Introduction
Chronic kidney disease is a common cause of
morbidity and mortality, and has a prevalence of 8–16% worldwide
(1). It is well established that
renal fibrosis, considered as the final stage of chronic kidney
disease, leads to the impairment of kidney function (2,3).
Myofibroblast accumulations, excessive deposition of extracellular
matrix (ECM) and renal tubule destruction have been demonstrated to
be typical hallmarks of tubule interstitial fibrosis (4,5).
Although recent advances in functional genomics have contributed to
the understanding of the pathophysiology of renal fibrosis,
definitive therapies are not yet available (5–7).
Therefore, the absence of a precise target and therapeutic strategy
renders this disease a considerable challenge.
Recent studies have demonstrated an association
between the expression of microRNAs (miRNAs or miRs) and fibrotic
diseases. miRNAs are a class of short noncoding RNAs that regulate
gene expression via post-translational modification and induction
of mRNA degradation, thus affecting numerous molecular and cellular
processes (8–12). Several miRNAs, including miR-29,
miR-192 and miR-21, have been reported to be associated with
fibrotic processes in various diseases (13–15).
These miRNAs can mediate transforming growth factor-β1 (TGF-β1)
signaling in renal cells, and normalization of their expression can
alleviate fibrosis in vitro and in vivo, thus making
miRNAs potential targets for therapy of renal fibrosis (15).
TGF-β1, as a major profibrotic agent, can trigger
renal fibrosis, and blockade of TGF-β1 suppresses progressive
kidney fibrosis (16,17). TGF-β1 can induce the transcription
of early growth response factor-1 (Egr1), which has been verified
to be correlated with excessive production of ECM (18,19).
Egr1, a zinc-finger transcription factor that encodes an 80–82 kDa
protein targeting a GCGGGGGCG binding site, regulates downstream
gene transcription and controls diverse cellular processes, such as
cell growth, proliferation and differentiation. It has been
reported that Egr1 serves a critical role during the fibrotic
process (20–22). However, the function of miR-181 in
renal fibrosis remains largely unknown, and the association between
miR-181 and Egr1 is not fully understood.
The present study aimed to investigate the role of
miR-181 in vitro and in vivo, and to further explore
its underlying mechanism in renal fibrosis. It was observed that
miR-181 functioned as an important factor in the modulation of the
fibrotic process in the kidneys, and Egr1 was identified as a
potential target of miR-181. Thus, the miR-181/Egr1 signaling
pathway may serve as a novel target for the diagnosis and prognosis
of renal fibrosis.
Materials and methods
Clinical samples
A total of 58 patients with renal fibrosis (46 males
and 12 females) with a mean age of 51.2±6.9 years (age range,
43.5–65.1 years), who were admitted to the Heping Hospital
Affiliated to Changzhi Medical College (Changzhi, China) to undergo
therapy, were enrolled into the present study. All patients were
diagnosed with renal fibrosis by biopsy (23). In addition, 10 normal control
subjects (8 males and 2 females) with a mean age of 54.9±7.1 years
were included in the study. Blood samples were collected from the
subjects and centrifuged at 1,000 × g for 10 min at 4°C to separate
the serum, which was stored at −80°C for subsequent assays. The
present study was approved by the Ethics Committee of Heping
Hospital Affiliated to Changzhi Medical College, and written
informed consent was provided by all the participants in the
study.
Experimental animals
A unilateral ureteral obstruction (UUO) kidney
disease model was established in C57Bl/6J mice (Jackson Laboratory,
Bar Harbor, ME, USA) by left ureteral ligation as previously
described (18). These mice were
housed five per cage under the following conditions: Constant
temperature, 22°C; humidity, 35–75%; free access to food and water;
12-h light/dark cycle. Briefly, male mice (age, 10–12 weeks;
weight, 30–40 g) were randomly divided into three groups, as
follows: Sham-operated group (n=6), in which mice underwent the
same surgical procedure as the observation group, with the
exception of ureteral ligation; UUO model group (n=8), in which
mice with UUO were injected with the negative control (NC) vector
(Biomics Biotechnologies, Guangzhou, China); and observation group
(n=8), in which mice with UUO were injected with a miR-181 agonist
(Biomics Biotechnologies). The NC vector and miR-181 agonist were
administered 1 week after surgery. Mice were sacrificed 14 days
after injection, and their kidney tissues were collected and
processed to evaluate the expression levels of collagen type I α1
(COL1A1), collagen type III α1 (COL3A1), α-smooth muscle actin
(α-SMA) and connective tissue growth factor (CTGF) by reverse
transcription-quantitative polymerase chain reaction (RT-qPCR). All
the procedures were performed in accordance with national and
international laws and policies, and were approved by the Heping
Hospital Affiliated to Changzhi Medical College Animal Care and Use
Ethics Committee.
Masson's trichrome staining
Changes in renal morphology were examined with
Masson's trichrome staining. Briefly, the kidney tissues were fixed
in 4% paraformaldehyde solution for 6–8 h at room temperature,
fully automatically dehydrated, paraffin embedded and sliced into
4-µm tissue sections. The slices were then stained with Masson's
trichrome (Beijing Solarbio Science & Technology Co., Ltd.,
Beijing, China) according to the manufacturer's protocol.
Bioinformatics analysis
The targets and binding sites of miR-181 were
predicted using numerous online databases with different
algorithms, TargetScan (http://www.targetscan.org/).
Cell culture and transfection
Rat kidney NRK49F cells (American Type Culture
Collection, Manassas, VA, USA) were cultured in Dulbecco's modified
Eagle's medium (Thermo Fisher Scientific, Inc., Waltham, MA, USA),
supplemented with 10% fetal calf serum (Gibco; Thermo Fisher
Scientific, Inc.), 1% glutamine (Gibco; Thermo Fisher Scientific,
Inc.), 1% nonessential amino acids (Gibco; Thermo Fisher
Scientific, Inc.) and 1% penicillin and streptomycin (Gibco; Thermo
Fisher Scientific, Inc.) in a humidified incubator containing 5%
CO2 at 37°C. Angiotensin II (10 µM; AngII; R&D
Systems, Inc., Minneapolis, MN, USA) was used to stimulate the
cells, in order to establish a fibrosis model (14). Subsequently, NRK49F cells
(5×105 cells/well) were transfected with 30 nM miR-181
mimics or NC miRNA mimics (both Ambion; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) in 6-well plates using siPORT NeoFX
Transfection Agent (Ambion; Thermo Fisher Scientific, Inc.), in
accordance with the manufacturer's protocol. Following transient
transfection for 24 h, the cells were synchronized by culture in
low-glucose medium without serum for 24 h.
RNA extraction and RT-qPCR
Total RNA from human blood samples, mouse renal
tissues and NRK49F cells was extracted using TRIzol®
reagent (Zoonbio Biotechnology Co., Ltd., Nanjing, China) in
accordance with the manufacturer's protocol. RNA concentration was
measured using a spectrophotometer (NanoDrop® ND-1000;
NanoDrop Technologies; Thermo Fisher Scientific, Inc., Wilmington,
DE, USA); RNA with an optical density (OD)260/OD280 range between
1.8 and 2.0 was used. Subsequently, cDNA was synthesized using the
TransScript miRNA RT Enzyme Mix (TransGen Biotech Co., Ltd.,
Beijing, China), according to the manufacturer's protocol, as
follows: RT at 50°C for 60 min and inactivation of reverse
transcriptase at 70°C for 15 min. For the detection of miR-181
expression, a miRNA-specific TaqMan MicroRNA assay (Applied
Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA) was
used. U6 was used as an internal control for normalization of miRNA
expression. GAPDH was used as an internal control for normalization
of mRNA expression. For the detection of mRNA expression, SYBR
Premix Ex Taq™ II assay (Takara Biotechnology Co., Ltd.,
Dalian, China) was conducted using a 20 µl reaction volume, under
the following conditions: First step, initial denaturation at 95°C
for 30 sec; second step, denaturation at 95°C for 30 sec and primer
annealing at 60°C for 30 sec, this step was repeated 35 times. The
experiments were performed in triplicate. Primer sequences are
provided in Table I. The
2−ΔΔCq method was used to calculate the results
(23).
 | Table I.Primer information. |
Table I.
Primer information.
| Gene | Sequence | Primer length
(bp) | Annealing
temperature (°C) |
|---|
| miR-181 | Forward,
5′-GTGGATCCGACATTCATTTGAGTCTGCTTGT-3′ | 31 | 62.0 |
|
| Reverse,
5′-GCGAATTCTCATCATGGACTGCTCTTAC-3′ | 28 | 60.2 |
| Egr1 | Forward,
5′-GCAGGCTCGCTCCCACGGTC-3′ | 20 | 62.0 |
|
| Reverse,
5′-GGGGTTGGCCGGGTTACATG-3′ | 20 | 58.8 |
| U6 | Forward, 5′-
GCTTCGGCAGCACATATACTAAAAT-3′ | 25 | 54.6 |
|
| Reverse,
5′-CGCTTCACGAACCGCGTGTCA-3′ | 21 | 58.3 |
| α-SMA | Forward,
5′-CATCACGAACTGGGATGACATG-3′ | 22 | 55.8 |
|
| Reverse,
5′-CATCTTCTCCCTGTTGGCTTTAG-3′ | 23 | 55.1 |
| CTGF | Forward,
5′-TCCTTTCTGAGCAATTCACCAAG-3′ | 23 | 60.7 |
|
| Reverse,
5′-GCACACTCCGTCTTTTTCCTC-3′ | 21 | 61.2 |
| COL1A1 | Forward,
5′-GAGGGCCAAGACGAAGACATC-3′ | 21 | 62.5 |
|
| Reverse,
5′-CAGATCACGTCATCGCACAAC-3′ | 21 | 61.6 |
| COL3A1 | Forward,
5′-GGAGCTGGCTACTTCTCGC-3′ | 19 | 62.2 |
|
| Reverse,
5′-GGGAACATCCTCCTTCAACAG-3′ | 21 | 60.0 |
| GAPDH | Forward,
5′-TGTGGGCATCAATGGATTTGG-3′ | 21 | 60.9 |
|
| Reverse,
5′-ACACCATGTATTCCGGGTCAAT-3′ | 22 | 61.4 |
Western blotting
Transfected NRK49F cells were collected according to
the previously described protocols (14). Subsequently, western blot analysis
was conducted as previously described (24). Total protein concentration was
quantified using a spectrophotometer (NanoDrop® ND-1000;
NanoDrop Technologies; Thermo Fisher Scientific, Inc.) to ensure
that sample quantity was consistent. Subsequently, proteins were
denatured at 95°C for 15 min in a water bath. The proteins (20
µg/well) were separated by 10% SDS-PAGE and transferred to a
polyvinylidene difluoride membrane at 200 mA for 3 h. The membranes
were blocked with 5% skimmed milk powder for 2 h at room
temperature, after which, they were incubated with the primary
antibody overnight at 4°C. Anti-Egr1 was used as the primary
antibody (1:1,000; cat. no. MAB2818; R&D Systems, Inc.) and
GAPDH (1:500; cat. no. AF5718; R&D Systems, Inc.) was used as a
protein loading control. Tris Buffered Saline with 0.1% Tween 20
(TBST; cat. no. 28358; Thermo Fisher, Scientific, Inc., Waltham,
MA, USA) was used to wash the membranes three times (10 min/wash),
and they were then incubated with a horseradish
peroxidase-conjugated goat anti-rabbit secondary antibody
(1:20,000; cat. no. HAF008; R&D Systems, Inc.) at room
temperature for 2 h. Finally, the membranes were washed with TBST
three times (10 min/wash). The images were developed with
SuperSignal West Pico Chemiluminescent Substrate (cat. no. 34080;
Pierce; Thermo Fisher Scientific, Inc.) using a ChemiDoc XRS (cat.
no. 1708265; Bio-Rad Laboratories, Inc., Hercules, CA, USA), and
analyzed with Gel-Pro analyzer (version 4.0, Media Cybernetics,
Inc., Rockville, MD, USA).
Dual-luciferase reporter assay
NRK49F cells were plated in a 24-well plate
(1×106 cells/well), and then co-transfected with a
luciferase reporter plasmid [pMIR-Egr1-wild-type (WT) or
pMIR-Egr1-mutant (Mut)] together with miR-181 or NC mimics and the
pRL-TK vector encoding Renilla luciferase (Promega
Corporation, Madison, WI, USA) using Lipofectamine® 2000
(Invitrogen; Thermo Fisher Scientific, Inc.). After 48 h, the
original culture medium was discarded and the cells were washed
gently three times with PBS (cat. no. AM9624; Thermo Fisher
Scientific, Inc.), after which, 20 µl passive lysis buffer (cat.
no. E1910; Promega Corporation) was added and the cells were
agitated at room temperature for 20 min. The luciferase activity
was analyzed using a Dual-Luciferase Reporter Assay system (Promega
Corporation) according to the manufacturer's recommendations.
Statistical analyses
All quantitative data for statistical analyses were
obtained from at least three independent experiments. Data are
presented as the mean ± standard deviation. Comparisons between two
groups were performed using Student's t-test, while paired
Student's t-test was used to analyze paired data. Comparisons among
three or more groups were performed by analysis of variance test,
with Bonferroni correction used as a post hoc test. Statistical
analyses were performed with IBM SPSS version 22.0 software (IBM
Corp., Armonk, NY, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
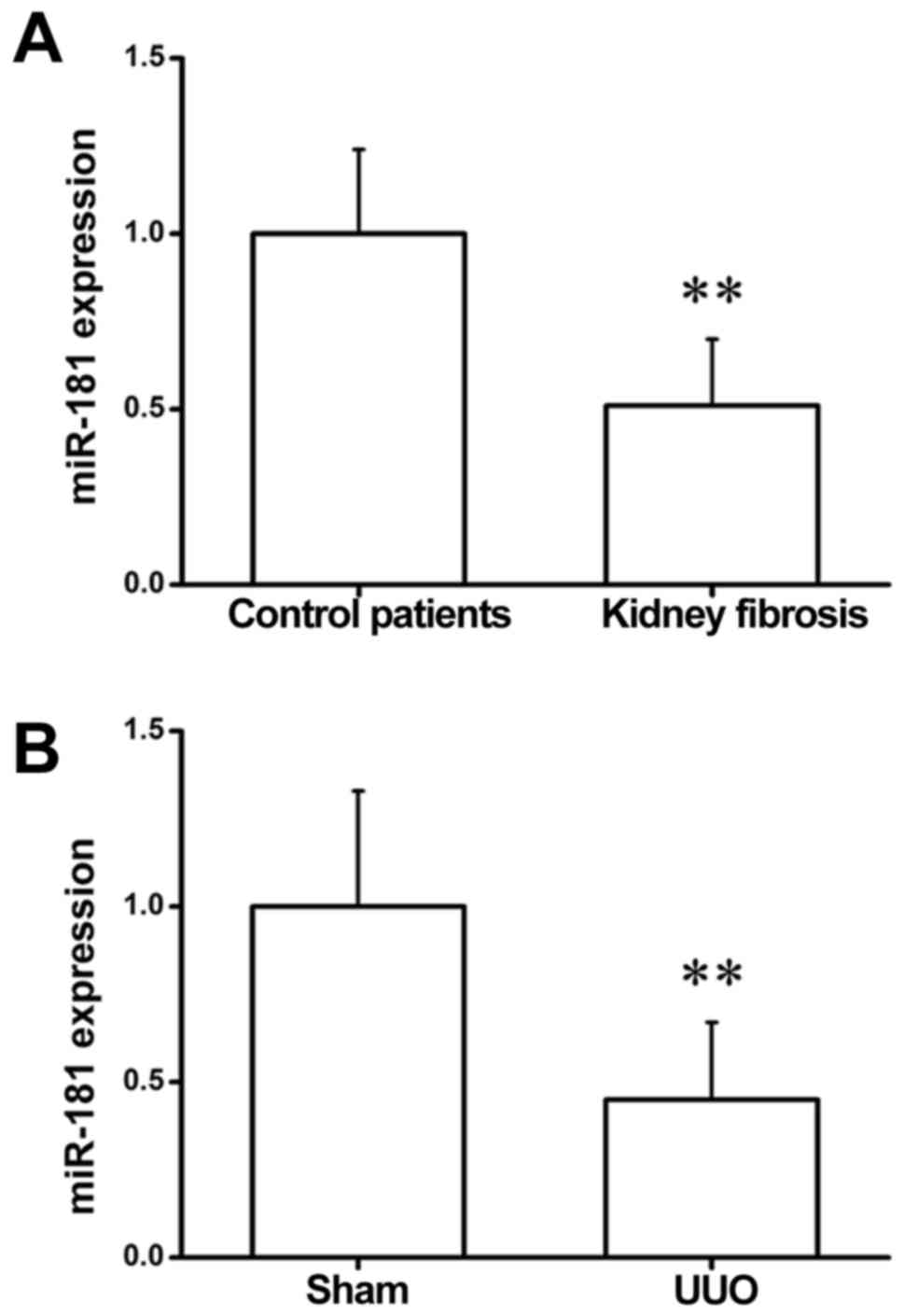
miR-181 expression is downregulated in
patients and mice with renal fibrosis
To explore whether miR-181 affects the progression
of kidney fibrosis, miR-181 expression was detected in clinical
blood samples by RT-qPCR. As shown in Fig. 1A, the expression of miR-181 was
significantly reduced in the serum of patients with renal fibrosis
as compared with the controls. Consistent with this observation,
the results of RT-qPCR assay in kidney samples of sham-operated and
UUO mice revealed that miR-181 exhibited a markedly lower
expression in UUO kidneys in comparison with the sham-operated
group (Fig. 1B). These
observations suggest that miR-181 may serve a critical role in the
development of fibrotic responses in the kidney.
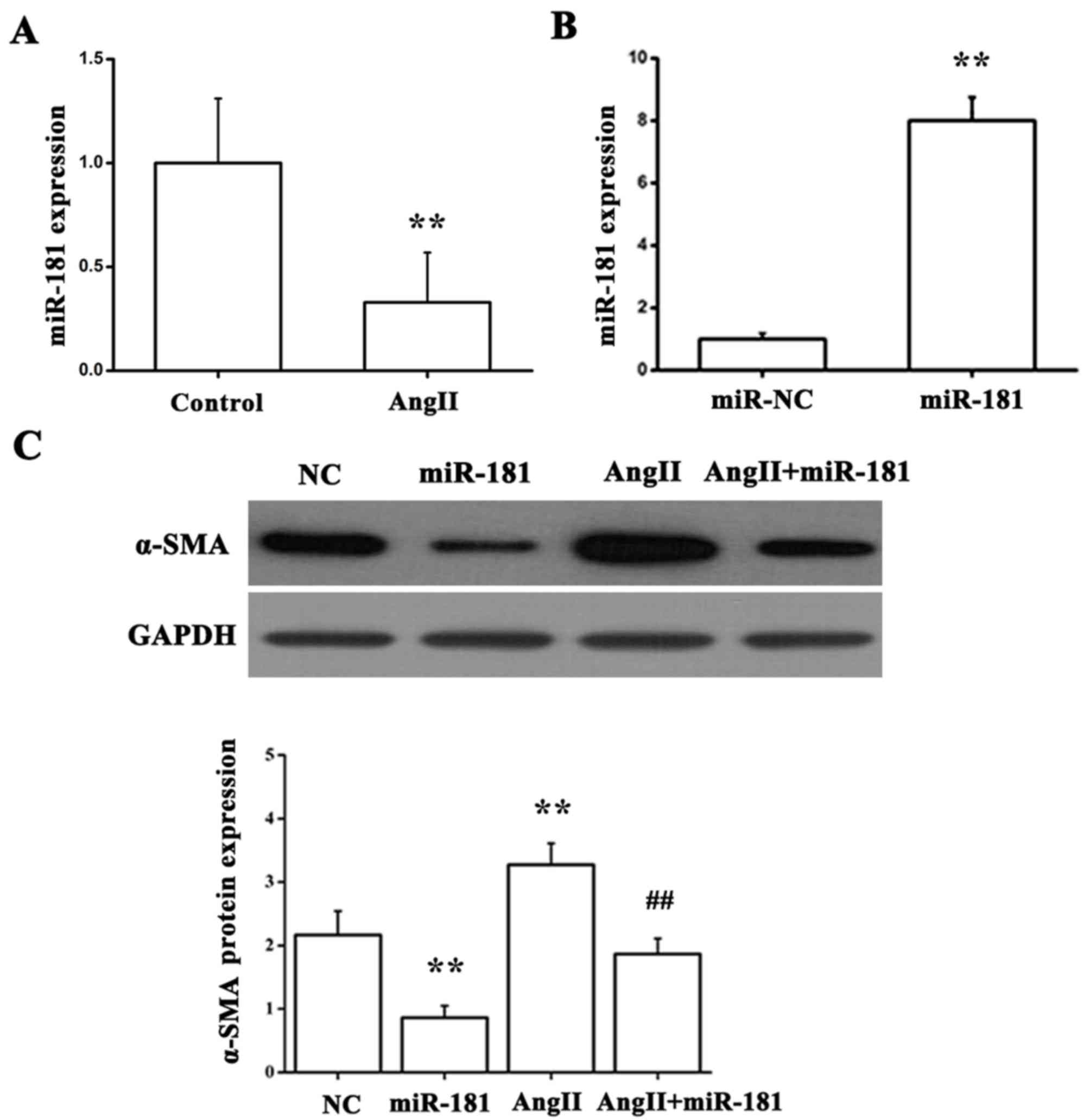
miR-181 downregulates α-SMA expression
in vitro
NRK49F, a normal rat renal fibroblast cell line, was
employed in the present study to gain insights into the functional
significance of miR-181 in renal fibrosis. It is well established
that AngII contributes to tubule interstitial injury and fibrosis
in kidney diseases (25,26). To identify whether the AngII
stimulation has an effect on miR-181 expression during renal
fibrotic disease, NRK49F cells were exposed to AngII for 36 h.
Using RT-qPCR, miR-181 expression was determined and was observed
to be significantly reduced in response to AngII treatment in
comparison with the control group (Fig. 2A). Additionally, NRK49F cells were
transfected with miR-181 mimics (Fig.
2B) and then stimulated with AngII. Compared with the group
treated with NC miRNA mimic, the group treated with miR-181 mimic
exhibited downregulation of α-SMA protein expression; however, the
group treated with AngII exhibited upregulation of α-SMA protein
expression; this effect was suppressed by miR-181 (Fig. 2C). These data suggest that miR-181
can suppress α-SMA expression induced by AngII in fibrotic kidney
progression.
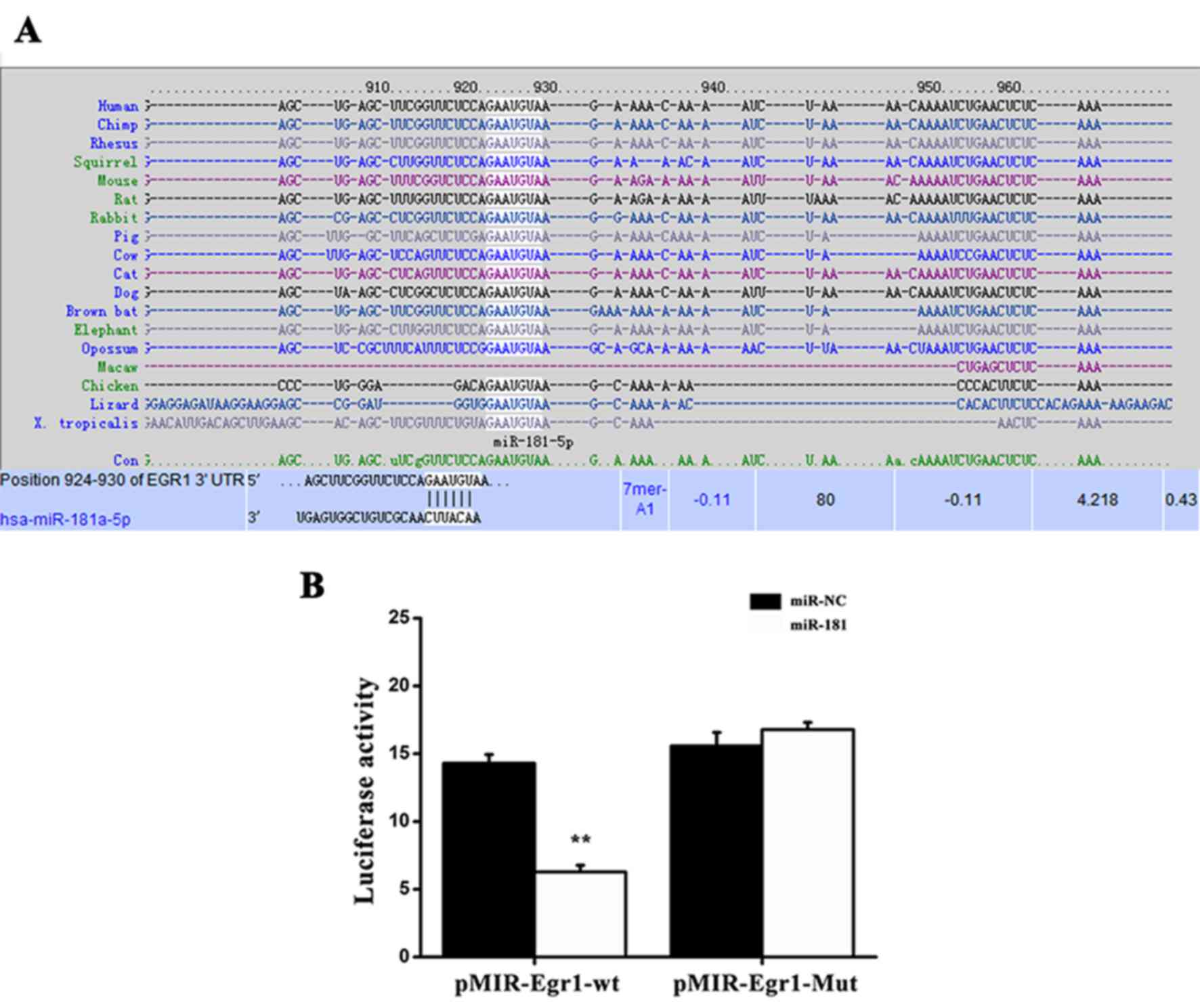
Egr1 is a direct target of
miR-181
The predicted target sequence of miR-181 was
evaluated using the accessible TargetScan Human database
(http://www.targetscan.org/vert_72/).
As shown in Fig. 3A, a potential
binding site for miR-181 was identified in the 3′-untranslated
region (UTR) of Egr1. To further investigate whether Egr1 is a
direct binding target of miR-181, the WT or Mut miR-181 target
sequences in the 3′-UTR of Egr1 were fused into a luciferase
reporter gene. Subsequently, pMIR-Egr1-WT or pMIR-Egr1-Mut reporter
was co-transfected into NRK49F cells together with miR-181 mimics
or miR-NC, and a luciferase reporter assay was performed. As
presented in Fig. 3B, NRK49F cells
transfected with miR-181 mimics exhibited markedly reduced
luciferase activity induced by pMIR-Egr1-WT compared with the
control vector. However, transfection with miR-181 mimics failed to
inhibit the luciferase activity of the Mut reporter gene. Taken
together, these observations indicate that miR-181 can directly
bind to the 3′-UTR of Egr1.
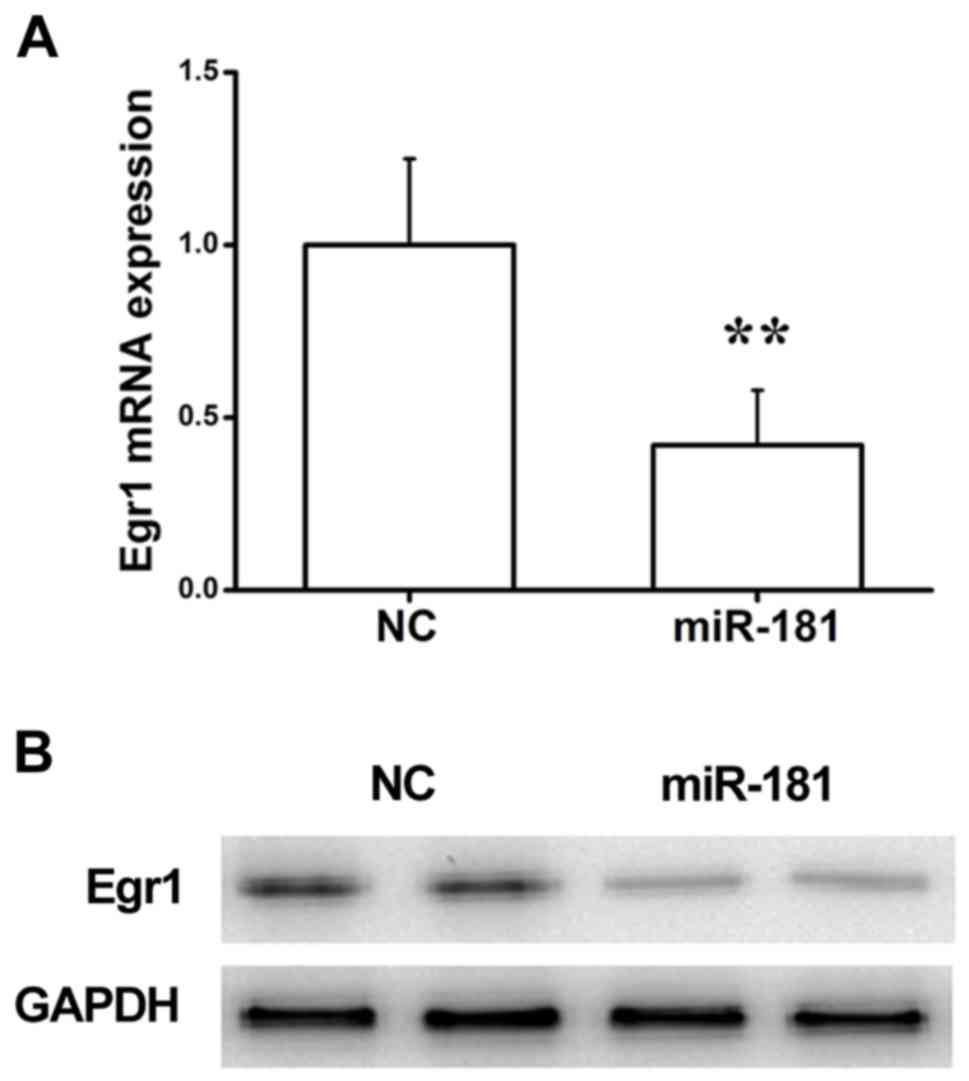
miR-181 regulates Egr1 expression at
the mRNA and protein level
To further investigate whether miR-181 successfully
regulated Egr1 expression in NRK49F cells, Egr1 expression in cells
transfected with miR-181 mimics or miR-NC was examined. As shown in
Fig. 4A, RT-qPCR assay indicated
that transfection with miR-181 significantly inhibited the
expression of Egr1 at the mRNA level compared with the NC group. In
addition, western blot analysis revealed that NRK49F cells
transfected with miR-181 mimics exhibited a significant reduction
in comparison with the NC group (Fig.
4B). Taken together, these results suggest that miR-181
suppresses Egr1 expression at the mRNA and protein levels in NRK49F
cells by directly binding to the 3′-UTR of Egr1.
miR-181 inhibits the progression of
renal fibrosis via downregulation of profibrotic markers
A previous study demonstrated that accumulation of
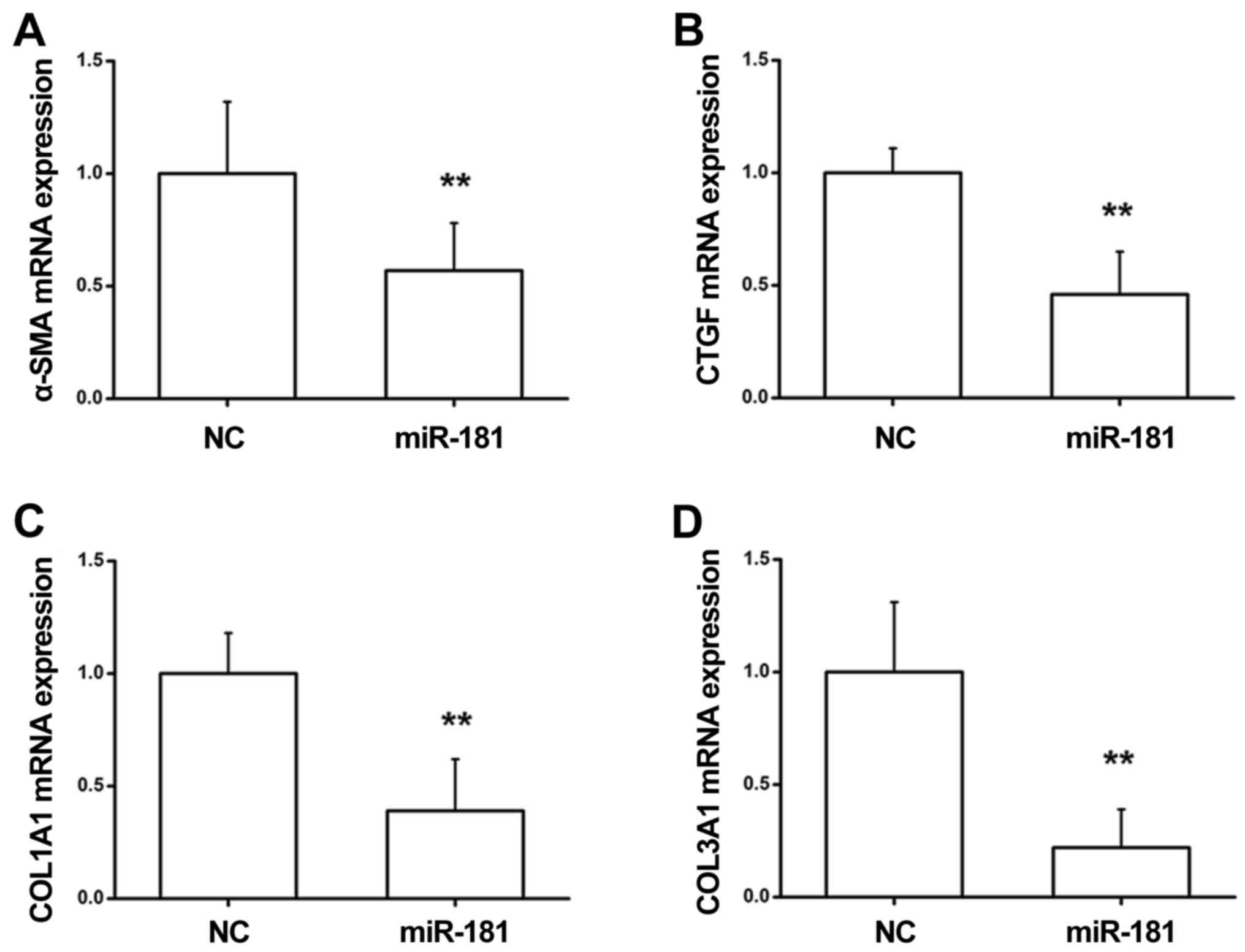
α-SMA, collagen and CTGF can contribute to renal fibrosis (27). To investigate the role of miR-181
in the renal fibrotic response, RT-qPCR was conducted to determine
the expression levels of α-SMA, CTGF, COL1A1 and COL3A1 in NRK49F
cells. As presented in Fig. 5A-D,
NRK49F cells transfected with miR-181 mimics exhibited a
significant reduction in the expression levels of α-SMA, CTGF,
COL1A1 and COL3A1 at the mRNA level as compared with the NC group.
These data indicated that miR-181 exerts an inhibitory effect
during renal fibrosis by suppressing the levels of profibrotic
markers.
Renal fibrosis morphology in UUO mice
is associated with miR-181 regulation
Based on the in vitro data indicating that
miR-181 serves a suppressive role in the progression of renal
fibrosis, an in vivo study was conducted to validate whether
miR-181 is able to mediate the consequent fibrosis. Upon
establishment of a UUO model, an miR-181 agonist was delivered into
the mice through an injection in the tail vein, and the
histopathological morphology of the kidneys was observed by
Masson's trichrome staining. As indicated in Fig. 6A, the expected clear structure of
renal tubules and glomerulus was identified in the sham-operated
mice, with no evident interstitial cell infiltration observed.
However, renal glomerular dilatation, and epithelial cell
degeneration and necrosis were detected in the UUO mice, although
this response was rescued by treatment with miR-181 agonist. The
injection of miR-181 agonist into the tail vein of mice in the UUO
model improved renal tubular dilatation, and reduced the
degeneration and necrosis of epithelial cells, suggesting that
miR-181 can alleviate kidney impairment during the fibrotic process
in vivo. In agreement with previous studies on collagen
expression mediated by miR-181, the present study revealed that
treatment with miR-181 agonist markedly suppressed COL1A1 and
COL3A1 mRNA expression levels in UUO kidneys (Fig. 6B and C), suggesting that miR-181
participates in kidney fibrotic disease via inhibition of collagen
deposition.
Discussion
It is widely accepted that renal fibrosis is the
final common stage of numerous forms of progressive kidney disease,
which leads to impairment of the renal function (28). At present, a definite therapy for
renal fibrosis is missing, which indicates that the precise
pathogenesis of this fibrotic disease is not yet fully understood.
Notably, a series of miRNAs have emerged as critical regulators of
numerous cellular processes, such as proliferation, migration,
apoptosis, differentiation and cell cycle progression (29–33).
Mounting evidence indicated that several miRNAs are implicated in
the development of certain renal disorders, including acute kidney
impairment (34), diabetic kidney
disease (35) and renal fibrosis
(36). However, the association
between miR-181 and fibrotic kidney disease remains unclear. In the
current study, the functional relevance of miR-181 in the fibrotic
response in kidney disease was identified. Additionally, miR-181
was able to halt the progression of renal fibrosis in an
established UUO model. Targeting Egr1 may be a mechanism by which
miR-181 mediates renal injury. Therefore, miR-181 may contribute to
the prognosis and therapy of renal fibrosis.
The present study confirmed an association between
miR-181 expression and renal fibrosis. It has been demonstrated
that aberrant expression of miRNAs is closely associated with
inflammatory responses. Previous studies revealed that miRNA
profiling of rodents displayed increased expression of miR-181 in
aged kidneys, indicating that epigenetic modulation of renal ageing
likely occurred via repressing miR-181-targeted genes (37,38).
Accumulation of miRNA-433 can drive renal fibrosis by activation of
the TGF-β signaling pathway (39).
In addition, miRNA-21 has been demonstrated to exhibit a marked
increase in expression in UUO kidneys, suggesting that miRNA-21 may
exert an important activity during renal fibrosis (40). The present study further revealed
that miR-181 is critical for mediating the fibrotic process in
experimental models and in human kidney diseases. Using RT-qPCR,
the expression of miR-181 was determined in the serum of renal
fibrosis patients and in renal tissues of UUO mice. Downregulation
of miR-181 expression occurred in patients with renal fibrosis.
Consistently, in UUO kidneys, miR-181 displayed a reduced
expression, suggesting that miR-181 may be involved in the
progression of renal fibrosis. Thus, miR-181 was subjected to
further investigation due to its expression signature.
AngII, a key regulator of proteinuria and fibrosis,
can contribute to the release of inflammatory and fibrogenic
substances, which leads to the fibrotic response. Zanchi et
al (41) reported that AngII
failed to trigger miR-181 expression, since AngII does not exert a
direct effect on miR-181. To validate the functional implications
of AngII in miR-181 expression, NRK49F cells were exposed to AngII
in the current study. The data revealed that miR-181 exhibited a
significantly reduced expression in response to AngII, suggesting
that AngII is an effective agent for modulating miR-181 expression
in rat kidney fibroblasts. Furthermore, western blotting revealed
that miR-181 caused a sustained downregulation of α-SMA, which is a
characteristic of activation of fibrogenesis (42). Coinciding with the signature of
α-SMA, other fibrotic markers, such as collagens (43–45),
can be induced to activate a cascade signaling pathway to drive
fibrosis. Previous studies have reported that the accumulation of
α-SMA, COL1A1, COL3A1 and CTGF promoted renal fibrosis, which was
mediated by a miRNA-433-regulated feedback loop of TGF-β signaling
(46,47). During the fibrotic process,
elevated COL3A1 has been verified in early myocardial remodeling,
whereas COL1A1 deposition has been observed at a later stage
(48). In the present study,
RT-qPCR assay revealed that miR-181 exerted an inhibitory effect on
the mRNA expression levels of α-SMA, CTGF, COL1A1 and COL3A1. To
support this notion, an established UUO mouse model was used to
conduct relevant functional experiments in vivo. Masson's
trichrome staining indicated that miR-181 rescued the renal
impairment induced by ureteral occlusion, as demonstrated by the
reduced COL1A1 and COL3A1 levels detected using RT-qPCR analysis.
These results emphasize the significance of miR-181 in halting
renal fibrosis by downregulation of profibrotic markers.
One important finding of the present study is that
miR-181 serves a crucial role in the fibrotic response in kidney
disease via directly targeting the Egr1 gene. A recent study
reported that Egr1, which is regarded as an intracellular TGF-β
target, can induce the expression of type III and IV collagens
(49,50). In addition, TGF-β can induce the
transcription of genes such as collagens and Egr1 to promote the
biological processes that are responsible for fibrosis (51). Notably, miRNAs are able to suppress
their targeted genes though binding to their 3′-UTR to induce
inhibition of protein translation or mRNA degradation (52). To further investigate the molecular
mechanism by which miR-181 exerted an inhibitory activity during
renal fibrosis, the two putative miR-181 target sites in the 3′-UTR
of Egr1 were screened, and the results of luciferase reporter assay
indicated that miR-181 directly targeted the 3′-UTR of Egr1.
Furthermore, the mRNA and protein levels of Egr1 were attenuated by
miR-181 compared with the control group, suggesting that miR-181
negatively regulated Egr1 by directly binding to the Egr1 gene.
These in vitro and in vivo data clearly demonstrated
that miR-181 downregulated profibrotic indicators during renal
fibrosis by targeting Egr1.
Egr1 is a generally expressed member of the
zinc-finger family of transcription factors, which has been
reported to be a potential regulator of the connective tissue
factor CCN family (including the cysteine-rich 61, CTGF and
nephroblastoma overexpressed members) (53). In addition, Egr1−/− mice
exhibited a deficiency in the expression of tendon genes, including
scleraxis, COL1A1 and COL1A2, where Egr1 was recruited to the
COL1A1 and COL2A1 promoters, and affected their expression
(54). In the current study,
miR-181 negatively regulated Egr1 by directly binding to the Egr1
gene, and the mRNA expression levels of α-SMA, CTGF, COL1A1 and
COL3A1 were significantly reduced, suggesting that Egr1 may serve a
regulatory role in the transcription of α-SMA, CTGF, COL1A1 and
COL3A1. However, future experiments are required to verify this
hypothesis.
In conclusion, the present study has provided direct
evidence to demonstrate that miR-181 serves an important role
during renal fibrosis by targeting Egr1. By combining in
vitro and in vivo data, the present study demonstrated
that miR-181 serves a role as a downstream regulator of profibrotic
markers to alleviate fibrotic kidney processes. These observations
indicate that targeting miR-181 may exert an antifibrotic effect in
kidney disease. However, whether miR-181 is a biomarker or serves
an additional role in the progression of renal fibrosis requires
further investigation.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XZ designed and supervised the study, performed
experiments, analyzed the data, and wrote and edited the
manuscript. CM, ZY and YH performed the experiments. CM and XZ
analyzed the data and drafted the manuscript.
Ethics approval and consent to
participate
Written informed consent was obtained from all
patients and the experiment was approved by the Ethics Committee of
Heping Hospital Affiliated to Changzhi Medical College. All animal
experiments were performed in accordance with national and
international laws and policies, and were approved by the Animal
Experimental Ethics Committee, Heping Hospital Affiliated to
Changzhi Medical College.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Jha V, Garcia-Garcia G, Iseki K, Li Z,
Naicker S, Plattner B, Saran R, Wang AY and Yang CW: Chronic kidney
disease: Global dimension and perspectives. Lancet. 382:260–272.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Remuzzi G and Bertani T: Pathophysiology
of progressive nephropathies. N Engl J Med. 339:1448–1456. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Iwano M and Neilson EG: Mechanisms of
tubulointerstitial fibrosis. Curr Opin Nephrol Hypertens.
13:279–284. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Müller GA, Zeisberg M and Strutz F: The
importance of tubulointerstitial damage in progressive renal
disease. Nephrol Dial Transplant. 15 (Suppl 6):S76–S77. 2000.
View Article : Google Scholar
|
|
5
|
Deng C, Zheng J, Wan W, Zhang S, Ding Z,
Mao G and Yang S: Suppression of cell proliferation and collagen
production in cultured human hypertrophic scar fibroblasts by Sp1
decoy oligodeoxynucleotide. Mol Med Rep. 7:785–790. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Boor P, Ostendorf T and Floege J: Renal
fibrosis: Novel insights into mechanisms and therapeutic targets.
Nat Rev Nephrol. 6:643–656. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Vilayur E and Harris DC: Emerging
therapies for chronic kidney disease: What is their role? Nat Rev
Nephrol. 5:375–383. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chung AC, Dong Y, Yang W, Zhong X, Li R
and Lan HY: Smad7 suppresses renal fibrosis via altering expression
of TGF-β/Smad3-regulated microRNAs. Mol Ther. 21:388–398. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lorenzen JM, Haller H and Thum T:
MicroRNAs as mediators and therapeutic targets in chronic kidney
disease. Nat Rev Nephrol. 7:286–294. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kato M and Natarajan R: MicroRNAs in
diabetic nephropathy: Functions, biomarkers, and therapeutic
targets. Ann N Y Acad Sci. 1353:72–88. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhong X, Chung AC, Chen HY, Dong Y, Meng
XM, Li R, Yang W, Hou FF and Lan HY: miR-21 is a key therapeutic
target for renal injury in a mouse model of type 2 diabetes.
Diabetologia. 56:663–674. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zavadil J, Narasimhan M, Blumenberg M and
Schneider RJ: Transforming growth factor-beta and microRNA: mRNA
regulatory networks in epithelial plasticity. Cells Tissues Organs.
185:157–161. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kato M and Natarajan R: Diabetic
nephropathy-emerging epigenetic mechanisms. Nat Rev Nephrol.
10:517–530. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kato M, Zhang J, Wang M, Lanting L, Yuan
H, Rossi JJ and Natarajan R: MicroRNA-192 in diabetic kidney
glomeruli and its function in TGF-beta-induced collagen expression
via inhibition of E-box repressors. Proc Natl Acad Sci USA.
104:3432–3437. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
McClelland A, Hagiwara S and Kantharidis
P: Where are we in diabetic nephropathy: MicroRNAs and biomarkers?
Curr Opin Nephrol Hypertens. 23:80–86. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Khan R: Examining potential therapies
targeting myocardial fibrosis through the inhibition of
transforming growth factor-beta 1. Cardiology. 108:368–380. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Murray LA, Chen Q, Kramer MS, Hesson DP,
Argentieri RL, Peng X, Gulati M, Homer RJ, Russell T, van Rooijen
N, et al: TGF-beta driven lung fibrosis is macrophage dependent and
blocked by Serum amyloid P. Int J Biochem Cell Biol. 43:154–162.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Guerquin MJ, Charvet B, Nourissat G, Havis
E, Ronsin O, Bonnin MA, Ruggiu M, Olivera-Martinez I, Robert N, Lu
Y, et al: Transcription factor EGR1 directs tendon differentiation
and promotes tendon repair. J Clin Invest. 123:3564–3576. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lv ZM, Wang Q, Wan Q, Lin JG, Hu MS, Liu
YX and Wang R: The role of the p38 MAPK signaling pathway in high
glucose-induced epithelial-mesenchymal transition of cultured human
renal tubular epithelial cells. PLoS One. 6:e228062011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lee CG, Cho SJ, Kang MJ, Chapoval SP, Lee
PJ, Noble PW, Yehualaeshet T, Lu B, Flavell RA, Milbrandt J, et al:
Early growth response gene 1-mediated apoptosis is essential for
transforming growth factor beta1-induced pulmonary fibrosis. J Exp
Med. 200:377–389. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Saadane N, Alpert L and Chalifour LE:
Altered molecular response to adrenoreceptor-induced cardiac
hypertrophy in Egr-1-deficient mice. Am J Physiol Heart Circ
Physiol. 278:H796–H805. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pritchard MT and Nagy LE: Ethanol-induced
liver injury: Potential roles for egr-1. Alcohol Clin Exp Res 29
(11 Suppl). 146S–150S. 2005. View Article : Google Scholar
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Meas-Yedid V, Servais A, Noël LH, Panterne
C, Landais P, Hervé N, Brousse N, Kreis H, Legendre C, Thervet E,
et al: New computerized color image analysis for the quantification
of interstitial fibrosis in renal transplantation. Transplantation.
92:890–899. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Arora MK and Singh UK: Molecular
mechanisms in the pathogenesis of diabetic nephropathy: An update.
Vascul Pharmacol. 58:259–271. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Macconi D, Remuzzi G and Benigni A: Key
fibrogenic mediators: Old players. Renin-angiotensin system. Kidney
Int Suppl (2011). 4:58–64. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ding H, Yang Q, Wang Z, Min R and Li Y:
Effects of sulfotanshinone IIA sodium on murine renal interstitial
fibrosis and CTGF level. Immunol J. 27:398–423. 2011.
|
|
28
|
Zeisberg M and Neilson EG: Mechanisms of
tubulointerstitial fibrosis. J Am Soc Nephrol. 21:1819–1834. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kota J, Chivukula RR, O'Donnell KA,
Wentzel EA, Montgomery CL, Hwang HW, Chang TC, Vivekanandan P,
Torbenson M, Clark KR, et al: Therapeutic microRNA delivery
suppresses tumorigenesis in a murine liver cancer model. Cell.
137:1005–1017. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ladeiro Y, Couchy G, Balabaud C,
Bioulac-Sage P, Pelletier L, Rebouissou S and Zucman-Rossi J:
MicroRNA profiling in hepatocellular tumors is associated with
clinical features and oncogene/tumor suppressor gene mutations.
Hepatology. 47:1955–1963. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cheng AM, Byrom MW, Shelton J and Ford LP:
Antisense inhibition of human miRNAs and indications for an
involvement of miRNA in cell growth and apoptosis. Nucleic Acids
Res. 33:1290–1297. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Croce CM and Calin GA: miRNAs, cancer, and
stem cell division. Cell. 122:6–7. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Karp X and Ambros V: Developmental
biology. Encountering microRNAs in cell fate signaling. Science.
310:1288–1289. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lorenzen JM, Kaucsar T, Schauerte C,
Schmitt R, Rong S, Hübner A, Scherf K, Fiedler J, Martino F,
Kumarswamy R, et al: MicroRNA-24 antagonism prevents renal ischemia
reperfusion injury. J Am Soc Nephrol. 25:2717–2729. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chien HY, Chen CY, Chiu YH, Lin YC and Li
WC: Differential microRNA profiles predict diabetic nephropathy
progression in Taiwan. Int J Med Sci. 13:457–465. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jiang F, Liu GS, Dusting GJ and Chan EC:
NADPH oxidase-dependent redox signaling in TGF-β-mediated fibrotic
responses. Redox Biol. 2:267–272. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bai XY, Ma Y, Ding R, Fu B, Shi S and Chen
XM: miR-335 and miR-34a promote renal senescence by suppressing
mitochondrial antioxidative enzymes. J Am Soc Nephrol.
22:1252–1561. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Liu X, Fu B, Chen D, Hong Q, Cui J, Li J,
Bai X and Chen X: miR-184 and miR-150 promote renal glomerular
mesangial cell aging by targeting Rab1a and Rab31. Exp Cell Res.
336:192–203. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li R, Chung AC, Dong Y, Yang W, Zhong X
and Lan HY: The microRNA miR-433 promotes renal fibrosis by
amplifying the TGF-β/Smad3-Azin1 pathway. Kidney Int. 84:1129–1144.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Thum T, Gross C, Fiedler J, Fischer T,
Kissler S, Bussen M, Galuppo P, Just S, Rottbauer W, Frantz S, et
al: MicroRNA-21 contributes to myocardial disease by stimulating
MAP kinase signalling in fibroblasts. Nature. 456:980–984. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zanchi C, Macconi D, Trionfini P, Tomasoni
S, Rottoli D, Locatelli M, Rudnicki M, Vandesompele J, Mestdagh P,
Remuzzi G, et al: MicroRNA-184 is a downstream effector of
albuminuria driving renal fibrosis in rats with diabetic
nephropathy. Diabetologia. 60:1114–1125. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chebotareva NV, Bobkova IN, Varshavskiĭ
VA, Golitsyna EP and Kozlovskaia LV: The role of smooth muscle
alpha-actin in development of renal fibrosis in patients with
chronic glomerulonephritis. Ter Arkh. 78:17–21. 2006.(In Russian).
PubMed/NCBI
|
|
43
|
Li ZI, Chung AC, Zhou L, Huang XR, Liu F,
Fu P, Fan JM, Szalai AJ and Lan HY: C-reactive protein promotes
acute renal inflammation and fibrosis in unilateral ureteral
obstructive nephropathy in mice. Lab Invest. 91:837–851. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Liu F, Chen HY, Huang XR, Chung AC, Zhou
L, Fu P, Szalai AJ and Lan HY: C-reactive protein promotes diabetic
kidney disease in a mouse model of type 1 diabetes. Diabetologia.
54:2713–2723. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Landau G, Bercovich Z, Park MH and Kahana
C: The role of polyamines in supporting growth of mammalian cells
is mediated through their requirement for translation initiation
and elongation. J Biol Chem. 285:12474–12481. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Cucoranu I, Clempus R, Dikalova A, Phelan
PJ, Ariyan S, Dikalov S and Sorescu D: NAD(P)H oxidase 4 mediates
transforming growth factor-beta1-induced differentiation of cardiac
fibroblasts into myofibroblasts. Circ Res. 97:900–907. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhang R, Zhang YY, Huang XR, Wu Y, Chung
AC, Wu EX, Szalai AJ, Wong BC, Lau CP and Lan HY: C-reactive
protein promotes cardiac fibrosis and inflammation in angiotensin
II-induced hypertensive cardiac disease. Hypertension. 55:953–960.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Graham HK, Horn M and Trafford AW:
Extracellular matrix profiles in the progression to heart failure.
European young physiologists symposium keynote lecture-bratislava
2007. Acta Physiol (Oxf). 194:3–21. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Alexander D, Judex M, Meyringer R,
Weis-Klemm M, Gay S, Müller-Ladner U and Aicher WK: Transcription
factor Egr-1 activates collagen expression in immortalized
fibroblasts or fibrosarcoma cells. Biol Chem. 383:1845–1853. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Aicher WK, Alexander D, Haas C, Kuchen S,
Pagenstecher A, Gay S, Peter HH and Eibel H: Transcription factor
early growth response 1 activity up-regulates expression of tissue
inhibitor of metalloproteinases 1 in human synovial fibroblasts.
Arthritis Rheum. 48:348–359. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Friedrich B, Janessa A, Artunc F, Aicher
WK, Müller GA, Lang F, Risler T and Alexander D: DOCA and TGF-beta
induce early growth response gene-1 (Egr-1) expression. Cell
Physiol Biochem. 22:465–474. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Lando D, Peet DJ, Gorman JJ, Whelan DA,
Whitelaw ML and Bruick RK: FIH-1 is an asparaginyl hydroxylase
enzyme that regulates the transcriptional activity of
hypoxia-inducible factor. Genes Dev. 16:1466–1471. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kim JN, Kim HJ, Jeong SH, Kye YC and Son
SW: Cigarette smoke-induced early growth response-1 regulates the
expression of the cysteine-rich 61 in human skin dermal
fibroblasts. Exp Dermatol. 20:992–997. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Guerquin MJ, Charvet B, Nourissat G, Havis
E, Ronsin O, Bonnin MA, Ruggiu M, Olivera-Martinez I, Robert N, Lu
Y, et al: Transcription factor EGR1 directs tendon differentiation
and promotes tendon repair. J Clin Invest. 123:3564–3576. 2013.
View Article : Google Scholar : PubMed/NCBI
|