Introduction
Endometrial cancer (EC) is one of the three most
common malignancies in the female reproductive system. With the
application of estrogen, increased obesity and dietary changes in
recent years, the incidence of EC has increased (1). For patients with metastatic tumor,
there is no effective treatment until now, Until now, there is no
new treatment available. Standard treatment consists of primary
hysterectomy and bilateral salpingo-oophorectomy, often using
minimally invasive approaches (laparoscopic or robotic). Lymph node
surgical strategy is contingent on histological factors (subtype,
tumor grade, involvement of lymphovascular space), disease stage
(including myometrial invasion), patients' characteristics (age and
comorbidities), and national and international guidelines. Adjuvant
treatment is tailored according to histology and stage. Various
classifications are used to assess the risks of recurrence and to
determine optimum postoperative management (2). Therefore, an in-depth study of the
role and mechanism of EC metastasis is very important for the
effective treatment of EC. Tumor-associated macrophages (TAMs) are
a group of cells with immunosuppressive functions, and are
important components of the tumor microenvironment. TAMs regulate
tumorigenesis, development and metastasis in a variety of ways
(3,4). The proliferation of TAMs and the
secretion of inflammatory factors have promoted the development of
type I EC (5). The density of TAMs
in type II endometrial carcinoma is almost twice that of type I EC,
and this difference may be due to the predominant result of M1
macrophages in the type II EC (6).
The chemokine (C-X-C motif) ligand 8 secreted by TAM downregulates
estrogen receptor alpha expression in EC cells by acting on
homeobox B 13 protein may is involved in cancer invasion (7). TAMs also is involved in the
production of neovascularization of EC and the infiltration of the
myometrium by cancer cells (8). In
a study of macrophage reactivity in 98 patients with primary EC,
~40% of colony stimulating factor-1 (CSF-1)-expressing EC cells
underwent infiltration of a large number of macrophages, suggesting
that CSF-1 expression promotes the progression of endometrial
cancer, and CSF-1 levels are closely associated with the degree of
malignancy of primary tumors and their corresponding lymph node
metastases (9). Therefore, CSF-1
may be involved in macrophage infiltration and the development of
EC.
CSF-1 binds to the CSF-1 receptor (CSF-1R), which is
expressed on the surface of macrophages, induces proliferation and
infiltration of TAMs, and is involved in tumor cell proliferation,
invasion and migration (10). A
previous study found that CSF-1 is highly expressed in multiple
types of tumors, and CSF-1 levels in the blood circulation could be
used as a molecular marker for lung cancer, breast cancer, prostate
cancer and lymphoma (11). A
previous study reported that CSF-1R inhibitors can significantly
reduce the volume of glioblastoma and tumor invasion by inhibiting
TAMs, which indicates that there is an inflammatory cytokine
interaction between TAMs and glioblastoma, and the role of mutual
protection is presence between EC cells and TAMs (12). Thus, overexpression of CSF-1 or
CSF-1R is positively associated with aggressive and poor prognosis
of EC, and blocking CSF-1 or CSF-1R may inhibit EC progression.
However, the mechanism of CSF-1 and CSF-1R pathway that regulates
tumor invasion, tumor immunity and tumor angiogenesis is unclear.
In the present study, through inhibiting the expression of CSF-1
and blocking CSF-1R, it was demonstrated that CSF-1 or CSF-1R
inhibition may serve a similar role in blocking macrophage
migration and EC cell proliferation.
Materials and methods
Patients
The records of 10 patients with endometrial
carcinoma and 10 patients with endometrial benign lesions who
underwent primary surgery at Huai'an First People's Hospital,
Nanjing Medical University (Huai'an, China) between January 2016
and December 2016 were reviewed retrospectively. Inclusion
criteria: Age, 35–65 years old; no hormone drug treatment for the
past three months. Patients with non-endometrioid carcinoma (clear
cell carcinoma and serous adenocarcinoma) and those who had
received preoperative chemotherapy were excluded. The present study
was approved by the Medical Ethics Committee of the Nanjing Medical
University and all patients provided their written consent.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from endometrial tissue or
EC cell lines (Cell bank, Shanghai Institute for Biological
Science, China) using TRIzol® reagent (Life
Technologies; Thermo Fisher Scientific, Inc., Waltham, MA, USA).
After the RNA pellet was resuspended in diethyl
pyrocarbonate-treated H2O, the purity and concentration
were determined by spectrophotometry, and the final concentration
was adjusted to 1 µg/µl. Reverse transcription was performed using
a PrimeScript™ II 1st Strand cDNA Synthesis kit (Takara Bio, Inc.,
Otsu, Japan) according to the manufacturer's instructions (5XRT
Master Mix, 1 µg RNA and Nuclease-free Water). The thermocycling
conditions were 37°C, 15 min; 50°C, 5 min and 98°C, 5 min (then
held at 4°C), and cDNA was used as a template for PCR amplification
of the target mRNA fragments. In order to detect the relative
expression levels of CSF-1, CSF-1R, Janus kinase 1 (JAK-1),
phosphoinositide 3-kinase (PI3K), AKT, cyclin-dependent kinase
(CDK) 2, CDK4 and retinoblastoma-associated protein (Rb),
SYBR® Premix Ex Taq™ (Tli RNaseH Plus) kit
(Takara Biotechnology, Co., Ltd., Dalian, China) was used for the
RT-qPCR reaction, and the thermocycling conditions were 60 sec at
95°C, followed by 40 cycles of 15 sec at 95°C, 15 sec at 60°C and
45 sec at 72°C. A comparative quantification cycle (ΔCq) method was
used to quantify target mRNAs (13) and GAPDH was used as internal
reference gene. PCR primers were: CSF-1, forward
5′-TGTGGTTTGTGGGAAAGCAG-3′, reverse 5′-CTTCAGGCTCCTCTCTCTGG-3′;
CSF-1R, forward 5′-GAGGATGCTGTCCTGAAGGT-3′, reverse
5′-GTACAGGCTCCCAGAAGGTT-3′; JAK-1, forward
5′-ATGGCCAGATGACAGTCACA-3′, reverse 5′-TGTCCGATTGGATGGTTGGA-3′;
PI3K, forward 5′-AGAAGCCTTCCTCTGTGTCC-3′, reverse
5′-TCTTGCACAGCATCTCGTTG-3′; AKT, forward
5′-CTTTCGGCAAGGTGATCCTG-3′, reverse 5′-GTACTTCAGGGCTGTGAGGA-3′;
CDK2, forward 5′-GCCTTATGAGGCAGGTGAGA-3′, reverse
5′-GTAGGAGGTGGACGTCAGAG-3′; CDK4, forward
5′-ACCGTTTACAAGGCCAGAGA-3′, reverse 5′-ATCATGGGCCTCAGGTGAAA-3′; Rb,
forward 5′-TTCCAGACCCAGAAGCCATT-3′, reverse
5′-TCTGGGTGCTCAGACAGAAG-3′; and GAPDH, forward
5′-CTTTGTCAAGCTCATTTCCTGG-3′, reverse
5′-TCTTGCTCAGTGTCCTTGC-3′.
ELISA
Endometrial tissue (1–2 cm3) was
collected from 10 patients with endometrial benign lesions and from
10 patients with EC. Supernatants from the 20 cases of endometrial
tissues (PBS and protease inhibitor were added to the tissue, and
tissue homogenizer and sonicator were used to obtain supernatants)
and cultured T-HESC, ECC-1, HEC-1A cells (density >90%) which
were all cultured in Dulbecco's modified Eagle's medium (DMEM;
Invitrogen; Thermo Fisher Scientific, Inc.) supplemented with 10%
heat-inactivated fetal bovine serum (FBS; Sigma-Aldrich; Merck
KGaA) at 37°C in a 95% humidified atmosphere of 95% air and 5%
CO2 were collected to measure the concentrations of
CSF-1 by microplate reader, according to the manufacturer's
protocol (DY216; R&D systems, Inc., Minneapolis, MN, USA).
Briefly, after normal endometrial tissue and EC tissue were
homogenized and cultured cell supernatants were collected, the
total protein concentration was determined by BCA Protein Assay kit
(P0010S; Beyotime Institute of Biotechnology, Shanghai, China).
According to the manufacturer's protocol, the protein supernatant
was diluted at different dilutions (1:1, 1:5 and 1:10), and the
concentration of CSF-1 was calculated in different tissues and
cells on the basis of the concentration of protein standards.
Western blot analysis
Western blot analysis was performed as previously
described (14). Briefly, total
protein (5 mg) was extracted from endometrial tissue or cultured
cells (2×106) using radioimmunoprecipitation lysis
buffer (Sangon Biotech, Co., Ltd., Shanghai, China) and total
protein concentration was determined by BCA Protein Assay kit
(P0010S; Beyotime Institute of Biotechnology). Primary polyclonal
anti-rabbit CSF-1R (67455, 1:2,000), anti-rabbit JAK-1 (29261,
1:2,000), anti-rabbit PI3K (4249, 1:1,500), anti-phosphorylated
rabbit (p)-AKT (4060, 1:2,000), anti-rabbit CDK2 (2546, 1:1,000),
anti-rabbit CDK4 (12790, 1:1,000) and anti-rabbit p-Rb (8516,
1:1,000) were all purchased from Cell Signaling Technology Inc.
(Danvers, MA, USA), and anti-GAPDH antibodies (AF0006, 1:5,000)
were purchased from Beyotime Institute of Biotechnology. The
primary antibody was added to the membrane for incubation overnight
at 4°C. The membrane was washed three times for 5 min with TBST at
room temperature. Following a secondary horseradish peroxidase
(HRP)-conjugated goat anti-rabbit (A0208) or goat anti-mouse
(A0216) immunoglobulin (Ig)G antibody (1:5,000, Beyotime Institute
of Biotechnology) incubation for 2 h at room temperature, signals
were detected by the EasyBlot Chemiluminescence kit (Sangon
Biotech, Co., Ltd.) and quantified using Image-Pro Plus 6.0
software (Media Cybernetics, Inc, Rockville, MD, USA).
Cell culture and immunocytochemistry
staining
U937, T-HESC, HEC-1A and ECC-1 cell lines (Cell
bank, Shanghai Institute for Biological Science, China) were all
cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with
10% heat-inactivated fetal bovine serum (FBS; Sigma-Aldrich; Merck
KGaA) at 37°C in a 95% humidified atmosphere of 95% air and 5%
CO2. In U937 and EC co-culture system, 100 U/ml M-CSF
(Beyotime Institute of Biotechnology) were added to the culture
medium at 37°C for 24 h to induce M2 type macrophages. Once U937
cells were successfully induced to M2 macrophages (expressing Arg-1
and CD206), they were used in subsequent experiments. Macrophage
subtype was confirmed by inducible nitric oxide synthase (iNOS;
610328; BD Biosciences, Franklin Lakes, NJ, USA) and CD86 (6553689;
BD Biosciences; double positive of M1 macrophage) and Arginase
(Arg-1; PA5-29645; Thermo Fisher Scientific, Inc.) and CD206
(MA5-16868; Thermo Fisher Scientific, Inc.; double positive of M2
macrophage). Prior to CSF-1R immunofluorescence staining or crystal
violet staining, cells were fixed with 4% paraformaldehyde for 15
min at room temperature. Primary mouse CSF-1R monoclonal antibody
(sc46662, Santa Cruz Biotechnology, Inc., Dallas, TX, USA; 1:100)
were revealed with specific goat anti-mouse Alexa Fluor®
555 (IgG H&L)-conjugated secondary antibodies (A0473; 1:50;
Beyotime Institute of Biotechnology) for 1 h at 37°C. Ki67 antibody
(AF1738; 1:100; Beyotime Institute of Biotechnology) was used to
examine the proliferation of EC cells. According to the
conventional fluorescent staining process, goat anti-mouse Alexa
Fluor® 555 secary antibodies were used to examine the
numbers of Ki67-positive EC cells. Subsequently, the cell
morphology of U937 or EC cells were observed under a fluorescence
microscope. Crystal violet purchased from Beyotime Institute of
Biotechnology and was used to stain U937 cells for 15 min at room
temperature.
ECC-1 or HEC-1A cell transfection
CSF-1-specific small interfering (si)RNA (siCSF-1;
sense, 5′-ACGUGGCUAAAGUGUUAAAG-3′; antisense,
5′-CCUGUUCUGCAGUUCCUUCCUUGU-3′), and its corresponding
non-silencing negative control siRNA (siNeg; sense,
5′-GGCAAAUUGCCCUUAUCCA-3′; antisense, 5′-AACGUUUAAACCGGUUACGUA3′)
were designed and synthesized by GeneChem, Inc. (Shanghai, China).
ECC-1 or HEC-1A cells (50% seeding density) were cultured in DMEM
supplemented with 10% heat-inactivated fetal calf serum (Thermo
Fisher Scientific, Inc.) at 37°C under 5% CO2 in a
humidified incubator. Cells in the exponential growth phase were
grown for 24 h and then transfected using Lipofectamine®
3000 (Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocol. The concentration of siCSF-1 and siNeg
were maintained at 100 nM. The culture medium was replaced with
DMEM plus 10% FBS after 6 h of transfection, then 10 µg/ml of
puromycin was added at 37°C for 24 h for screening positive cells,
and 2 ug/ml puromycin were added to the cell culture medium to
maintain positive cell selection. ECC-1 or HEC-1A cells transfected
with siCSF-1 and siNeg were used for wound healing assay and
Chemotactic migration assay.
CCK-8 assay
Cell proliferation or growth rate was evaluated with
the CCK-8 kit (Beyotime Institute of Biotechnology). U937 cells
(1×104/well) were cultured in 96-well plates, or U937
cells (5×104/well) were co-cultured with T-HESC
(5×104/well), ECC-1 (5×104/well) or HEC-1A
(5×104/well) which were seeded in 24-well Transwell
plates (Corning Inc., Corning, NY, USA), with U937 cells cultured
on the upper chamber. After adding different concentrations of
CSF-1 (10, 50, 100, 250 and 500 U/ml) and 10 µM PLX3397 into the
culture medium on the upper chamber, cells were incubated with 20
µl CCK-8 for 1 h at 37°C, according to the manufacturer's protocol.
Cell proliferation or growth rate was analyzed using a microplate
reader to determine the absorbance at 450 nm (Biotec, Dresden,
Germany).
Wound-healing assay
U937 cell migratory ability was measured using the
wound-healing assay. Briefly, U937 cells were seeded in 6-well
plates and grown to 90% confluency. The medium was replaced with
cell (T-HESC, ECC-1 or HEC-1A) culture supernatant or fresh medium
containing 100 U/ml CSF-1 or 10 µM PLX3397; PBS was used as a
vehicle control. Following the fresh medium exchange, a scratch was
created with a 20 µl pipette tip and an image was captured
immediately. After 24 h incubation at 37°C, images were captured
again and the cell migration was quantified randomly by counting
cells that had moved above the reference line. The control group
was calculated as 100 and the experimental group compared with the
control group to ascertain the difference between the groups.
Chemotactic migration assay
Chemotactic migration of U937 cells was measured
using a Transwell chamber with 6.5 mm polycarbonate membrane (8 µm
pore size) (Corning Inc.). Normal U937 cells
(5×104/well) with fresh medium containing 10 µM PLX3397
or 100 U/ml CSF-1 were plated on the upper chamber, ECC-1 or HEC-1A
cells (5×104/well) that were stably transfected with
CSF-1 interference plasmid (siCSF-1) or control plasmid (siNeg),
were cultured on the lower chamber. The U937 cells were loaded at
1×105 cells/well to the upper chambers and allowed to
migrate for 16 h at 37°C. The cells were subsequently fixed with 4%
paraformaldehyde at room temperature for 15 min and stained with
crystal violet at room temperature for 10 min, non-migrating cells
on the upper surface of the membrane were removed using a cotton
swab. The cells that had migrated to the lower surface of the
membrane were randomly counted (5×103/well) under a
brightfield microscope.
Statistical analysis
Statistical analysis was performed using SPSS 18.0
software (SPSS Inc., Chicago, IL, USA). Data are presented as the
mean ± standard deviation of three independent experiments. One-way
analysis of variance was used to determine statistical significance
of differences between groups followed by Turkey post hoc test for
comparison of two or more groups. P<0.05 was considered to
indicate a statistically significant difference.
Results
Expression of CSF-1 and CSF-1R in EC
cells and macrophages in vivo and in vitro
In order to investigate CSF-1 expression in
endometrial tissue and corresponding cells, RT-qPCR and ELISA were
used to detect the RNA and protein expression levels, respectively,
of CSF-1 in normal endometrial tissue, EC tissue and T-HESC, ECC-1
and HEC-1A EC cells. It was found that not only RNA but also
protein expression levels in EC tissues and cell lines were
significantly higher compared with the respective expression levels
in normal endometrial tissue and T-HESC cells (Fig. 1A-D). Similar CSF-1, the protein
expression levels of CSF-1R were significantly higher in EC tissue
compared with normal tissue (Fig. 1E
and F). In order to further investigate the expression of
CSF-1R in macrophages, immunofluorescence staining was used to
observe the expression of CSF-1R in U937 cell lines (Fig. 1G), which demonstrated that CSF-1R
was highly positive on U937 cell membrane. CSF-1 and CSF-1R were
both expressed in normal endometrial tissue and corresponding
cells, especially in EC tissue. These results suggested that EC
cells may recruit macrophages by secreting CSF-1. In order to
examine the function of CSF-1, different concentrations of CSF-1
(10, 50, 100 and 500 U/ml) were used to treat U937 cell, and
proliferation was investigated. The result demonstrated that CSF-1
concentrations >250 U/ml promoted the proliferation of U937
cells (Fig. 1H). Therefore 100
U/ml CSF-1 which did not promote cell proliferation was used in
order to further observe the induction of macrophage migration by
EC cells.
EC cells promote macrophage migration
by secreting CSF-1
In order to observe whether EC cells could promote
macrophage migration, EC cell supernatant was used for
wound-healing and migration assay. In a wound-healing assay, the
U937 culture medium was replaced with the supernatant from T-HESC,
ECC-1 or HEC-1A cell cultures following a scratch on U937 cells.
ECC-1 and HEC-1A supernatant treatment promoted more U937 cells to
migrate compare with normal endometrial cell line T-HESC (Fig. 2A), which indicated that the
supernatant of ECC-1 and HEC-1A cells may contain some components
that are capable of promoting the migration of U937 cells. The
CSF-1R inhibitor PLX3397 was added to the culture supernatant of
U937 cells to observe whether CSF-1R is a key protein affecting
U937 cell migration. It was found that the number of migrated U937
cells decreased when CSF-1R was blocked (Fig. 2A), which suggested that CSF-1 in
ECC-1 and HEC-1A culture supernatant may be the key molecule that
induces U937 cell migration. Additionally, a chemotactic migration
assay was used to confirm the role of CSF-1 in promoting U937 cell
migration, and it was demonstrated that EC cells induced U937 cell
migration, which was significantly reduced by blocking the CSF-1R
on macrophages (Fig. 2B).
To further confirm the role of CSF-1 in inducing
macrophage migration, 100 U/ml CSF-1 was directly added to U937
cell culture supernatant, and it was shown that CSF-1 may promote
U937 cell migration, as measured by a wound healing (Fig. 3A) and a chemotactic migration assay
(Fig. 3B). In order to further
confirm the role of CSF-1, the expression of CSF-1 in ECC-1 and
HEC-1A cells was reduced by siCSF-1 transfection; RT-qPCR and ELISA
assay were used to examine the transfection efficiency of the
siCSF-1 plasmid and confirmed the expression of CSF-1 was
significantly decreased in transfected ECC-1 and HEC-1A cells
(Fig. 3C). When the expression of
CSF-1 in EC cells was silenced, the supernatant of ECC-1 and HEC-1A
cells also failed to promote U937 cell migration (Fig. 3D and E). These results indicated
that CSF-1 secreted by EC cells may be a key factor that promotes
macrophage migration.
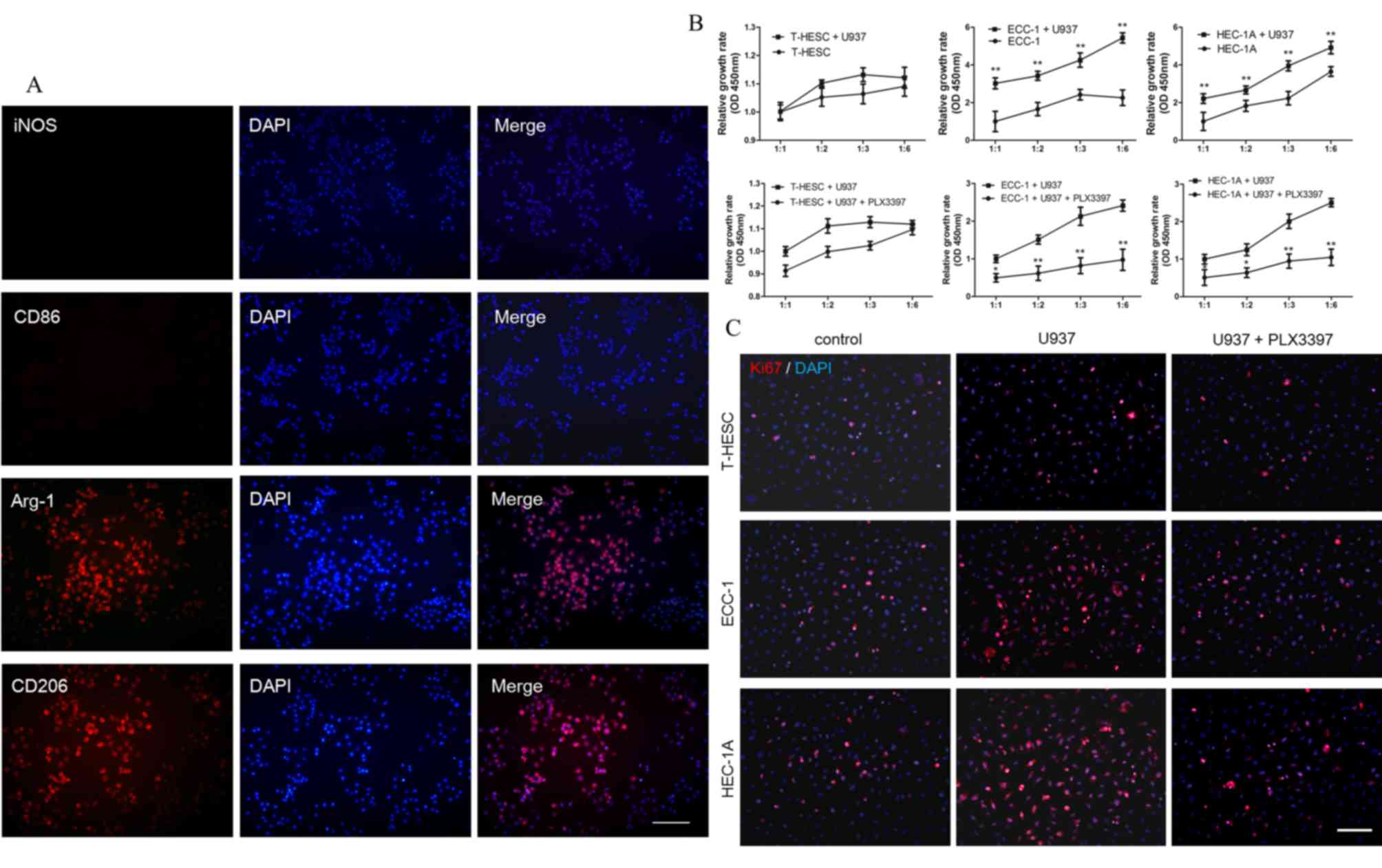
EC cells induce U937 cells into M2
macrophages (TAMs), which promote the proliferation of EC
cells
In order to investigate the effects of macrophages
migrating to EC tissue and whether macrophages affected the
proliferation of EC cells, macrophages and EC cells were
co-cultured. In the co-cultured system, EC cells were first
cultured in 24-well plates and U937 cells cultured on coverslips
respectively. When cell density reached the experimental
requirements, the coverslip was moved into the 24-well to establish
a co-culture system. U937 and ECC-1 cell lines were co-cultured
in vitro for 24 h, and makers of M1 macrophage [inducible
nitric oxide synthase (iNOS) and CD86] and M2 macrophage [Arginase
(Arg-1) and CD206] in U937 cell lines were investigated. iNOS and
CD86 expressions in U937 cell lines were low, whereas Arg-1 and
CD206 showed high expression in U937 cell lines (Fig. 4A). These data indicated that U937
were induced into M2 macrophages at 24 h culture. Subsequently,
whether TAM had a role of promoting EC cell proliferation in this
co-culture system was investigated, and it was found that the
proliferation rate of EC cells (ECC-1 and HEC-1A) was increased,
whereas U937 cells did not promote normal endometrial cell (T-HESC)
proliferation (Fig. 4B). When
PLX3397 was added to U937 culture system, the proliferation rate of
endometrial cancer cells decreased, without affecting the
proliferation of normal endometrial cells (Fig. 4B). Additionally, the proliferation
of EC cells in the co-culture system was investigated by Ki67
immunofluorescence staining. Consistent with the above conclusions,
it was found that the proliferation of EC cells was increased in
the co-culture system, whereas it was inhibited by the CSF-1R
inhibitor PLX3397 (Fig. 4C).
Therefore, it was speculated that CSF-1 secreted by EC cells may
promote migration of macrophages, transforming them to
tumor-associated macrophages and that some growth factors secreted
by tumor-associated macrophages promoted EC cells
proliferation.
In order to further clarify the role of macrophages
in promoting the proliferation of EC cells by CSF-1 and CSF-1R
binding, the expression of proliferation-associated molecules was
investigated at the mRNA and protein expression levels. It was
found that U937 co-cultured with EC cells significantly increased
the mRNA expression levels of JAK-1, PI3K, AKT, CDK2, CDK4 and Rb,
however, their expression levels, apart from that of CDK2 (ECC-1
cells only) and Rb (ECC-1 and HEC-1A cells), were decreased when
PLX3397 was pre-added in the co-culture system (Fig. 5A and B). Additionally, the protein
expression levels of JAK-1, PI3K, p-AKT, CDK2, CDK4 and p-Rb were
all increased in the co-culture system, and, apart from p-Rb and
CDK2 they all decreased when the CSF-1R was blocked (Fig. 5C-F). However, in the ECC-1 and U937
co-culture system, PLX3397 did not inhibit CDK2 expression at the
mRNA or protein levels, whereas PLX3397 did not affect the
expression of Rb at the mRNA level either in ECC-1 and U937
co-culture system or in HEC-1A and U937 co-culture system.
Consequently, it may be concluded that EC cells secreted CSF-1 to
promote macrophage migration, which would then promote the
proliferation of EC cells. On the other hand, when CSF-1R was
blocked, the migration of macrophages and the proliferation of EC
cells were both attenuated. However, this needs to be validated
further.
 | Figure 5.CSF-1R inhibitor influences
proliferation-associated protein expression. (A and B) mRNA
expression levels of JAK-1, PI3K, AKT, CDK2, CDK4 and Rb, in (A)
ECC-1 and (B) HEC-1A cells and their inhibition by the CSF-1R
inhibitor PLX3397 (10 µM), as measured by reverse
transcription-quantitative polymerase chain reaction. (C) Protein
expression of JAK-1, PI3K, p-AKT, CDK2, CDK4 and p-Rb and (D)
relative quantification of their expression levels in ECC-1 cells
and their inhibition by the CSF-1R inhibitor PLX3397 (10 µM), as
measured by western blotting. (E) Protein expression of JAK-1,
PI3K, p-AKT, CDK2, CDK4 and p-Rb and (F) relative quantification of
their expression levels in HEC-1A cells and their inhibition by the
CSF-1R inhibitor PLX3397 (10 µM), as measured by western blotting.
Data are presented as the mean ± standard deviation from 5
independent experiments; *P<0.05 vs. Control;
#P<0.05 vs. U937 cells and ECC-1 or HEC-1A cells
co-culture group. CDK4, cyclin-dependent kinase 4; CSF,
colony-stimulating factor; CSF-1R, colony-stimulating factor 1
receptor; EC, endometrial cancer; JAK, Janus kinase; p,
phosphorylated; PI3K, phosphoinositide 3-kinase; Rb,
retinoblastoma-associated protein. |
Discussion
Although macrophages and other mononuclear
phagocytes are regulated by a variety of growth factors, CSF-1 is
still the most important regulator of TAM (15,16).
Previous studies found that CSF-1 and its receptors are mainly
involved in the induction of monocyte development, and CSF-1
secreted by tumor (breast, ovarian and endometrial cancer) cells
was found to promote tumor cell invasion (17). The present study found that CSF-1
secreted by EC cells binds to CSF-1R located on the surface of
macrophages, inducing macrophage infiltration, promoting the
proliferation of EC cells. Macrophages are divided into M1 type
(classic activated macrophages) and M2 type (alternatively
activated macrophages) (18). M1
macrophages are involved in the inflammatory response, clear the
body from pathogens, participate in antitumor immunity, whereas M2
macrophages have the function of anti-inflammatory, repair damage
and they promote tumor formation (19). Macrophages infiltrate into
malignant tumor tissues in high numbers and it has been
demonstrated that macrophages have pro-tumor functions and are
closely related to tumor progression. Consistent with these
functions, studies using human tumor samples have demonstrated that
a higher density of macrophages, especially macrophages with the M2
phenotype, is closely associated with worse clinical prognosis in
many kinds of malignant tumors (20–22).
In the early stages of tumorigenesis, TAM takes M1 macrophages as
the main body and serves the role of killing tumor cells, whereas
in the late stages of tumor development, TAM is mainly consisted by
M2 macrophages, which could promote tumor growth, invasion and
metastasis (23). CSF-1 can
promote macrophage survival and differentiation, and macrophages
can be converted to TAM in its presence (12). Therefore, in the present study,
when exogenous CSF-1 was added to the macrophage culture medium, or
macrophages and EC cells were co-cultured, macrophages were
transformed into TAM favoring the growth of EC cells, therefore,
macrophages seem to have induced the proliferation of EC cells in
the co-culture system, but this needs to be verified in future
studies.
In the present study, EC cells promoted the
migration of macrophages by secreting CSF-1, and macrophages did
not promote cell proliferation after blocking the CSF-1R. The above
results demonstrated that under the presence of CSF-1, macrophages
undergo phenotypic conversion, as EC cells convert macrophages to
TAM which prompting EC cells to proliferate more. Therefore,
targeting CSF-1 to reduce the number of TAMs can be used as a
cancer treatment. Zeisberger et al (24) used liposome-coated clodronate to
remove macrophages from the whole body of tumor-bearing mice,
showing that tumor growth was effectively controlled. In a study of
murine breast cancer induced by spontaneous polyoma virus middle T
oncoprotein, Lin et al (25) demonstrated that transgenic mice
deficient in CSF-1 could significantly reduce the number of TAMs,
reduce tumor angiogenesis and delay tumor growth progression
reducing lung metastases. Previous studies have pointed out that
the removal of CSF-1 and other factors from the ascites of ovarian
cancer would affect the macrophage-induced TAM (26,27).
Other studies have used transgenic technology to overexpress CSF-1
in mammary glands, showing that the branches of breast ducts
increased and precancerous lesions appeared, suggesting that CSF-1
promotes tumorigenesis (28).
Other studies have indicated that CSF-1/CSF-1R-induced activation
of nuclear factor-κB in TAM is required for tumor progression in
inflammation-induced murine tumor models (29,30).
Staining of CSF-1 and its receptor showed strong positive
immunostaining in metastatic ovarian carcinoma, while benign
ovarian tissue showed a little expression of CSF-1/CSF-1R (31). Therefore, high expression of CSF-1
and its receptor regulates the proliferation of tumor cells. At
present, there are many strategies and methods for tumor therapy by
targeting TAM; however, the role of TAM in EC and by which way EC
cells recruit macrophage is still unclear. The present study
demonstrated that EC cells can recruit macrophages by secreting
CSF-1, and macrophages stimulated by CSF-1 can promote the
proliferation of EC cells. The development of drugs targeting TAM,
especially humanized antibodies or inhibitors targeting CSF-1 or
its receptor, promise benefits in cancers such as EC.
Acknowledgements
The authors would like to thank Professor Ren Mulan,
the Dean of Gynecology, Southeast University (Jiangsu, China), for
his individual support and help to this study.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
FH and YT performed the experiments and analyzed the
data. YG analyzed the data. CL and XL collected EC tissues of
patients and FH prepared the manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Nanjing Medical University and written informed
consent was obtained from each patient prior to surgery and
enrolment in the study.
Patient consent for publication
Patients consented to the publication of their
clinical data.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
van Niekerk CC, van Dijck JAAM and Verbeek
ALM: The impact of histological subtype in developing both ovarian
and endometrial cancer: A longstanding nationwide incidence study.
Eur J Obstet Gynecol Reprod Biol. 221:17–22. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Morice P, Leary A, Creutzberg C,
Abu-Rustum N and Darai E: Endometrial cancer. Lancet.
387:1094–1108. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lewis CE and Pollard JW: Distinct role of
macrophages in different tumor microenvironments. Cancer Res.
66:605–612. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yuan A, Chen JJ and Yang PC:
Pathophysiology of tumor-associated macrophages. Adv Clin Chem.
45:199–223. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hu HL, Bai HS and Pan HX: Correlation
between TAMs and proliferation and invasion of type I endometrial
carcinoma. Asian Pac J Trop Med. 8:643–650. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kelly MG, Francisco AM, Cimic A, Wofford
A, Fitzgerald NC, Yu J and Taylor RN: Type 2 endometrial cancer is
associated with a high density of tumor-associated macrophages in
the stromal compartment. Reprod Sci. 22:948–953. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tong H, Ke JQ, Jiang FZ, Wang XJ, Wang FY,
Li YR, Lu W and Wan XP: Tumor-associated macrophage-derived CXCL8
could induce ERα suppression via HOXB13 in endometrial cancer.
Cancer Lett. 376:127–136. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Soeda S, Nakamura N, Ozeki T, Nishiyama H,
Hojo H, Yamada H, Abe M and Sato A: Tumor-associated macrophages
correlate with vascular space invasion and myometrial invasion in
endometrial carcinoma. Gynecol Oncol. 109:122–128. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Espinosa I, Catasus L, D' Angelo E, Mozos
A, Pedrola N, Bértolo C, Ferrer I, Zannoni GF, West RB, van de Rijn
M, et al: Stromal signatures in endometrioid endometrial
carcinomas. Mod Pathol. 27:631–639. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mantovani A and Sica A: Macrophages,
innate immunity and cancer: Balance, tolerance, and diversity. Curr
Opin Immunol. 22:231–237. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
O'Brien J, Lyons T, Monks J, Lucia MS,
Wilson RS, Hines L, Man YG, Borges V and Schedin P: Alternatively
activated macrophages and collagen remodeling characterize the
postpartum involuting mammary gland across species. Am J Pathol.
176:1241–1255. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pyonteck SM, Akkari L, Schuhmacher AJ,
Bowman RL, Sevenich L, Quail DF, Olson OC, Quick ML, Huse JT,
Teijeiro V, et al: CSF-1R inhibition alters macrophage polarization
and blocks glioma progression. Nat Med. 19:1264–1272. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dou F, Huang L, Yu P, Zhu H, Wang X, Zou
J, Lu P and Xu XM: Temporospatial expression and cellular
localization of oligodendrocyte myelin glycoprotein (OMgp) after
traumatic spinal cord injury in adult rats. J Neurotrauma.
26:2299–2311. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Murray PJ and Wynn TA: Protective and
pathogenic functions of macrophage subsets. Nat Rev Immunol.
11:723–737. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hamilton JA: Colony-stimulating factors in
inflammation and autoimmunity. Nat Rev Immunol. 8:533–544. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kacinski BM: CSF-1 and its receptor in
ovarian, endometrial and breast cancer. Ann Med. 27:79–85. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dun EC, Hanley K, Wieser F, Bohman S, Yu J
and Taylor RN: Infiltration of tumor-associated macrophages is
increased in the epithelial and stromal compartments of endometrial
carcinomas. Int J Gynecol Pathol. 32:576–584. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sica A and Mantovani A: Macrophage
plasticity and polarization: In vivo veritas. J Clin Invest.
122:787–795. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Weigert A, Sekar D and Brune B:
Tumor-associated macrophages as targets for tumor immunotherapy.
Immunotherapy. 1:83–95. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jinushi M and Komohara Y: Tumor-associated
macrophages as an emerging target against tumors: Creating a new
path from bench to bedside. Biochim Biophys Acta. 1855:123–130.
2015.PubMed/NCBI
|
|
22
|
Komohara Y, Fujiwara Y, Ohnishi K and
Takeya M: Tumor-associated macrophages: Potential therapeutic
targets for anti-cancer therapy. Adv Drug Deliv Rev. 99:180–185.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ruffell B, Affara NI and Coussens LM:
Differential macrophage programming in the tumor microenvironment.
Trends Immunol. 33:119–126. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zeisberger SM, Odermatt B, Marty C,
Zehnder-Fjällman AH, Ballmer-Hofer K and Schwendener RA:
Clodronate-liposome-mediated depletion of tumour-associated
macrophages: A new and highly effective antiangiogenic therapy
approach. Br J Cancer. 95:272–281. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lin EY, Nguyen AV, Russell RG and Pollard
JW: Colony-stimulating factor 1 promotes progression of mammary
tumors to malignancy. J Exp Med. 193:727–740. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Duluc D, Delneste Y, Tan F, Moles MP,
Grimaud L, Lenoir J, Preisser L, Anegon I, Catala L, Ifrah N, et
al: Tumor-associated leukemia inhibitory factor and IL-6 skew
monocyte differentiation into tumor-associated macrophage-like
cells. Blood. 110:4319–4330. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jeannin P, Duluc D and Delneste Y: IL-6
and leukemia-inhibitory factor are involved in the generation of
tumor-associated macrophage: Regulation by IFN-γ. Immunotherapy 3
(4 Suppl). S23–S26. 2011. View Article : Google Scholar
|
|
28
|
Kirma N, Hammes LS, Liu YG, Nair HB,
Valente PT, Kumar S, Flowers LC and Tekmal RR: Elevated expression
of the oncogene c-fms and its ligand, the macrophage
colony-stimulating factor-1, in cervical cancer and the role of
transforming growth factor-beta1 in inducing c-fms expression.
Cancer Res. 67:1918–1926. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mancino A and Lawrence T: Nuclear
factor-kappaB and tumor-associated macrophages. Clin Cancer Res.
16:784–789. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Biswas SK and Lewis CE: NF-κB as a central
regulator of macrophage function in tumors. J Leukoc Biol.
88:877–884. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chambers SK: Role of CSF-1 in progression
of epithelial ovarian cancer. Future Oncol. 5:1429–1440. 2009.
View Article : Google Scholar : PubMed/NCBI
|