Introduction
Acute lung injury (ALI) or acute respiratory
distress syndrome (ARDS) refer to a life-threatening clinical
syndrome that is characterized by severe hypoxemia and acute
respiratory failure, caused by alveolar epithelial injury and
accumulation of protein-rich fluid in the alveolar spaces, with
in-hospital mortality of 40% (1,2).
Improving the outcome of ALI/ARDS requires the clearance of
excessive edema in the alveolar spaces and repair of the alveolar
epithelium (3).
The epithelial sodium channel (ENaC), which mainly
determines the alveolar fluid volume in vivo, provides the
driving force for the removal of edema from the alveolar spaces,
(4–6). ENaC is expressed in the apical
membrane of alveolar epithelial type II cells and has been shown to
participate in amiloride-sensitive sodium influx. ENaC comprises
three homologous subunits, namely α-, β- and γ-ENaC, which share a
structure consisting of two hydrophobic membrane-spanning regions,
intracellular amino and carboxy termini, two transmembrane spanning
domains and a large extracellular loop with highly conserved
cysteine residues (7). Previous
studies have reported that alveolar edema could not be cleared in
mice lacking the α-, β- or γ-ENaC genes, which proves the important
role of ENaC in alveolar fluid clearance (AFC) (8–10).
Phosphatidylinositol 3-kinase (PI3K), which contains
a catalytic and a regulatory domain, participates in cell survival,
migration, metabolism, cytoskeletal rearrangement and vesicular
trafficking (11). The PI3K
signaling pathway was demonstrated to be integral for the
regulation of ENaC by insulin stimulation in kidney cells (12–14).
The serum/glucocorticoid-inducible kinase-1 (SGK1), a critical
regulatory protein of ENaC, is activated by insulin through the
PI3K signaling pathway (15,16).
Insulin is considered to dominate the anti-inflammatory function
during ALI (17); however, its
effect on AFC remains unclear. Our previous study revealed that
insulin upregulated α-ENaC, possibly via the activation of SGK1, in
the lung (18). However, whether
the PI3K/SGK1 signaling pathway participated in the regulation of
ENaC-mediated AFC in ALI has yet to be elucidated.
In the present study, the aim was to investigate the
effect of insulin on ENaC expression and the involvement of the
PI3K/SGK1 pathway in lipopolysaccharide (LPS)-induced lung
injury.
Materials and methods
Animals and materials
A total of 24 C3H/HeN mice (Charles River
Laboratories, Inc., Wilmington, MA, USA) aged 7–9 weeks were housed
under specific pathogen-free conditions in a temperature- and
humidity-controlled environment (temperature: 22±1°C and humidity:
70±10%) with a 12-h light/dark cycle, and given free access to food
and water. PI3K p85α small interfering RNA (siRNA) and a scramble
non-targeting siRNA (serving as the control) were purchased from
Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). SGK1 siRNA was
designed and synthesized by Shanghai Gene Pharma Co., Ltd.
(Shanghai, China). All animal experiments were conducted in
accordance with the Guide for the Care and Use of Laboratory
Animals, and approved by the Ethics Committee of the Second
Affiliated Hospital of Chongqing Medical University (Chongqing,
China).
Animal model and intervention
Mice were anesthetized by intraperitoneal
administration of sodium pentobarbital (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) at a dose of 50 mg/kg. The ALI model was
established using LPS (Escherichia coli serotype O111:B4;
Sigma-Aldrich; Merck KGaA) at a dose of 5 mg/kg in 50 µl sterile
phosphate-buffered saline by a one-time intratracheal injection
(19). Human insulin (Eli Lilly
& Co., Indianapolis, IN, USA) was administered at a dose of 0.1
U/kg/h at a rate of 2.5 mU/h/mouse via an internal jugular vein
catheter attached to micro-osmotic pumps at 16 h prior to exposure
to LPS. Mice were randomly divided into four groups: i) Control
group (n=6); ii) LPS group (n=6); iii) LPS + insulin group (n=6);
and, iv) LPS + insulin + SGK1 siRNA group (n=6). Mice in the
control group received an equivalent volume of saline. Blood
samples were collected from the catheter by centrifugation at 1,500
× g at 4°C for 15 min. Glucose levels in the plasma were recorded
at different time points by a glucometer (Johnson & Johnson
Medical Ltd., Shanghai, China). Mice were sacrificed by
exsanguination from the carotid artery 24 h after LPS
administration. Next, bronchoalveolar lavage fluid (BALF) and lung
tissue were obtained for analysis.
Primary cell isolation and
culture
Alveolar epithelial type II cells were isolated from
healthy untreated male C3H/HeN mice, as previously described
(20). The cells were cultured in
Dulbecco's modified Eagle's medium containing 10% fetal bovine
serum, 100 U/ml penicillin and 0.1 mg/ml streptomycin in an
atmosphere with 5% CO2 and 95% air.
siRNA transfection
Alveolar epithelial cells isolated from healthy
untreated male C3H/HeN mice were transfected with PI3K siRNA or
SGK1siRNA using Lipofectamine® 2000 (Invitrogen; Thermo
Fisher Scientific, Inc., Waltham, MA, USA), according to the
manufacturer's protocol. At 72 h after transfection, cells were
incubated with or without 200 mU/l insulin for 2 h. They were
divided into 4 groups: Control group, insulin group, insulin+PI3K
siRNA group and insulin+SGK1 siRNA group. Then the alveolar
epithelial cells were processed for western blotting. In the mice
experiments, intratracheal delivery of siRNA was performed in the
mice, as previously described (21). Mice were anesthetized and their
tongues were gently pulled out. Then 75 µg siRNA specific for SGK1
was pipetted in vehicle, 100 µl saline, directly into the throat,
momentarily blocking the mouse's airway 2 h after LPS exposure in
the mice treated with LPS + insulin. An equivalent dose of
non-targeting siRNA served as the control in the other mice groups.
Inspiration carried the siRNA and vehicle into the lung.
BALF analysis
Normal saline (5 ml) was instilled into the right
lung three times. The lavage fluid from the right lung was
carefully pooled and centrifuged at 1,700 × g for 30 min at 4°C.
The total cell count was then determined by a hemocytometer, and
differential cell counts were assessed on cytocentrifuge
preparations stained with Diff-Quik (Sigma-Aldrich; Merck KGaA).
Total protein in the BALF was determined using a KeyGen assay kit
(Nanjing KeyGen Biotech Co., Ltd., Nanjing, China).
Bronchoalveolar epithelial
permeability analysis
At 24 h after LPS-induced lung injury, mice were
injected with fluorescein isothiocyanate-conjugated dextran 4000
(FD4; Sigma-Aldrich; Merck KGaA) solution in PBS (10 mg/kg) via the
internal jugular vein. Normal saline (1 ml) was instilled into the
lungs three times after 50 min. Subsequently, BALF was carefully
collected, and serum was collected from the internal jugular vein
blood by centrifugation at 1,500 × g for 5 min. The concentrations
of FD4 in the BALF and serum were determined by a
spectrofluorometer (BD Biosciences, Franklin Lakes, NJ, USA) using
an excitation wavelength of 492 nm and an emission wavelength of
515 nm. Bronchoalveolar epithelial permeability was determined
based on the BALF/serum fluorescence ratio.
Hematoxylin and eosin staining
The unlavaged left lungs were harvested 24 h after
LPS administration and fixed with 10% neutral buffered formalin for
24 h. The fixed tissues were embedded in paraffin and cut into 5-µm
section. Next, the sections were mounted onto glass slides and then
stained with hematoxylin for 10 min and eosin for 2 min. Then, 5
random areas were examined at a magnification of ×100 for each
section. Lung injury was evaluated according to a new histologic
lung injury scoring system (22).
Wet-to-dry ratio
The left lungs were isolated to determine the
wet-to-dry ratio of the tissue. After the wet weight of the lungs
was measured, the lungs were placed in an oven at 80°C for 48 h and
then weighed again in order to obtain their dry weight. Pulmonary
edema was determined based on the wet-to-dry ratio; a high ratio
indicated more pulmonary edema while low ratio indicated less
pulmonary edema compared with normal lungs.
AFC analysis
AFC was measured as previously described (23). Briefly, the isolated right lungs
were placed in a humidified incubator at 37°C and ventilated with
100% nitrogen to remove oxygen from the alveolar spaces.
Physiological saline solution (5 ml/kg) containing 5% albumin and
0.15 mg/ml Evans blue dye (Sigma-Aldrich; Merck KGaA) was injected
into the alveolar spaces at an airway pressure of 7 cm
H2O. Alveolar fluid was aspirated 1 h after
instillation, and the concentrations of Evans blue-labeled albumin
in the injected and aspirated solutions were measured using a
spectrophotometer (BD Bioscience). AFC was calculated as
follows:
AFC=[(Vi-Vf)/Vi]x100%Vf=(VixPi)/Pf
Vi and Vf represent the injected and final alveolar
fluid volume, respectively. Pi and Pf represent the injected and
final concentrations of Evans blue-labeled 5% albumin solution,
respectively. Albumin solution containing amiloride
(5×10−4 M; Sigma-Aldrich; Merck KGaA) was injected into
the alveolar spaces.
Terminal deoxynucleotidyl transferase
dUTP nick end labeling (TUNEL) assay
Detection of apoptotic cells in the lung epithelium
was performed on paraffin-embedded 5-µm lung sections using an
in situ cell death detection kit (Roche Diagnostics,
Indianapolis, IN, USA) by TUNEL reaction. The extent of apoptosis
was evaluated by counting the TUNEL-positive cells (brown-stained).
TUNEL positive cells were counted in five randomly selected fields
(magnification, ×400) for each section.
Reverse transcription-polymerase chain
reaction (RT-PCR)
Total RNA was extracted from alveolar epithelial
cells following the manufacturer's protocol (Takara Bio, Inc.,
Otsu, Japan). The concentration and purity of the RNA were
estimated using a spectrophotometer. (DU730; Beckman Coulter, Inc.,
Brea, CA, USA). RT and PCR amplification were performed using an
RT-PCR kit (Takara Bio, Inc.). The RT reaction was conducted at
65°C for 5 min, 42°C for 30 min, 95°C for 5 min and 4°C for 5 min.
The PCR amplification reaction was conducted at 94°C for 60 sec,
followed by 30 cycles at 94°C for 30 sec, then 53°C (α-ENaC), 53°C
(β-ENaC), 55°C (γ-ENaC) or 55°C (β-actin) for 30 sec, and finally
72°C for 60 sec. The primer sequences used were: α-ENaC (509 bp),
5′-TACCCTTCCAAGTATACACAGC-3′ (forward) and
5′-CAGAAGGAGACTCCGAATTAGT-3′ (reverse); β-ENaC (406 bp),
5′-GCTAAAGAGCTAGCAGTAATGG-3′ (forward) and
5′-CTGGTGTTTGTTATGCCTAGAG-3′ (reverse); γ-ENaC (363 bp),
5′-GGATCCTGAGAGAGAATCATGC-3′ (forward) and
5′-GTGTCCAGCTATGCCCTTTAAC-3′ (reverse); β-actin (871 bp),
5′-GTACAACCTTCTTGCAGCTCCT-3′ (forward) and
5′-ACAGGATTCCATACCCAGGAAG-3′ (reverse). PCR products were
electrophoresed on 1.0% agarose gels containing ethidium bromide,
and images of the gels were captured using a gel imaging system
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). The expressions of
α-, β- and γ-ENaC were quantified by normalizing the band intensity
to β-actin using Quantity One software (version 4.4, Bio-Rad
Laboratories, Inc.).
Immunofluorescence assay
Lungs were fixed by immersion in 10%
neutral-buffered formalin and embedded in paraffin. The slides with
lung sections (5 µm) were soaked in xylene and rehydrated in 100,
95, 85 and 70% solutions of ethanol for 5 min. Following blocking
with 10% fetal calf serum for 1 h at room temperature, the sections
were incubated with rabbit polyclonal primary antibodies against
α-, β- or γ-ENaC (1:200; sc-21012, sc-21013 and sc-21014
respectively; Santa Cruz Biotechnology, Inc.) overnight at 4°C. The
tissues were then incubated with a goat anti-rabbit secondary
antibody labeled with Alexa Fluor 594 (1:400; A-11012; Thermo
Fisher Scientific, Inc.) at 37°C for 1 h. Subsequently, nuclei were
stained with DAPI (1:2,000; Sigma-Aldrich; Merck KGaA). Images were
captured by confocal laser scanning microscopy (Bio-Rad
Laboratories, Inc.) and analyzed using Image-Pro Plus software,
version 6.0 (Media Cybernetics, Inc., Rockville, MD, USA).
Western blotting
Total proteins were obtained by incubation with 1 ml
lysis buffer and 1 ml extraction buffer using the KeyGen protein
extraction kit (Nanjing KeyGen Biotech Co., Ltd., Nanjing, China).
The concentration of each protein sample was determined using a BCA
protein assay kit (KeyGen Biotech Co., Ltd.). Proteins were then
separated by 10% SDS-PAGE and transferred to polyvinylidene
fluoride membranes. Following blocking with 5% nonfat dried milk in
Tris-buffered saline containing 0.05% Tween 20 at room temperature
for 1 h, the membranes were incubated with primary antibodies
against α-ENaC (1:300; sc-21012), β-ENaC (1:300; sc-21013), γ-ENaC
(1:300; sc-21014), PI3K p85α (1:1,000; sc-71892), SGK1 (1:500;
sc-33774), phosphorylated-SGK1 (Ser422; 1:500; sc-16745) all from
Santa Cruz Biotechnology, Inc. and β-actin (1:500, bsm-33036M,
BIOSS, Beijing, China) overnight at 4°C, and then incubated with
horseradish peroxidase-conjugated secondary antibody (1:5,000) at
room temperature for 1.5 h. Using enhanced chemiluminescence
(KeyGen Biotech Co., Ltd.), the protein bands were visualized by a
UVP gel imaging system (Analytik Jena, Upland, CA, USA) and
analyzed by Labworks software (version 4.6; Labworks LLC, Lehi, UT,
USA).
Statistical analysis
All data are presented as the mean ± standard error
of the mean. Student's t-test and one-way analysis of variance were
performed. SPSS version 12.0 software (SPSS, Inc., Chicago, IL,
USA) was used for the statistical analysis. P<0.05 was
considered to indicate a statistically significant difference.
Results
The effect of siRNA transfection in
mouse lung tissues and primary alveolar epithelial cells
As observed in Fig.
1A, transfection with SGK1 siRNA resulted in evident
downregulation of SGK1 expression in mouse lung tissues, as
compared with the control and scramble siRNA groups. As shown in
Fig. 1B, PI3K siRNA and SGK1 siRNA
also significantly decreased PI3K and SGK1 protein expression
levels in primary alveolar epithelial cells, respectively.
Effect of exogenous insulin on plasma
glucose levels
No significant differences were observed in the
plasma glucose levels of normal mice treated with insulin for 0,
30, 60, 120 and 240 min (Table I).
In addition, no significant differences were observed in the plasma
glucose levels of insulin- and saline-treated mice at 0, 1, 4 and 8
h in the LPS-induced lung injury group. The results indicated that
insulin did not alter the glucose level during ALI (Table II).
 | Table I.Effect of exogenous insulin (0.1
U/kg) on plasma glucose levels in normal mice. |
Table I.
Effect of exogenous insulin (0.1
U/kg) on plasma glucose levels in normal mice.
| Time after insulin
administration (min) | Plasma glucose
(mmol/l) |
|---|
| 0 | 4.8±0.5 |
| 30 | 4.9±0.4 |
| 60 | 5.1±0.6 |
| 120 | 5.1±0.5 |
| 240 | 5.3±0.6 |
| Table II.Effect of exogenous insulin on plasma
glucose levels in mice with LPS-induced lung injury. |
Table II.
Effect of exogenous insulin on plasma
glucose levels in mice with LPS-induced lung injury.
|
| Plasma glucose
level (mmol/l) |
|---|
|
|
|
|---|
| Time (h) | LPS | LPS + insulin |
|---|
| 0 | 7.6±0.7 | 7.1±0.8 |
| 1 | 6.8±0.7 | 6.5±0.5 |
| 4 | 6.3±0.4 | 6.2±0.3 |
| 8 | 5.9±0.5 | 5.7±0.6 |
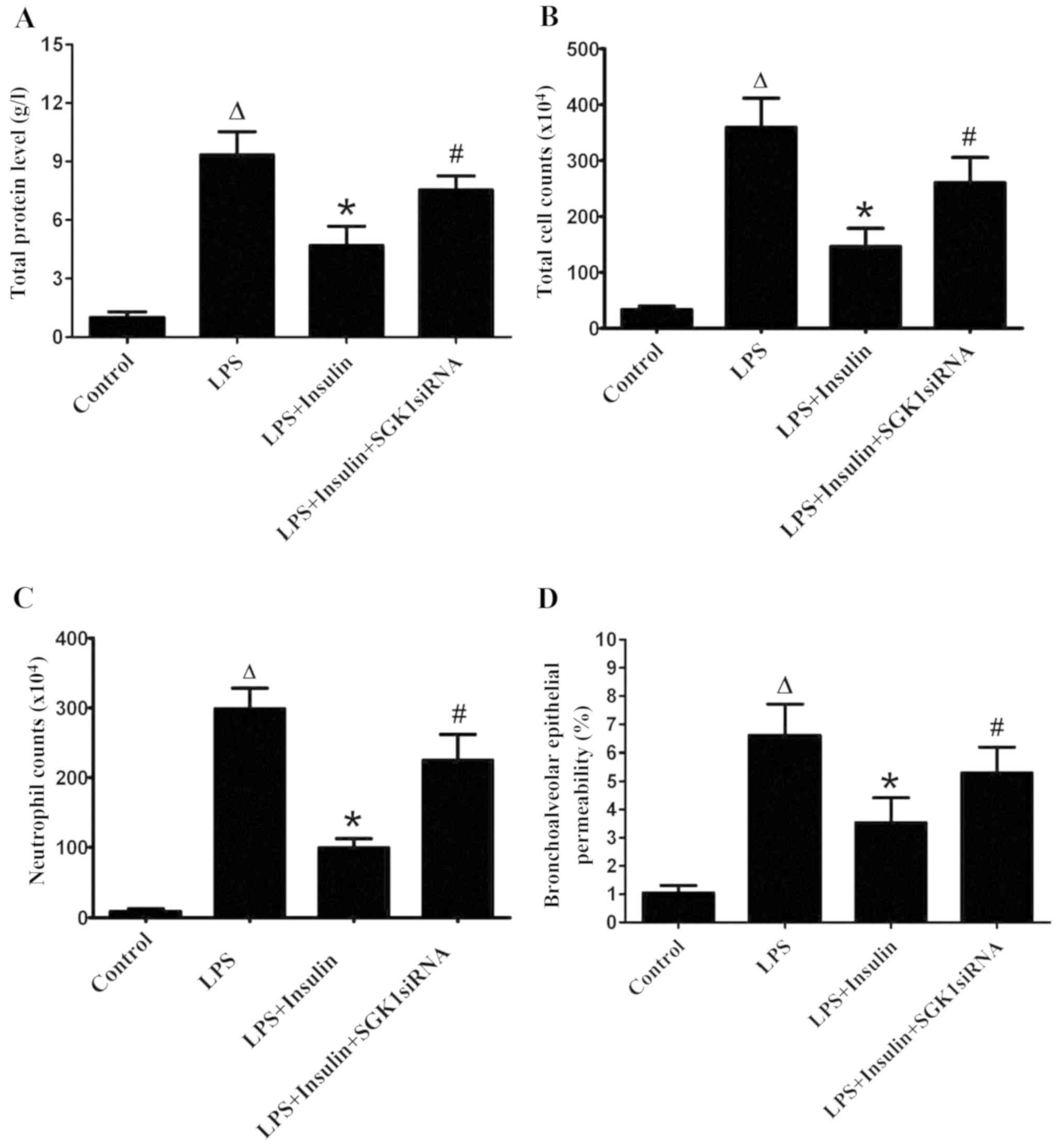
Exogenous insulin protects the
pulmonary epithelial barrier in LPS-induced lung injury
Insulin treatment significantly decreased the
LPS-induced increase in total protein, total cell numbers and
neutrophils in BALF (Fig. 2A-C).
However, the delivery of SGK1 siRNA significantly blocked the
effect of insulin in the LPS-induced lung injury group (Fig. 2A-C). The pulmonary epithelial
barrier was injured in mice with LPS-induced lung injury, as
indicated by the increase in the BALF/serum fluorescence ratio,
which was a result of high bronchoalveolar epithelial permeability
(Fig. 2D). The BALF/serum
fluorescence ratio was significantly decreased by insulin,
indicating a decreased bronchoalveolar epithelial permeability;
thus, insulin exerted a protective effect on pulmonary epithelial
barrier function in ALI. However, SGK1 siRNA clearly blocked the
effect of insulin, as evidenced by the marked increase in the
BALF/serum fluorescence ratio (Fig.
2D).
Exogenous insulin attenuates
LPS-induced lung injury
The pulmonary morphology of ALI mice demonstrated
significant injury, with the presence of severe interstitial edema,
thickened alveolar septa, inflammatory cell infiltration and
formation of proteinaceous debris in the alveolar spaces, as
compared with the control mice (Fig.
3A and B). Insulin treatment significantly attenuated the
LPS-induced lung injury (Fig. 3C),
while the lung injury was significantly aggravated with the
co-administration of SGK1 siRNA (Fig.
3D). The lung injury score supported the observation that
insulin ameliorated LPS-induced lung injury and that SGK1 siRNA
blocked the effect of insulin in ALI mice (Fig. 3E).
 | Figure 3.Effect of exogenous insulin on the
pulmonary morphology at 24 h after LPS-induced lung injury in mice.
(A) Control group, indicating normal pulmonary morphology. (B) LPS
group, demonstrating severe interstitial edema, thickened alveolar
septa, inflammatory cells and red blood cell infiltration in the
alveolar spaces with proteinaceous debris filling the airspaces.
(C) LPS + insulin group, demonstrating evident alleviation of
interstitial edema, inflammatory cells and red blood cell
infiltration in the alveolar space, and of thickened alveolar
septa. (D) LPS + insulin + SGK1 siRNA group, demonstrating
interstitial edema, inflammatory cells and red blood cell
infiltration in the alveolar space, and aggravation of
proteinaceous debris. (E) Lung injury score, indicating that
insulin significantly attenuated LPS-induced lung injury, while
SGK1 siRNA significantly blocked the effect of insulin (n=6 per
group). Original magnification, ×100. Data are presented as the
mean ± standard error. ΔP<0.01 vs. control group;
*P<0.01 vs. LPS group; #P<0.01 vs. LPS + insulin
group. LPS, lipopolysaccharide; SGK1,
serum/glucocorticoid-inducible kinase-1; siRNA, small interfering
RNA. |
Exogenous insulin attenuates pulmonary
edema and improves AFC in LPS-induced lung injury
Insulin induced a decrease in the wet-to-dry ratio
and increase in AFC in mice with LPS-induced lung injury at 6, 12
and 24 h (Fig. 4A and B). In
addition, exposure to amiloride, a sodium channel inhibitor,
inhibited the insulin-induced AFC in mice with LPS-induced lung
injury (Fig. 4B). It was also
observed that SGK1 siRNA significantly blocked the insulin-induced
decrease in the wet-to-dry ratio (Fig.
4C). Furthermore, insulin significantly increased AFC (by 93%)
in the LPS-induced lung injury group; however, this effect was
significantly inhibited by treatment with amiloride or SGK1 siRNA,
which resulted in a 60 and 33% decrease in AFC, respectively, as
compared with the LPS + insulin group (Fig. 4D).
Effect of exogenous insulin on
apoptosis in LPS-induced lung injury
The total number of apoptotic cells in the
LPS-induced lung injury group was significantly higher as compared
with that in the control group. Treatment with insulin reduced the
number of apoptotic cells in the ALI group. However, SGK1 siRNA
reversed the effect of insulin on the apoptosis of alveolar
epithelial cells (Fig. 5).
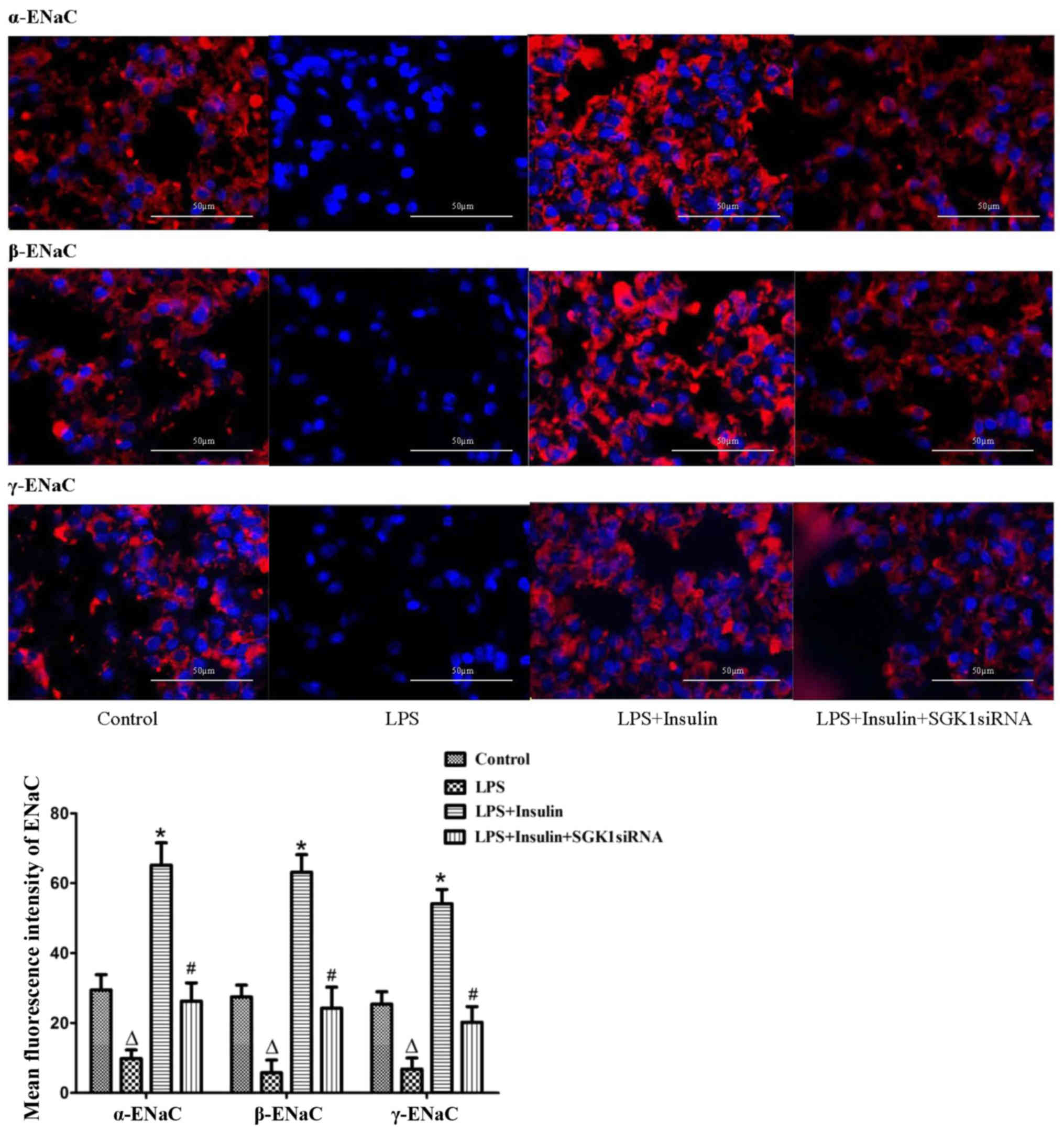
Exogenous insulin increases the
expression of ENaC in vivo and in vitro
α-, β- and γ-ENaC expression was examined by
immunofluorescence in the LPS-induced lung injury group (Fig. 6). In normal mice, α-, β- and γ-ENaC
were localized in the alveolar epithelium. However, the
distribution and expression of α-, β- and γ-ENaC were clearly
reduced in the LPS-induced lung injury group. Immunostaining of α-,
β- and γ-ENaC were significantly increased by insulin treatment,
while the insulin-induced expression of α-, β- and γ-ENaC was
clearly blocked by SGK1 siRNA (Fig.
6). Furthermore, in the primary alveolar epithelial cells,
insulin significantly increased the mRNA and protein expression
levels of α-, β- and γ-ENaC; however, this insulin-induced effect
was inhibited in cells transfected with PI3K siRNA or SGK1 siRNA
(Fig. 7). These results indicated
that insulin improved AFC by upregulating ENaC expression via the
PI3K/SGK1 pathway.
 | Figure 7.Effect of exogenous insulin on the
expression levels of α-, β- and γ-ENaC in alveolar epithelial cells
following transfection with control siRNA, PI3K siRNA or SGK1
siRNA, examined by (A) RT-PCR and (B) western blotting. At 72 h
after transfection, primary alveolar epithelial cells were
incubated with or without 200 mU/l insulin for 2 h, and then RT-PCR
and western blotting were performed. The band intensity of α-, β-
and γ-ENaC was quantified by normalizing it to β-actin. Data are
presented as the mean ± standard error. ΔP<0.01 vs.
control group; *P<0.01 vs. insulin group. ENaC, epithelial
sodium channel; PI3K, phosphatidylinositol 3-kinase; SGK1,
serum/glucocorticoid-inducible kinase-1; siRNA, small interfering
RNA; RT-PCR, reverse transcription polymerase chain reaction. |
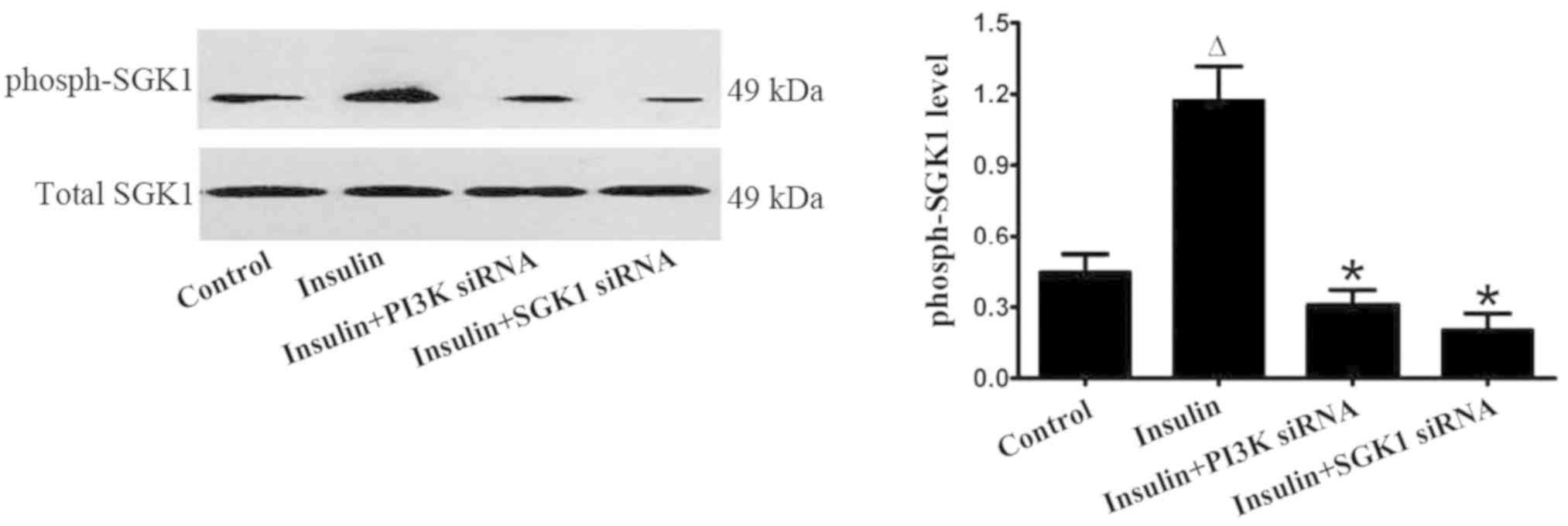
Exogenous insulin activates the
P13K/SGK1 pathway in vivo and in vitro
The level of phosphorylated SGK1 was markedly
increased by insulin in the LPS-induced lung injury group; however,
SGK1 siRNA reversed the insulin-induced increase in the level of
phosphorylated SGK1 (Fig. 8). In
primary alveolar epithelial cells, insulin significantly increased
the level of phosphorylated SGK1; however, the effect of insulin
was markedly inhibited by the transfection with PI3K siRNA or SGK1
siRNA (Fig. 9). These findings
strongly indicated that insulin regulated the ENaC expression via
the P13K/SGK1 pathway in LPS-induced lung injury.
Discussion
The present study results demonstrated that insulin
protected the lung epithelium and attenuated pulmonary edema by
improving AFC through the upregulation of ENaC via the PI3K/SGK1
pathway in LPS-induced lung injury. A model of ALI without
hyperglycemia was used to maintain the glucose levels within the
normal range since hyperglycemia contributes to the inflammatory
response, and lung injury is attenuated by insulin treatment in
euglycemia (24–26). Previous results from clinical
studies have confirmed that glucose control is very important in
critically ill patients, leading to decreased mortality and
morbidity (27,28). In addition, the dose and rate of
human insulin infused by micro-osmotic pumps in the present study
had an anti-inflammatory effect rather than an effect on the
modulation of glucose metabolism, since it was observed that
insulin treatment did not affect the glucose levels in the
LPS-induced lung injury group.
Toll-like receptor 4 (TLR4) is detected in a number
of immune cells, including monocytes, macrophages, dendritic cells
and several T cell populations, and is part of the receptor complex
that binds LPS from the Gram-negative bacterial cell wall and
endogenous ligands, including heat-shock proteins and other
inflammatory mediators (29).
LPS-induced activation of TLR4 leads to the dimerization of TLR4
monomers and the generation of nucleus-seeking nuclear factor
(NF)-κB transcription factors. Numerous pro-inflammatory cytokine
and chemokine genes contain NF-κB response elements in their
promoter regions, such as tumor necrosis factor-α and interleukin-6
(30). LPS-stimulated cytokine
release is achieved through the TLR4-mediated NF-κB signaling
pathway (31).
Different inflammatory mediators, such as cytokines,
are released following LPS stimulation to recruit activated
neutrophils into the injured lung, which is the main cause of
pulmonary edema and the development of ALI (32,33).
Activated neutrophils migrate into alveolar spaces and pulmonary
epithelium by CXC chemokines, which lead to alveolar capillary
barrier leakage, and interstitial and alveolar edema by reactive
oxygen species (33,34). In the present study, LPS was
administered to establish a well-characterized model of ALI,
resulting in an increase in protein, total cell count and
neutrophils in the BALF, as well as an elevation in bronchoalveolar
epithelial permeability, which indicated alveolar capillary barrier
damage. Increased alveolar capillary permeability results in edema
fluid accumulation in the alveolar spaces. Treatment with insulin
protected the alveolar capillary barrier that was damaged as a
result of the LPS-induced lung injury; however, SGK1 siRNA reversed
the protective effect of insulin. In addition, LPS-treated mice
exhibited typical pathological alterations, such as interstitial
edema, thickened alveolar septa, proteinaceous debris and
inflammatory cell infiltration in the alveolar space. Insulin
treatment alleviated these pathological changes, whereas this
alleviation was inhibited by SGK1 siRNA. The results indicated that
insulin attenuated pulmonary edema with the involvement of
SGK1.
It is well recognized that AFC is an effective way
to remove edema from the alveolar spaces (35). In the present study, insulin
improved AFC and decreased pulmonary edema in the LPS-induced lung
injury group. However, SGK1 siRNA blocked the effect of insulin on
AFC in the LPS-treated group, which indicated the essential
involvement of SGK1. In addition, the use of amiloride, a sodium
channel inhibitor, inhibited the insulin-stimulated AFC. This
supported the result that ENaC may participate in AFC, which is
consistent with the findings of previous studies (24,36).
Lung epithelium injury was also reported to be a
determinant for the development of protein-rich pulmonary edema in
ARDS (37). In particular, lung
epithelial apoptosis is considered to be an important pathogenetic
mechanism in ALI (38,39). An acceleration of lung epithelial
cell apoptosis may contribute to the destruction of the alveolar
epithelial barrier, a promotion of protein-rich edema in the
alveoli and altered fluid clearance from the alveolar space in
ALI/ARDS (1,40). In the present study, insulin
exerted a protective effect on the lung epithelium by reducing the
apoptosis of alveolar epithelial cells, indicating the alleviation
of pulmonary edema and the important role of SGK1 in the protection
against cell apoptosis, as described previously (41).
We previously reported that ENaC served an important
role in the regulation of sodium and water balance in AFC (24,42,43).
Therefore, the regulation of ENaC by insulin, possibly via the
PI3K/SGK1 signaling pathway, was further investigated in the
current study. SGK1, a serine/threonine protein kinase, was
reported to be a key regulator of sodium transport by hormones,
such as insulin, in mammalian epithelia (44–47).
Furthermore, the activation of SGK1 was dependent on the
phosphorylation of S422 in the hydrophobic motif at its COOH
terminus by the PI3K-dependent pyruvate dehydrogenase kinase 2
pathway (47,48). It has been recognized that a
central function of activated SGK1 was to increase the expression
of ENaC in the cell surface by inhibiting the ubiquitin ligase
Nedd4-2 (49,50). In the present study, the expression
levels of α-, β- and γ-ENaC, and the level of phosphorylated SGK1
were increased by insulin treatment, whereas these were decreased
by co-treatment with SGK1 siRNA in the LPS-induced lung injury
group. In vitro, PI3K siRNA or SGK1 siRNA prevented the
insulin-induced increase in the expression levels of α-, β- and
γ-ENaC, and phosphorylated SGK1. The increased expression of ENaC
by insulin-induced SGK1 was possibly dependent on the
phosphorylation of Nedd4-2 in a PY motif-dependent manner (50,51).
Taken together, in the present study, the effect of insulin on AFC
was further illustrated through the upregulation of α-, β- and
γ-ENaC via the PI3K/SGK1 signaling pathway, which was a further
investigation of our previous findings (18).
In conclusion, the present results demonstrated that
insulin protected the lung epithelium and attenuated pulmonary
edema in mice with LPS-induced lung injury, without affecting the
blood glucose levels, and this effect was achieved through the
upregulation of ENaC via the PI3K/SGK1 pathway. While further
research is required to fully understand the role of insulin in the
complex mechanisms of ALI/ARDS, the present study provides novel
evidence of the protective role of insulin in AFC associated with
ALI.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural
Science Foundation of China (grant no. 81600058).
Availability of data and material
All data analyzed during the present study are
included in the published article. The raw datasets used for the
analysis are available from the corresponding author on reasonable
request.
Authors' contributions
WD and DW participated in the design of the study.
CL and JT performed the animal experiments. JH and YZ performed the
immunocytochemistry, RT-qPCR and western blotting assays. WD and DW
performed the statistical analysis, interpreted the data and
drafted the manuscript. WD and DW are accountable for all aspects
of the work. All authors contributed to the interpretation of the
data and critical revision of the manuscript. Each individual
author read and approved the final manuscript.
Ethics approval and consent to
participate
All animal experiments were approved by the Ethics
Committee of the Second Affiliated Hospital of Chongqing Medical
University (Chongqing, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ENaC
|
epithelial sodium channel
|
|
ALI
|
acute lung injury
|
|
ARDS
|
acute respiratory distress
syndrome
|
|
AFC
|
alveolar fluid clearance
|
|
LPS
|
lipopolysaccharide
|
|
PCR
|
polymerase chain reaction
|
|
PI3K
|
phosphatidylinositol 3-kinase
|
|
SGK1
|
serum/glucocorticoid-inducible
kinase-1
|
|
TUNEL
|
terminal deoxynucleotidyl transferase
dUTP nick end labeling
|
|
BALF
|
bronchoalveolar lavage fluid
|
|
TLR4
|
Toll-like receptor 4
|
References
|
1
|
Máca J, Jor O, Holub M, Sklienka P, Burša
F, Burda M, Janout V and Ševčík P: Past and present ARDS mortality
rates: A systematic review. Respir Care. 62:113–122. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Villar J, Sulemanji D and Kacmarek RM: The
acute respiratory distress syndrome: Incidence and mortality, has
it changed? Curr Opin Crit Care. 20:3–9. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Azzam ZS and Sznajder JI: Lung edema
clearance: Relevance to patients with lung injury. Rambam
Maimonides Med J. 6:2015. View Article : Google Scholar
|
|
4
|
Berthiaume Y and Matthay MA: Alveolar
edema fluid clearance and acute lung injury. Respir Physiol
Neurobiol. 159:350–359. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Matalon S, Bartoszewski R and Collawn JF:
Role of epithelial sodium channels in the regulation of lung fluid
homeostasis. Am J Physiol Lung Cell Mol Physiol. 309:L1229–L1238.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lee JW, Krasnodembskaya A, McKenna DH,
Song Y, Abbott J and Matthay MA: Therapeutic effects of human
mesenchymal stem cells in ex vivo human lungs injured with live
bacteria. Am J Respir Crit Care Med. 187:751–760. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Folkesson HG and Matthay MA: Alveolar
epithelial ion and fluid transport: Recent progress. Am J Respir
Cell Mol Biol. 35:10–19. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hummler E, Barker P, Gatzy J, Beermann F,
Verdumo C, Schmidt A, Boucher R and Rossier BC: Early death due to
defective neonatal lung liquid clearance in alpha-ENaC-deficient
mice. Nat Genet. 12:325–328. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Randrianarison N, Clerici C, Ferreira C,
Fontayne A, Pradervand S, Fowler-Jaeger N, Hummler E, Rossier BC
and Planès C: Low expression of the beta-ENaC subunit impairs lung
fluid clearance in the mouse. Am J Physiol Lung Cell Mol Physiol.
294:L409–L416. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Elias N, Rafii B, Rahman M, Otulakowski G,
Cutz E and O'Brodovich H: The role of alpha-, beta-, and gamma-ENaC
subunits in distal lung epithelial fluid absorption induced by
pulmonary edema fluid. Am J Physiol Lung Cell Mol Physiol.
293:L537–L545. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Marone R, Cmiljanovic V, Giese B and
Wymann MP: Targeting phosphoinositide 3-kinase: Moving towards
therapy. Biochim Biophys Acta 1784. 159–185. 2008.
|
|
12
|
Record RD, Froelich LL, Vlahos CJ and
Blazer-Yost BL: Phosphatidylinositol 3-kinase activation is
required for insulin-stimulated sodium transport in A6 cells. Am J
Physiol. 274:E611–E617. 1998.PubMed/NCBI
|
|
13
|
Blazer-Yost BL, Esterman MA and Vlahos CJ:
Insulin-stimulated trafficking of ENaC in renal cells requires
PI3-kinase activity. Am J Physiol Cell Physiol. 284:C1645–C1653.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cohen P: The origins of protein
phosphorylation. Nat Cell Biol. 4:E127–E130. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Soundararajan R, Melters D, Shih IC, Wang
J and Pearce D: Epithelial sodium channel regulated by differential
composition of a signaling complex. Proc Natl Acad Sci USA.
106:7804–7809. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Perrotti N, He RA, Phillips SA, Haft CR
and Taylor SI: Activation of serum- and glucocorticoid-induced
protein kinase (Sgk) by cyclic AMP and insulin. J Biol Chem.
276:9406–9412. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu ML, Dong HY, Zhang B, Zheng WS, Zhao
PT, Liu Y, Niu W, Xu DQ and Li ZC: Insulin reduces LPS-induced
lethality and lung injury in rats. Pulm Pharmacol Ther. 25:472–477.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhu T, Zhang W and Wang DX: Insulin
up-regulates epithelial sodium channel in LPS-induced acute lung
injury model in rats by SGK1 activation. Injury. 43:1277–1283.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang X, Huang H, Yang T, Ye Y, Shan J,
Yin Z and Luo L: Chlorogenic acid protects mice against
lipopolysaccharide-induced acute lung injury. Injury. 41:746–752.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dobbs LG: Isolation and culture of
alveolar type II cells. Am J Physiol. 258:L134–L147.
1990.PubMed/NCBI
|
|
21
|
Lomas-Neira JL, Chung CS, Wesche DE, Perl
M and Ayala A: In vivo gene silencing (with siRNA) of pulmonary
expression of MIP-2 versus KC results in divergent effects on
hemorrhage-induced, neutrophil-mediated septic acute lung injury. J
Leukoc Biol. 77:846–853. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Matute-Bello G, Downey G, Moore BB,
Groshong SD, Matthay MA, Slutsky AS and Kuebler WM; Acute Lung
Injury in Animals Study Group, : An official American Thoracic
Society workshop report: Features and measurements of experimental
acute lung injury in animals. Am J Respir Cell Mol Biol.
44:725–738. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sakuma T, Hida M, Nambu Y, Osanai K, Toga
H, Takahashi K, Ohya N, Inoue M and Watanabe Y: Effects of hypoxia
on alveolar fluid transport capacity in rat lungs. J Appl Physiol
(1985). 91:1766–1774. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Deng W, Li CY, Tong J, Zhang W and Wang
DX: Regulation of ENaC-mediated alveolar fluid clearance by insulin
via PI3K/Akt pathway in LPS-induced acute lung injury. Respir Res.
13:292012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hagiwara S, Iwasaka H, Hasegawa A, Koga H
and Noguchi T: Effects of hyperglycemia and insulin therapy on high
mobility group box 1 in endotoxin-induced acute lung injury in a
rat model. Crit Care Med. 36:2407–2413. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen HI, Yeh DY, Liou HL and Kao SJ:
Insulin attenuates endotoxin-induced acute lung injury in conscious
rats. Crit Care Med. 34:758–764. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fahy BG, Sheehy AM and Coursin DB: Glucose
control in the intensive care unit. Crit Care Med. 37:1769–1776.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
van den Berghe G, Wouters P, Weekers F,
Verwaest C, Bruyninckx F, Schetz M, Vlasselaers D, Ferdinande P,
Lauwers P and Bouillon R: Intensive insulin therapy in critically
ill patients. N Engl J Med. 345:1359–1367. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Stevens CW, Aravind S, Das S and Davis RL:
Pharmacological characterization of LPS and opioid interactions at
the toll-like receptor 4. Br J Pharmacol. 168:1421–1429. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kenny EF and O'Neill LA: Signaling
adaptors used by Toll-like receptors: An update. Cytokine.
43:342–349. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Guijarro-Muñoz I, Compte M,
Álvarez-Cienfuegos A, Álvarez-Vallina L and Sanz L:
Lpopolysaccharide activates Toll-like receptor 4 (TLR4)-mediated
NF-κB signaling pathway and proinflammatory response in human
pericytes. J Biol Chem. 289:2457–2468. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Abraham E: Neutrophils and acute lung
injury. Crit Care Med. 31:S195–S199. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Reutershan J and Ley K: Bench-to-bedside
review: Acute respiratory distress syndrome-how neutrophils migrate
into the lung. Crit Care. 8:453–461. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fialkow L, Wang Y and Downey GP: Reactive
oxygen and nitrogen species as signaling molecules regulating
neutrophil function. Free Radic Biol Med. 42:153–164. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Eaton DC, Helms MN, Koval M, Bao HF and
Jain L: The contribution of epithelial sodium channels to alveolar
function in health and disease. Annu Rev Physiol. 71:403–423. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bellmeyer A, Martino JM, Chandel NS, Scott
Budinger GR, Dean DA and Mutlu GM: Leptin resistance protects mice
from hyperoxia-induced acute lung injury. Am J Respir Crit Care
Med. 175:587–594. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Matthay MA and Zemans RL: The acute
respiratory distress syndrome: Pathogenesis and treatment. Annu Rev
Pathol. 6:147–163. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Martin TR, Nakamura M and Matute-Bello G:
The role of apoptosis in acute lung injury. Crit Care Med.
31:S184–S188. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Perl M, Lomas-Neira J, Chung CS and Ayala
A: Epithelial cell apoptosis and neutrophil recruitment in acute
lung injury-a unifying hypothesis? What we have learned from small
interfering RNAs. Mol Med. 14:465–475. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Gropper MA and Wiener-Kronish J: The
epithelium in acute lung injury/acute respiratory distress
syndrome. Curr Opin Crit Care. 14:11–15. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Mikosz CA, Brickley DR, Sharkey MS, Moran
TW and Conzen SD: Glucocorticoid receptor-mediated protection from
apoptosis is associated with induction of the serine/threonine
survival kinase gene, sgk-1. J Biol Chem. 276:16649–16654. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Deng J, Wang DX, Deng W, Li CY, Tong J and
Ma H: Regulation of alveolar fluid clearance and ENaC expression in
lung by exogenous angiotensin II. Respir Physiol Neurobiol.
181:53–61. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Deng W, Wang DX, Zhang W and Li CY:
Regulation of epithelial sodium channel α-subunit expression by
adenosine receptor A2a in alveolar epithelial cells.
Chin Med J (Engl). 124:1551–1555. 2011.PubMed/NCBI
|
|
44
|
Lang F, Bohmer C, Palmada M, Seebohm G,
Strutz-Seebohm N and Vallon V: (Patho)physiological significance of
the serum- and glucocorticoid-inducible kinase isoforms. Physiol
Rev. 86:1151–1178. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Loffing J, Flores SY and Staub O: Sgk
kinases and their role in epithelial transport. Annu Rev Physiol.
68:461–490. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wulff P, Vallon V, Huang DY, Völkl H, Yu
F, Richter K, Jansen M, Schlünz M, Klingel K, Loffing J, et al:
Impaired renal Na(+) retention in the sgk1-knockout mouse. J Clin
Invest. 110:1263–1268. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Webster MK, Goya L, Ge Y, Maiyar AC and
Firestone GL: Characterization of sgk, a novel member of the
serine/threonine protein kinase gene family which is
transcriptionally induced by glucocorticoids and serum. Mol Cell
Biol. 13:2031–2040. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Park J, Leong ML, Buse P, Maiyar AC,
Firestone GL and Hemmings BA: Serum and glucocorticoid-inducible
kinase (SGK) is a target of the PI3-kinase-stimulated signaling
pathway. EMBO J. 18:3024–3033. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Snyder PM, Olson DR and Thomas BC: Serum
and glucocorticoid-regulated kinase modulates Nedd4-2-mediated
inhibition of the epithelial Na+ channel. J Biol Chem. 277:5–8.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Debonneville C, Flores SY, Kamynina E,
Plant PJ, Tauxe C, Thomas MA, Münster C, Chraïbi A, Pratt JH,
Horisberger JD, et al: Phosphorylation of Nedd4-2 by Sgk1 regulates
epithelial Na(+) channel cell surface expression. EMBO J.
20:7052–7059. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wiemuth D, Lott JS, Ly K, Ke Y,
Teesdale-Spittle P, Snyder PM and McDonald FJ: Interaction of
serum- and glucocorticoid regulated kinase 1 (SGK1) with the
WW-domains of Nedd4-2 is required for epithelial sodium channel
regulation. PLoS One. 5:e121632010. View Article : Google Scholar : PubMed/NCBI
|