, 1:1,000; Cell Signaling Technology,
Inc.), pro-caspase-3 (catalogue number 06–735, 1:1,000; EMD
Millipore), cleaved caspase-3 (catalogue number C8487, 1:1,000;
Sigma-Aldrich; Merck KGaA), Bcl-2 (catalogue number 658702,
1:1,000; BioLegend, Inc.), Bax (catalogue number 5023, 1:1,000;
Cell Signaling Technology, Inc.), cytochrome c (catalogue
number 05–479, 1:500; EMD Millipore), Fas ligand (FasL; catalogue
number 68405, 1:500), apoptosis-inducing factor (AIF; catalogue
number 4642, 1:1,000), pro-caspase-8 (catalogue number 9746S,
1:1,000) and cleaved caspase-8 (catalogue number 9496S, 1:1,000;
all from Cell Signaling Technology, Inc.). Then, the membranes were
incubated with goat anti-mouse IgG horseradish peroxidase
(HRP)-conjugated antibodies (catalogue number 405306, 1:5,000) or
anti-rabbit IgG HRP-conjugated antibodies (catalogue number 410406,
1:10,000; both from BioLegend, Inc.) at room temperature for 1 h.
β-actin (catalog number MAB1501, 1:1,000; Chemicon; EMD Millipore)
was used as an endogenous control and detected by the same
secondary antibodies as aforementioned. Target proteins were
visualized using Clarity™ Western ECL Substrate (Bio-Rad
Laboratories, Inc.) and HyBlot CL film (Denville Scientific, Inc.).
The intensities of the bands were quantified with ImageJ software
1.52a (NIH).
Statistical analysis
All results were expressed as the mean ± standard
deviation (SD) of at least three independent experiments. Data were
analyzed by analysis of variance (ANOVA) using SPSS20 software (IBM
Corp.). Scheffe's test was used for post hoc analysis to compare
all pairs of groups of the ANOVA test. Results were considered
statistically significant at a value of P<0.05.
Results
Cr(VI) exposure decreases cell
viability and alters the morphology of HK-2 cells
To evaluate the toxic effect of Cr(VI) on renal
tubular cells, a normal human proximal tubule epithelial cell line
HK-2 was treated with different concentrations of
K2Cr2O7, which produced Cr(VI) in
aqueous solution. Various concentrations of
K2Cr2O7 were added into culture
medium and the cell viability was evaluated at 24 and 48 h
post-K2Cr2O7 intoxication. The
viability of 10-µM K2Cr2O7-treated
cells was approximately 50–60 and 20–30%, at 24 and 48 h,
respectively (Fig. 1A). To
evaluate the toxic effect of
K2Cr2O7 for 24–72 h, similar
viability was obtained and is presented in Fig. 1B, compared to the control cells.
The morphology of 10-µM
K2Cr2O7-treated cells was altered.
Significant cell shrinkage was observed when compared to the
control cells (Fig. 1C). These
results indicated that the morphology and viability of HK-2 cells
were significantly affected upon exposure to 10 µM
K2Cr2O7 for 24–72 h.
Cr(VI) exposure increases
intracellular ROS production in HK-2 cells
Previous studies have suggested that the production
of ROS during the reduction of Cr(VI) to Cr(III) promoted apoptosis
(5,6). Thus, the intracellular level of ROS
was assessed in HK-2 cells exposed to 10 and 100 µM of
K2Cr2O7 for 30 min (Fig. 2A). The results revealed that the
intracellular ROS level in 10 and 100 µM
K2Cr2O7-exposed HK-2 cells was
greater than that in control cells. Quantification of data also
revealed similar results (Fig.
2B). A high level of ROS may be a factor that triggers cell
death in K2Cr2O7-exposed HK-2
cells.
Cr(VI) exposure increases apoptosis in
HK-2 cells
The results revealed that cells treated with 10 µM
K2Cr2O7 for 48 h exhibited a
viability of ~20–30%. Since a significant reduction in cell
viability from 24 to 48 h suggested that apoptosis-regulating
proteins were induced, the protein lysates were collected during a
24 to 48-h time frame. The protein lysates of HK-2 cells exposed to
K2Cr2O7 were harvested at 24, 36,
40, and 45 h (Fig. 3A) and the
quantitative results are presented in Fig. 3B-E. The activation of caspase-3 and
cleavage of PARP indicated activation of the apoptotic pathway
(22). After 24 h of
K2Cr2O7 exposure, the expression
levels of cleaved PARP and cleaved caspase-3 were significantly
upregulated. In addition, the expression of Bcl-2, which prevents
the release of mitochondrial apoptogenic factors such as cytochrome
c and AIF (23), was
significantly suppressed in
K2Cr2O7-exposed HK-2 cells,
indicating that 10 µM K2Cr2O7
exposure induced apoptotic pathways at 24 h
post-K2Cr2O7 treatment. At other
time-points, similar expression patterns were observed. However,
there was no significant difference between the control and 10-µM
K2Cr2O7-exposed cells at 36, 40
and 45 h due to high standard deviation.
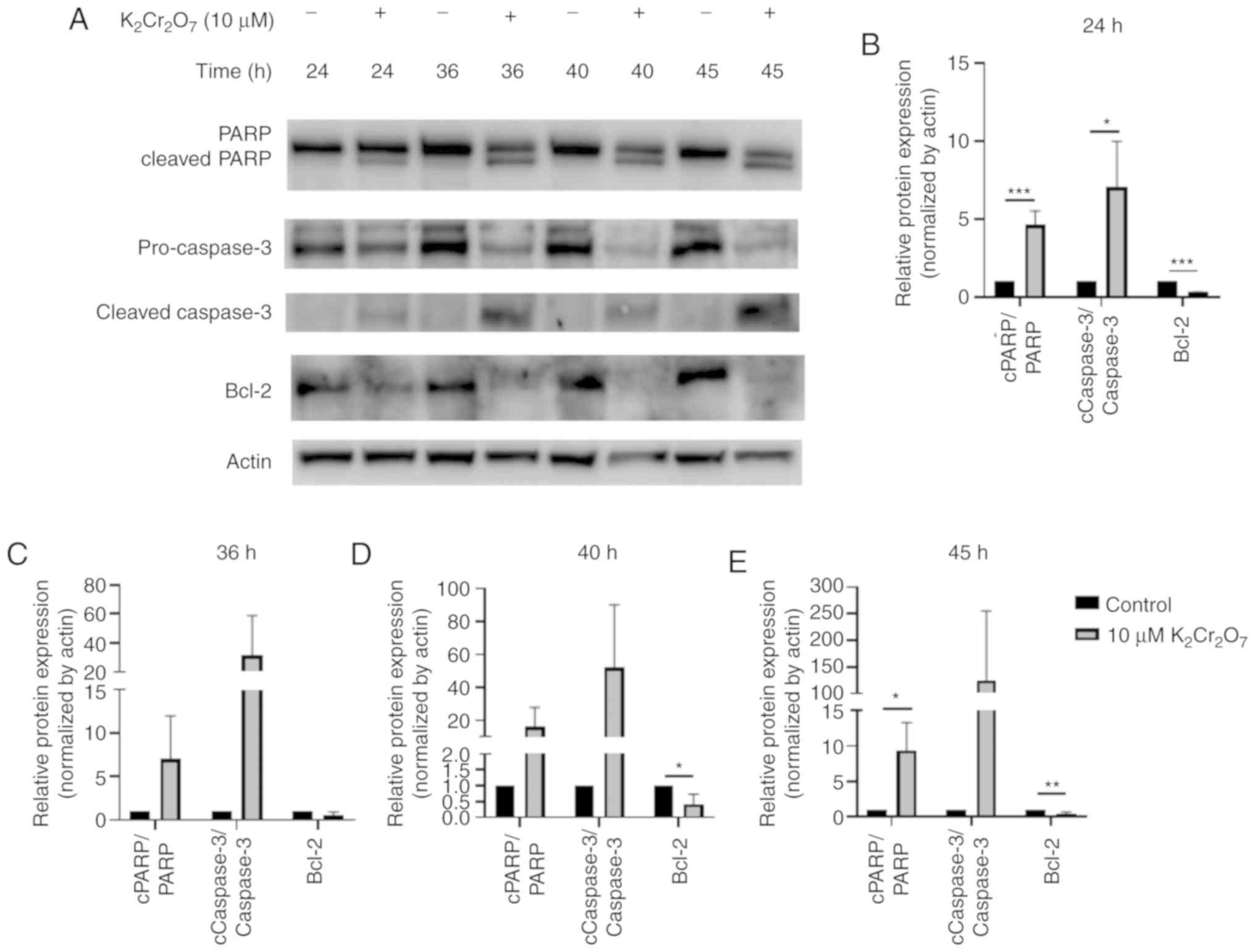 | Figure 3.Evaluation of apoptotic markers. HK-2
cells were exposed to 10 µM
K2Cr2O7 and protein lysates were
collected at different time-points (24, 36, 40, 45 h after
K2Cr2O7 treatment). (A) The
expression of PARP, cleaved PARP, pro-caspase-3, cleaved caspase-3,
and Bcl-2 in HK-2 cells. The relative protein expression levels at
(B) 24 (C) 36 (D) 40 and (E) 45 h are presented. *P<0.05,
**P< 0.01 and ***P<0.001. 10 µM
K2Cr2O7 exposure vs. the control
at various time-points. K2Cr2O7,
potassium dichromate; PARP, poly (ADP-ribose) polymerase; AIF,
apoptosis-inducing factor. |
Cr(VI) exposure increases not only the
intrinsic, but also the extrinsic and caspase-independent apoptosis
in HK-2 cells
Previous studies have revealed that Cr(VI) toxicity
induced cell apoptosis mainly via intrinsic mitochondrial pathways
(5). Our results showed that the
expression levels of molecules involved in the intrinsic pathway
(BAX, cytochrome c), caspase-independent pathway (AIF), and
extrinsic pathway (soluble FasL and cleaved caspase-8) were
significantly induced after 45 h of incubation in the presence of
10 µM K2Cr2O7 (Fig. 4). These findings indicated that
these pathways contributed to Cr(VI)-mediated human renal cell
death. The summary of this study is outlined in Fig. 5.
 | Figure 4.Evaluation of apoptotic pathways. (A)
The expression levels of PARP, cleaved PARP, cleaved caspase-3, and
Bax are presented. (B) The expression levels of AIF and cytochrome
c are presented. (C) The expression levels of soluble FasL,
pro-caspase-8, and cleaved caspase-8 were assessed. (D)
Quantification of the western blot assay. The data represent the
mean ± SD of three independent experiments. *P<0.05, **P<0.01
and ***P<0.001, 10 µM K2Cr2O7
exposure vs. the control. PARP, poly (ADP-ribose) polymerase; AIF,
apoptosis-inducing factor; FasL, Fas ligand;
K2Cr2O7, potassium dichromate. |
Discussion
Exposure to Cr(VI) affects various physiological
events, and Cr(VI)-induced apoptosis has been reported by previous
studies. In human diploid and normal fibroblasts and in HeLa human
cervical cancer cells, reduced form of Cr(VI) caused DNA strand
breaks (24–26). In U937 cells, exposure to 20 µM
Cr(VI) for 24 h induced intrinsic but not extrinsic apoptosis
(27). Exposure to 30 µM Cr(VI)
induced both p53-dependent and independent intrinsic mitochondrial
apoptosis in HCT116 human colon carcinoma cells (28). Activation of ribosomal protein L3
(rpL3) plays a role in chemotherapeutic drug-induced
p53-independent apoptosis in colon and lung cancer cells (29,30).
However, the role of rpL3 in Cr(VI)-induced apoptotic processes is
unclear. Another study revealed that Bcl-2 and p21 were
downregulated after HCT-116 cells were exposed to 30 µM Cr(VI)
(5). In a rat model, a single
subcutaneous injection of K2Cr2O7
at 15 mg/kg induced the intrinsic mitochondrial pathway in kidneys
(19). Clinically, Cr(VI) exposure
often causes acute renal failure that may result in permanent
hemodialysis or even death, and renal injury has mainly been
detected in renal proximal tubular epithelial cells (31). Therefore, investigating the
mechanism of Cr(VI)-induced apoptosis in renal cells is an
important issue. To develop an optimal strategy to treat acute
Cr(VI) intoxication, the apoptosis mechanism in human kidney cells
under a curable dose was evaluated. According to a clinical study,
chromium at a concentration of 10 mg/l (approximate 34 µM) or
greater in human blood is considered to be lethal for human life
(32). A case study indicated that
a patient with chromium blood concentration of 3.4 mg/l fortunately
survived after intermittent hemodialysis treatment (33). Compared to cells exposed to 10 µM
of Cr(VI), low cell viability was observed when cells were exposed
to 30 and 100 µM for 24 and 48 h (Fig.
1A). Conversely, cell viability was relatively high when cells
were exposed to low concentrations of Cr (VI) (<10 µM).
Therefore, the present results also indicated that higher
concentrations were lethal and lower doses may not induce
apoptosis. Cr(VI) toxicity at 10 µM (2.95 mg/l) was selected to
induce apoptosis in the HK-2 human proximal tubular epithelial cell
line.
The intrinsic pathway is regulated by pro-apoptotic
and anti-apoptotic proteins such as Bax and Bcl-xL, respectively
(22). These proteins alter the
fate of cytochrome c release and activation of the caspase
cascade, which regulate the permeability of the outer mitochondrial
membrane (22). Increased Bax and
decreased Bcl-2 protein expression indicated that the intrinsic
mitochondrial apoptotic pathway was induced in HK-2 cells (Figs. 3 and 4). AIF is a pro-apoptotic protein
released from the mitochondria, and its nuclear translocation
caused DNA fragmentation and was involved in caspase-independent
apoptosis (34,35). Extrinsic apoptotic pathways
initiate apoptosis via the interaction between ligands and
transmembrane receptors that belong to the tumor necrosis factor
(TNF) receptor gene superfamily (22). Induction of extrinsic apoptosis was
observed in mouse glomerular podocytes when they were exposed to
toxic metals such as arsenite, cadmium, and mercury (36). Increased expression of soluble FasL
and cleaved caspase-8 was detected after 10 µM Cr(VI) toxicity.
Soluble FasL is well known as a degradation product of the membrane
form of FasL by matrix metalloproteinases (37,38).
Previous studies have documented that chromium had the potential to
enhance the function of matrix metalloproteinases (39,40),
which may explain the increased ratio between soluble and membrane
FasL in the present results. In addition, the prevalence of the
soluble form in association with the increased activation of
caspase-8 (cleaved caspase-8) indicated the possible active role of
soluble FasL in chromium-induced HK-2 cell apoptosis. Therefore,
the present results indicated that Cr(VI) toxicity for 48 h
triggered the extrinsic apoptotic pathway. To the best of our
knowledge, this study is the first to observe the induction of
extrinsic apoptotic markers in Cr(VI)-exposed human renal cells.
However, the mechanism of Cr(VI)-induced soluble FasL release is
still unknown. The crosstalk between tumor necrosis factors and ROS
may be a potential explanation (41), but further studies are required to
confirm this hypothesis.
The limitation of the present study is that the
conclusions were supported by one technique in one cell line. For
confirmation, more experiments are required in future studies. For
example, cell death can be also detected via BrdU staining assay
and Annexin V staining assay at each time-point. Inhibition of
intrinsic and extrinsic apoptosis-dependent pathways via specific
inhibitors on caspase-8 and caspase-9 may further confirm the most
critical pathways at specific time-points. To further investigate
the role of Cr(VI)-induced ROS production, treatments with
antioxidants or ROS scavengers are strategies for future
experiments. In addition, performing animal experiments may
strengthen our conclusion.
Collectively, not only intrinsic but also extrinsic
apoptotic pathway-related apoptotic markers were activated in an
immortalized proximal tubular epithelial cell line after exposure
to 10 µM activated Cr(VI). The experimental dosage of Cr(VI) was
similar to the Cr(VI) dosage that causes acute renal dysfunction.
It is anticipated that the present study may be beneficial in
designing optimal treatments for acute Cr(VI) toxicity in the
future.
Acknowledgements
The authors would like to thank Professor Jung-San
Chang (Department of Renal Care, College of Medicine, Kaohsiung
Medical University) for scientific discussions and technical
assistance.
Funding
The present study was supported by grants from the
Kaohsiung Medical University Hospital (grant no. KMUH103-3M52).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author upon reasonable
request.
Authors' contributions
YHW, JCL and IJY designed the study. PLW and FWC
performed the experiments. YHW, TYW, TJL, MCY, YLS, YHL, and IJY
analyzed the data and interpreted the results. All authors read and
approved the final version of the manuscript and agree to be
accountable for all aspects of the research in ensuring that the
accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
Cr(VI)
|
hexavalent chromium
|
|
ROS
|
reactive oxygen species
|
|
Cr(III)
|
trivalent chromium
|
|
PARP
|
poly (ADP-ribose) polymerase
|
|
AIF
|
apoptosis-inducing factor
|
References
|
1
|
Wang XF, Xing ML, Shen Y, Zhu X and Xu LH:
Oral administration of Cr(VI) induced oxidative stress, DNA damage
and apoptotic cell death in mice. Toxicology. 228:16–23. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sun H, Brocato J and Costa M: Oral
chromium exposure and toxicity. Curr Environ Health Rep. 2:295–303.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Izbicki JA, Wright MT, Seymour WA,
BlaineMcCleskey R, Fram MS, Belitz KF and Esser BK: Cr(VI)
occurrence and geochemistry in water from public-supply wells in
California. Appl Geochem. 63:203–217. 2015. View Article : Google Scholar
|
|
4
|
Chen Y, An D, Sun S, Gao J and Qian L:
Reduction and removal of chromium VI in water by powdered activated
carbon. Materials (Basel). 11(pii): E2692018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chiu A, Shi XL, Lee WK, Hill R, Wakeman
TP, Katz A, Xu B, Dalal NS, Robertson JD, Chen C, et al: Review of
chromium (VI) apoptosis, cell-cycle-arrest, and carcinogenesis. J
Environ Sci Health C Environ Carcinog Ecotoxicol Rev. 28:188–230.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hu G, Zheng P, Feng H and Jia G: Imbalance
of oxidative and reductive species involved in chromium(VI)-induced
toxic effects. Reactive Oxygen Species. 3:12017.
|
|
7
|
Liu KJ and Shi X: In vivo reduction of
chromium (VI) and its related free radical generation. Mol Cell
Biochem. 222:41–47. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pan TL, Wang PW, Chen CC, Fang JY and
Sintupisut N: Functional proteomics reveals hepatotoxicity and the
molecular mechanisms of different forms of chromium delivered by
skin administration. Proteomics. 12:477–489. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bright P, Burge PS, O'Hickey SP, Gannon
PF, Robertson AS and Boran A: Occupational asthma due to chrome and
nickel electroplating. Thorax. 52:28–32. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Costa M: Toxicity and carcinogenicity of
Cr(VI) in animal models and humans. Crit Rev Toxicol. 27:431–442.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Verschoor MA, Bragt PC, Herber RF,
Zielhuis RL and Zwennis WC: Renal function of chrome-plating
workers and welders. Int Arch Occup Environ Health. 60:67–70. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wedeen RP and Qian LF: Chromium-induced
kidney disease. Environ Health Perspect. 92:71–74. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sharma BK, Singhal PC and Chugh KS:
Intravascular haemolysis and acute renal failure following
potassium dichromate poisoning. Postgrad Med J. 54:414–415. 1978.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ellis EN, Brouhard BH, Lynch RE, Dawson
EB, Tisdell R, Nichols MM and Ramirez F: Effects of hemodialysis
and dimercaprol in acute dichromate poisoning. J Toxicol Clin
Toxicol. 19:249–258. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Leonard SS, Roberts JR, Antonini JM,
Castranova V and Shi X: PbCrO4 mediates cellular responses via
reactive oxygen species. Mol Cell Biochem. 255:171–179. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sinha K, Das J, Pal PB and Sil PC:
Oxidative stress: The mitochondria-dependent and
mitochondria-independent pathways of apoptosis. Arch Toxicol.
87:1157–1180. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Quinteros FA, Poliandri AH, Machiavelli
LI, Cabilla JP and Duvilanski BH: In vivo and in vitro effects of
chromium VI on anterior pituitary hormone release and cell
viability. Toxicol Appl Pharmacol. 218:79–87. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xiao F, Li Y, Dai L, Deng Y, Zou Y, Li P,
Yang Y and Zhong C: Hexavalent chromium targets mitochondrial
respiratory chain complex I to induce reactive oxygen
species-dependent caspase-3 activation in L-02 hepatocytes. Int J
Mol Med. 30:629–635. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Molina-Jijon E, Tapia E, Zazueta C, El
Hafidi M, Zatarain-Barrón ZL, Hernández-Pando R, Medina-Campos ON,
Zarco-Márquez G, Torres I and Pedraza-Chaverri J: Curcumin prevents
Cr(VI)-induced renal oxidant damage by a mitochondrial pathway.
Free Radic Biol Med. 51:1543–1557. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Redza-Dutordoir M and Averill-Bates DA:
Activation of apoptosis signalling pathways by reactive oxygen
species. Biochim Biophys Acta. 1863:2977–2992. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lin TJ, Huang YL, Chang JS, Liu KT, Yen
MC, Chen FW, Shih YL, Jao JC, Huang PC and Yeh IJ: Optimal dosage
and early intervention of L-ascorbic acid inhibiting
K2Cr2O7-induced renal tubular cell
damage. J Trace Elem Med Biol. 48:1–7. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Elmore S: Apoptosis: A review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tsujimoto Y: Role of Bcl-2 family proteins
in apoptosis: apoptosomes or mitochondria? Genes Cells. 3:697–707.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Snyder RD: Role of active oxygen species
in metal-induced DNA strand breakage in human diploid fibroblasts.
Mutat Res. 193:237–246. 1988.PubMed/NCBI
|
|
25
|
Wakeman TP, Kim WJ, Callens S, Chiu A,
Brown KD and Xu B: The ATM-SMC1 pathway is essential for activation
of the chromium[VI]-induced S-phase checkpoint. Mutat Res.
554:241–251. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ha L, Ceryak S and Patierno SR: Generation
of S phase-dependent DNA double-strand breaks by Cr(VI) exposure:
Involvement of ATM in Cr(VI) induction of gamma-H2AX.
Carcinogenesis. 25:2265–2274. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hayashi Y, Kondo T, Zhao QL, Ogawa R, Cui
ZG, Feril LB Jr, Teranishi H and Kasuya M: Signal transduction of
p53-independent apoptotic pathway induced by hexavalent chromium in
U937 cells. Toxicol Appl Pharmacol. 197:96–106. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hill R, Leidal AM, Madureira PA, Gillis
LD, Cochrane HK, Waisman DM, Chiu A and Lee PW: Hypersensitivity to
chromium-induced DNA damage correlates with constitutive
deregulation of upstream p53 kinases in p21-/-HCT116 colon cancer
cells. DNA Repair (Amst). 7:239–252. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pagliara V, Saide A, Mitidieri E,
d'Emmanuele di Villa Bianca R, Sorrentino R, Russo G and Russo A:
5-FU targets rpL3 to induce mitochondrial apoptosis via
cystathionine-beta-synthase in colon cancer cells lacking p53.
Oncotarget. 7:50333–50348. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Russo A, Saide A, Cagliani R, Cantile M,
Botti G and Russo G: rpL3 promotes the apoptosis of p53 mutated
lung cancer cells by down-regulating CBS and NFκB upon 5-FU
treatment. Sci Rep. 6:383692016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Parveen K, Khan MR and Siddiqui WA:
Pycnogenol prevents potassium dichromate K2Cr2O7-induced oxidative
damage and nephrotoxicity in rats. Chem Biol Interact. 181:343–350.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pedersen RS and Morch PT: Chromic acid
poisoning treated with acute hemodialysis. Nephron. 22:592–595.
1978. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
KhanSaif R, Aparna Q, Shahzad S and Haque
F: Chromium induced AKI: Case with protean implications. Annals
Tropical Medicine Public Health. 7:136–138. 2014.
|
|
34
|
Tait SW and Green DR: Caspase-independent
cell death: Leaving the set without the final cut. Oncogene.
27:6452–6461. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ranjan A and Iwakuma T: Non-canonical cell
death induced by p53. Int J Mol Sci. 17(pii): E20682016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Eichler T, Ma Q, Kelly C, Mishra J, Parikh
S, Ransom RF, Devarajan P and Smoyer WE: Single and combination
toxic metal exposures induce apoptosis in cultured murine podocytes
exclusively via the extrinsic caspase 8 pathway. Toxicol Sci.
90:392–399. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Vargo-Gogola T, Crawford HC, Fingleton B
and Matrisian LM: Identification of novel matrix
metalloproteinase-7 (matrilysin) cleavage sites in murine and human
Fas ligand. Arch Biochem Biophys. 408:155–161. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Musial K and Zwolinska D: Matrix
metalloproteinases and soluble Fas/FasL system as novel regulators
of apoptosis in children and young adults on chronic dialysis.
Apoptosis. 16:653–659. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Cammarota M, Lamberti M, Masella L,
Galletti P, De Rosa M, Sannolo N and Giuliano M: Matrix
metalloproteinases and their inhibitors as biomarkers for metal
toxicity in vitro. Toxicol In Vitro. 20:1125–1132. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wan R, Mo Y, Zhang X, Chien S, Tollerud DJ
and Zhang Q: Matrix metalloproteinase-2 and −9 are induced
differently by metal nanoparticles in human monocytes: The role of
oxidative stress and protein tyrosine kinase activation. Toxicol
Appl Pharmacol. 233:276–285. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Blaser H, Dostert C, Mak TW and Brenner D:
TNF and ROS crosstalk in inflammation. Trends Cell Biol.
26:249–261. 2016. View Article : Google Scholar : PubMed/NCBI
|

















