Introduction
Acute kidney injury (AKI) is a frequent complication
among hospitalized patients and is associated with poor clinical
outcomes (1,2). Major surgery, sepsis and
chemotherapeutic agent use are the leading causes of AKI (3–6).
Cisplatin (CP), one of the most commonly used chemotherapeutic
drugs, is used to treat a wide spectrum of human malignancies
(7). However, it has been reported
that 25–35% of patients experience nephrotoxicity, particularly
AKI, which is a major limitation for the clinical application of CP
(8,9). CP-based chemotherapy combined with
surgery is widely used to treat cancer and has been demonstrated to
improve postoperative survival rate (10). However, patients with cancer are
often required to undergo both surgery and chemotherapy, which can
exacerbate renal injury (11). A
previous study reported that the incidence of AKI in patients with
cancer was higher compared with in patients without cancer
(12). Therefore, effective
strategies to prevent AKI are essential for patients in the
perioperative period.
Previous studies have focused on the development of
anti-inflammatory and anti-apoptotic drugs to manage CP-induced
renal injury (8,13). The endoplasmic reticulum (ER) can
serve a critical role in CP cellular toxicity (13,14).
The ER is the organelle responsible for synthesis and maturation of
proteins, calcium storage and maintenance of cell homeostasis
(15). Cellular events, such as
oxidative stress, can trigger ER stress (ERS). As a protective
stress response, the homeostasis of the ER can be restored via the
unfolded protein response (UPR) (16). However, in cases of severe ERS,
accumulation of a large number of unfolded proteins results in the
dissociation of the 78-kDa glucose-regulated protein (GRP78) from
transmembrane proteins, and activation of the downstream signaling
molecules C/EBP homologous protein (CHOP) and caspase-12,
eventually leading to apoptosis (17). Previous studies have experimentally
shown amelioration of CP-induced nephrotoxicity mediated by
attenuating ERS-induced apoptosis (14,18,19).
In addition, the PI3K/AKT signaling pathway has a central role in
cell growth, differentiation and apoptosis (20). Using PI3K-knockout mice, it was
previously revealed that a conventional dose of CP was more lethal
in these knockout mice, indicating the importance of the PI3K/AKT
pathway in protecting the kidneys (21).
Dexmedetomidine (Dex), a highly selective
α2 adrenoreceptor (α2AR) agonist, has been
reported to be beneficial for the prevention of numerous types of
kidney diseases, including acute stress-induced kidney injury
(22), ischemia-reperfusion and
sepsis-induced kidney injuries (23–25).
It has also been demonstrated that Dex protects the kidney against
CP-induced AKI by regulating apoptosis and the inflammatory
response (26). However, the
molecular mechanisms underlying the effects of Dex against
CP-induced AKI have not been fully elucidated. As the pathogenesis
of CP-induced AKI is mediated by ERS-induced apoptosis, Dex may be
able to alleviate apoptosis by inhibiting ERS (27,28).
Therefore, the present study aimed to investigate the protective
effects of Dex against CP-induced AKI and assessed whether these
effects were mediated by attenuation of ERS-induced apoptosis via
the α2AR/PI3K/AKT pathway.
Materials and methods
Animals
Animal experiments were performed in accordance with
The Guiding Principles for the Care and Use of Laboratory Animals
(updated 2011; National Institutes of Health) (29) and were approved by The Animal Care
Committee of Hebei Medical University (Shijiazhuang, China). In
total, 40 male Sprague-Dawley rats (weight, 200–220 g; age, 6
weeks) were obtained from Liaoning Chengsheng Biotechnology Co.,
Ltd. [certificate of conformity no. SCXK (liao) 2015-0001]. Rats
were acclimated for 7 days prior to the start of the study. All
rats were kept under standard conditions: Temperature (22±2°C),
humidity (50–60%), with a 12-h light/dark cycle, and had free
access to food (standard pellet laboratory chow) and water. The
rats were randomly divided into the following four groups
(n=10/group): i) Control (Con) group, rats received intraperitoneal
(i.p.) injections of 0.9% saline (5 ml/kg) on days 1 and 3; ii) CP
group, rats received an i.p. injection of 5 mg/kg CP (1 mg/ml,
dissolved in 0.9% saline) on day 1 and an i.p. injection of 0.9%
saline (10 ml/kg) on day 3; iii) Dex + CP group, rats were
administered an i.p. injection of 25 µg/kg Dex immediately after CP
treatment; and iv) Dex + CP + atipamezole (Atip) group, rats
received an i.p. injection of atipamezole, the α2AR
antagonist, at a dose of 250 µg/kg and then received the same
treatment as the Dex + CP group. Dex and Atip were given once a day
for 3 days. The doses of Dex, CP and Atip were based on previous
studies (26,30). CP and Atip were purchased from
Sigma-Aldrich (Merck KGaA), and Dex was purchased from Jiangsu
Hengrui Medicine Co., Ltd. The rats were weighed daily and
monitored for food and water intake, and mental status. The rats
were anesthetized with 2% pentobarbital solution (60 mg/kg) 96 h
after treatment with CP. A total of 2 ml blood was withdrawn from
the abdominal aorta, and the serum was separated by centrifugation
(3,000 × g; 4°C for 20 min) and stored at −80°C. Subsequently,
animals were euthanized with 5% pentobarbital solution (150 mg/kg),
and both kidneys were immediately excised, weighed and cut into
coronal sections. In total, two pieces of the kidney were snap
frozen in liquid nitrogen and stored at −80°C for later use in
biochemical tests and western blot analysis. A small part of the
kidney was preserved in 10% neutral buffered-formalin for
histological evaluations, immunohistochemistry and terminal
deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL)
assay.
Biochemical analysis
Serum blood urea nitrogen (BUN) and serum creatinine
(Scr) levels were assessed using commercially supplied kits (cat.
nos. C010 and C074; Wuhan Servicebio Technology Co., Ltd.); samples
were analyzed according to the manufacturer's instructions. The
renal index (RI) was calculated as follows: Weight of both kidneys
(g)/animal weight (g) ×100.
Histopathological analysis
To evaluate histological changes, samples were fixed
with 10% formalin buffer for 24 h at 25°C and then embedded in
paraffin. Paraffin-embedded tissue samples were cut into 5-µm thick
sections. The tissue sections were subsequently deparaffinized in
xylene (22±2°C) twice for 15–20 min each and rehydrated in pure
ethanol for 10 min each, followed by a descending ethanol series
(95, 90, 80 and 70%) for 5 min each. Deparaffinized sections were
incubated in 3% hydrogen peroxide diluted in methanol for 10 min at
25°C in the dark to inhibit endogenous peroxidase activity and
subsequently with 3% BSA (cat. no. G5001; Wuhan Servicebio
Technology Co., Ltd.) for 30 min at 25°C to block any non-specific
binding. Tissue sections were then stained with 0.1% hematoxylin
for 10 min and with 0.5% eosin for 1 min (both at 22±2°C).
Histopathological changes were observed using a light microscope
(magnification, ×400; Leica Microsystems GmbH). Assessment of the
tubular damage score was performed in 10 fields in five sections
per group using the following index of renal tubular necrosis:
Score of 0 (absence of damage); score of 1 (<25% damage); score
of 2 (25–50% damage); score of 3 (50–75% damage); and score of 4
(>75% damage). Tubular necrosis was characterized by loss of the
proximal tubular brush border, blebbing of apical membranes,
epithelial detachment from the basement membrane or intraluminal
hyaline cast formation.
TUNEL staining analysis
The TUNEL assay (cat. no. 116848179; Roche
Diagnostics GmbH) was conducted according to the manufacturer's
instructions. Tissues were fixed and prepared in the same manner as
for histopathology and subsequently, deparaffinized tissue sections
were incubated with 3% hydrogen peroxide in methanol for 10 min at
15–25°C in the dark, washed three times with PBS and incubated with
0.1% Triton X-100 in freshly prepared 0.01% sodium citrate for 8
min at 25°C. Tissue sections were then incubated with proteinase K
working solution (cat. no. G1205; Wuhan Servicebio Technology Co.,
Ltd.) for 25 min at 37°C and washed three times with PBS (pH 7.4)
in a Rocker device for 5 min each. Reagent 1 (TdT) and reagent 2
(dUTP; both from the TUNEL assay kit) were mixed at a ratio of 1:9
and the mixture was incubated with the tissue sections at 37°C for
2 h. The sections were washed three times with PBS (pH 7.4) and
then cell nuclei were counterstained with 2 µg/ml DAPI solution
(cat. no. G1012; Wuhan Servicebio Technology Co., Ltd.) at room
temperature for 10 min in the dark and mounted with 50 µl anti-fade
mounting medium (cat. no. G1401; Wuhan Servicebio Technology Co.,
Ltd.). TUNEL-positive cells were observed in five randomly-selected
fields using a fluorescence microscope (magnification, ×400); the
nucleus is blue and positive apoptotic cells are green. The number
of positive cells per high-power field was calculated and analyzed
in a blinded manner using Image-Pro Plus 6.0 software (Media
Cybernetics, Inc.).
Immunohistochemical analysis
For immunohistochemical analysis, kidney tissues
were fixed with 10% formaldehyde solution for 24 h at 15–25°C.
Subsequently, tissues were embedded in paraffin and cut into 5-µm
sections. Deparaffinization of the sections was followed by heating
for 10 min at 95°C in citrate buffer solution (0.01 M; pH 6.0). The
slides were then washed with TBS (0.01 M; pH 7.4) and incubated
with 10% goat serum (Wuhan Servicebio Technology Co., Ltd.) for 60
min at 37°C and with primary antibodies at 4°C overnight. The
primary antibodies used were anti-GRP78 (1:200; cat. no. ab21685;
Abcam) and anti-caspase-12 (1:100; cat. no. ab62484; Abcam). After
three washes with PBS, the sections were incubated with a
horseradish peroxidase (HRP)-conjugated goat secondary antibody
(1:200; cat. no. G1213; Wuhan Servicebio Technology Co., Ltd.) for
30 min at 37°C. To count stained nuclei, 0.1% hematoxylin staining
for 10 min at 22±2°C was performed. Analysis was performed using
Leica QWin V3 image analysis software (Leica Microsystems GmbH).
Brown areas of staining were considered as positive. The intensity
of the staining was graded as follows: 0 (no color); 1 (light
yellow); 2 (light brown); and 3 (brown). The percentage of
positively stained areas was graded as follows: 0 (<5%); 1
(5–25%); 2 (25–50%); 3 (51–75%); and 4 (>75%). In total, ten
high-power fields (magnification, ×400) per section were randomly
selected using light microscopy, and two sections per kidney per
group were examined in each experiment.
Western blotting
Renal tissues were lysed with RIPA buffer (cat. no.
G2002; Wuhan Servicebio Technology Co., Ltd), supplemented with 1
mM PMSF and homogenized. The homogenate was centrifuged at 4,000 ×
g for 20 min at 4°C, and the supernatant was collected and measured
for protein concentration using a bicinchoninic acid protein assay
kit (cat. no. G2026; Wuhan Servicebio Technology Co., Ltd.). Equal
amounts of protein (40 µg) from each sample were separated by
10–12% SDS-PAGE and electrotransferred onto a PVDF membrane and
blocked with 5% skimmed milk at room temperature for 2 h on a
rotating shaker. Membranes were subsequently washed with TBS-0.1%
Tween (TBST). The primary antibodies used were as follows: GRP78
(1:1,000; rabbit; cat. no. ab21685; Abcam), CHOP (1:1,000; rabbit;
cat. no. 2895; Cell Signaling Technology, Inc.), caspase-12 (1:600;
rabbit; cat. no. ab62484; Abcam), AKT (1:1,000; rabbit; cat. no.
AF6261; Affinity Biosciences), phosphorylated (p)-AKT (1:1,000;
rabbit; cat. no. AF0832; Affinity Biosciences), PI3K (1:1,000;
rabbit; cat. no. AF6241; Affinity Biosciences), p-PI3K (1:1,000;
rabbit; cat. no. AF3241; Affinity Biosciences) and β-actin
(1:3,000; mouse; cat. no. GB12001; Wuhan Servicebio Technology Co.,
Ltd.). Membranes were incubated with antibodies overnight at 4°C.
After washing three times in TBST, the membranes were incubated
with HRP-conjugated secondary antibodies (1:3,000; cat. nos.
GB23302 and GB23303; Wuhan Servicebio Technology Co., Ltd.) for 2 h
at room temperature. Bands were detected using standard ECL (EMD
Millipore). The band intensity was measured using ImageJ version
8.0 software (National Institutes of Health).
Statistical analysis
The statistical analyses were performed, and the
graphs were created using GraphPad Prism (version 7.0; GraphPad
Software, Inc.). Data are presented as the mean ± SEM of between
3–10 experimental repeats. Variables were checked for normal
distribution using the Shapiro-Wilk test. Data were analyzed using
one-way ANOVA followed by Tukey's multiple comparisons testing as
appropriate. The histopathological and immunohistochemical data
were compared using Kruskal-Wallis test followed by Dunn's multiple
comparisons test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Dex treatment rescues the change in
body weight and RI
A previous study suggested that CP could reduce body
weight in rats (31). I n the
present study, animal models of CP-induced AKI were established by
i.p. injection of 5 mg/kg CP. It was revealed that CP-treated
animals lost a significant amount of weight and exhibited increased
RI compared with the Con group (Fig.
1A and B; P<0.001). However, administration of Dex in
combination with CP significantly rescued the alteration in body
weight and RI compared with the CP group (P<0.05). Moreover,
treatment with Atip in combination with Dex resulted in a similar
outcome to the CP group (Fig. 1A and
B; P<0.05).
 | Figure 1.Effect of Dex on renal function and
body weight. (A) Body weight, (B) RI, (C) BUN and (D) Scr. Data are
presented as the mean ± SEM (n=10). **P<0.01, ***P<0.001 vs.
Con; #P<0.05, ##P<0.01 vs. Dex + CP.
Atip, atipamezole; BUN, blood urea nitrogen; Con, control; CP,
cisplatin; Dex, dexmedetomidine; Scr, serum creatinine; RI, renal
index. |
Dex treatment ameliorates CP-induced
nephrotoxicity markers
Clinical markers for renal tissue injury, including
the levels of serum BUN and Scr, were investigated to evaluate
CP-induced nephrotoxicity. It has previously been reported that a
single dose of CP (3–8 mg/kg) can cause acute nephrotoxicity and
induce changes to kidney structure and function in rats (32). In the present study, animal models
of CP-induced AKI were established by i.p. injection of 5 mg/kg CP.
It was demonstrated that rats in the CP-treated group developed
renal dysfunction and exhibited significantly increased Scr and BUN
levels compared with the Con group (Fig. 1C and D; P<0.001). However, Scr
and BUN expression levels in the Dex + CP group were significantly
lower compared with the CP group (P<0.01). Furthermore, the
renoprotective effects of Dex were reversed by treatment with the
α2AR antagonist Atip (Fig. 1C and
D; P<0.01).
Dex treatment ameliorates CP-induced
kidney tubular damage
H&E staining was performed to investigate the
renoprotective effects of Dex following CP administration. The
histological examination results suggested that the kidneys from
the Con and Dex + CP groups exhibited normal tubular morphology,
whereas the kidneys from the CP group showed severe tubular damage
and widespread tubular necrosis, with dilatation and tubular
atrophy (Fig. 2A). In addition,
the extent of histological damage in the CP group was significantly
higher compared with the Con group (P<0.001; Fig. 2B). However, the extent of tubular
damage was significantly reduced in the Dex + CP group compared
with the CP group (P<0.05; Fig. 2A
and B). Furthermore, the α2 receptor blocker Atip abolished the
effects of Dex (P<0.05; Fig. 2A and
B).
Dex treatment ameliorates CP-induced
apoptosis of tubular epithelial cells
Apoptotic cell death is the major pathological
process which renal tissue undergoes as a result of CP toxicity
(32). To investigate whether Dex
protected renal tubular epithelial cells against apoptosis induced
by CP, tubular cell apoptosis was detected in the present
CP-induced AKI model using TUNEL assay. It was revealed that
apoptotic cell death, characterized by TUNEL-positive cells, was
significantly increased in the CP group compared with the Con group
(P<0.001; Fig. 3A and B). Dex
administration significantly reduced the number of TUNEL-positive
cells in the kidneys after CP treatment (P<0.05), indicating
reduced apoptosis in the Dex + CP group. In addition, Atip reversed
the renoprotective effects of Dex (P<0.05; Fig. 3B), indicating that the α2AR blocker
atipamezole abolished the protective effects of Dex on apoptotic
cell death.
 | Figure 3.Dex protects against proximal tubular
cell apoptosis induced by CP. (A) TUNEL staining assay
(magnification, ×400; scale bar, 50 µm). Green, TUNEL; blue, DAPI;
red arrows, TUNEL-positive cells. (B) Quantification of
TUNEL-positive cells. Data are presented as the mean ± SEM (n=10).
***P<0.001 vs. Con; #P<0.05 vs. Dex + CP. Atip,
atipamezole; Con, control; CP, cisplatin; Dex, dexmedetomidine;
HPF, high-power field; TUNEL, terminal deoxynucleotidyl transferase
dUTP nick-end labelling. |
Dex treatment ameliorates the
expression of GRP78, CHOP and caspase-12
A previous study has also demonstrated that
ERS-mediated apoptotic pathways are implicated in CP-induced AKI
(6). Therefore, to investigate the
mechanisms underlying the renoprotective effects of Dex, the
expression levels of GRP78, CHOP and caspase-12, which are major
proteins involved in ERS-mediated apoptosis (33), were examined. The present results
suggested that the protein expression levels of GRP78, CHOP and
caspase-12 were significantly increased in the CP group compared
with the Con group (P<0.01; Fig.
4A-D). However, the expression levels of these proteins were
significantly reduced in the Dex-treated group (P<0.05 and
P<0.01). The present study also investigated the expression
levels of GRP78 and caspase-12 using immunohistochemical staining
(Fig. 4H). The
immunohistochemistry results of GRP78 and caspase-12 protein
expression were consistent with the western blotting results, where
CP treatment increased the expression levels of GRP78 and
caspase-12 compared with the Con group (P<0.01 and P<0.05;
Fig. 4I and J). Moreover, Dex
treatment significantly reduced CP-induced GRP78 and caspase-12
expression, whereas Atip reversed the renoprotective effects of Dex
(P<0.01; Fig. 4I and J).
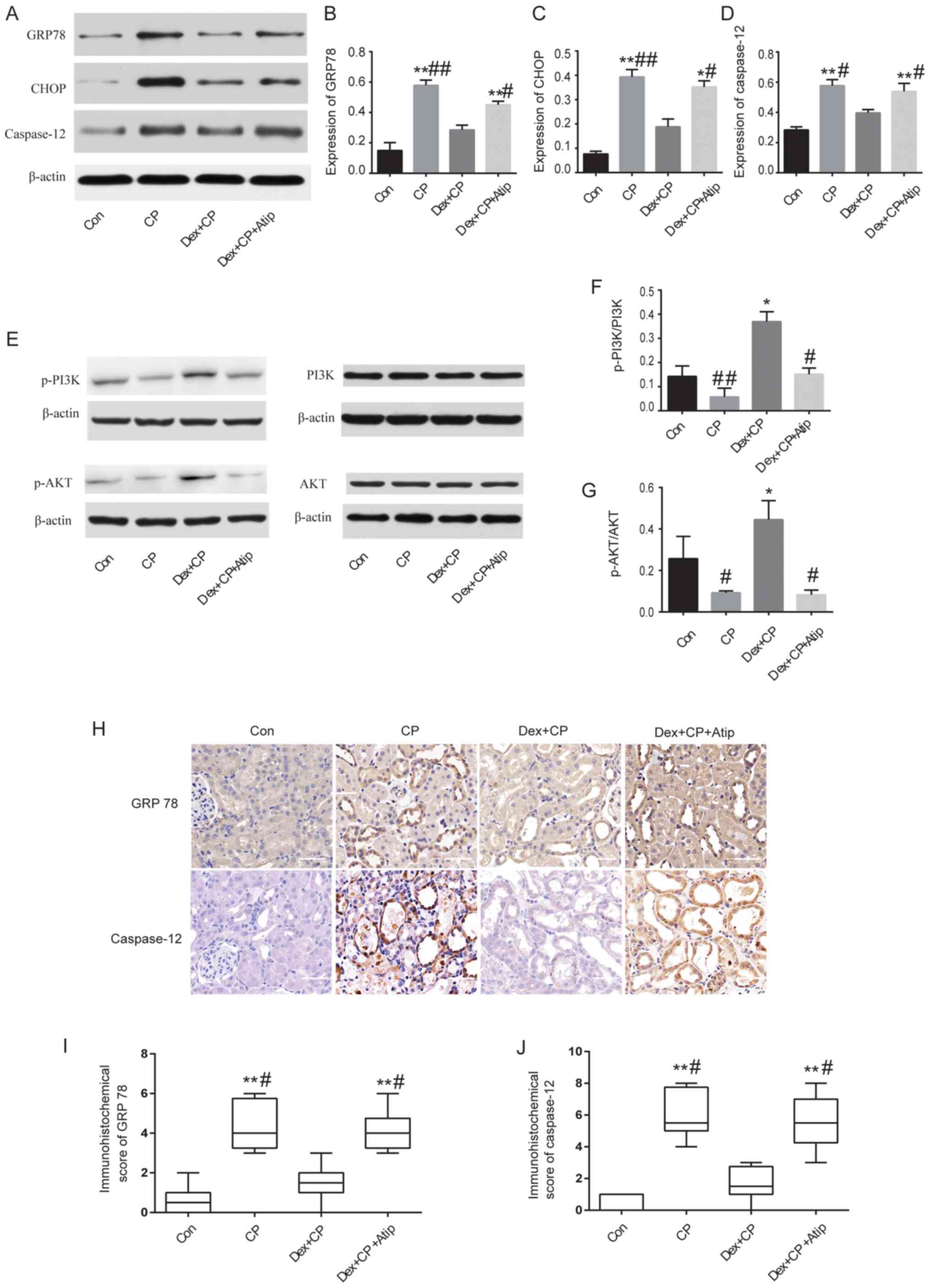 | Figure 4.Western blot analysis and
immunohistochemical staining of proteins in the kidney. (A) Western
blot analysis, and semi-quantification of protein expression levels
of (B) GRP78, (C) CHOP and (D) caspase-12. (E) Western blot
analysis, and semi-quantification of protein expression levels of
(F) p-PI3K/PI3K (G) and p-AKT/AKT relative to β-actin control. (H)
Expression levels of GRP78 and caspase-12 detected in damaged
tubules by immunohistochemistry (magnification, ×400; scale bar, 50
µm). (I) Semi-quantification of GRP78 in kidney sections. (J)
Semi-quantification of caspase-12 in kidney sections. Data are
presented as the mean ± SEM (n=3). *P<0.05, **P<0.01 vs. Con;
#P<0.05, ##P<0.01 vs. Dex + CP. Atip,
atipamezole; CHOP, C/EBP homologous protein; Con, control; CP,
cisplatin; Dex, dexmedetomidine; GRP78, 78-kDa glucose-regulated
protein; p-, phosphorylated. |
Dex treatment regulates the PI3K/AKT
signaling pathway
Activation of the PI3K/AKT pathway serves a key role
in cytoprotective signaling and has been demonstrated to exert
protective effects against CP-induced renal toxicity (34). A previous study reported that Dex
activates the cell survival PI3K/AKT signaling pathway via α2AR in
order to provide renoprotection (35). To investigate whether Dex activated
the PI3K/AKT signaling pathway, the present study measured the
expression levels of p-PI3K and p-AKT. The results revealed that
the protein expression levels of p-PI3K and p-AKT were
significantly decreased in the CP group compared with the Dex + CP
group (P<0.05 and P<0.01; Fig.
4E-G), indicating that Dex activated the PI3K/AKT signaling
pathway. Treatment with Atip in combination with Dex resulted in a
similar outcome to CP; Dex + CP + Atip decreased the protein
expression levels of p-PI3K and p-AKT compared with the CP + Dex
group (P<0.05; Fig. 4F and
G).
Discussion
CP is known to induce AKI as a result of renal
tubular epithelial cell injury. ERS-induced apoptosis of tubular
epithelium cells serves a pivotal role in the pathogenesis of
CP-induced nephrotoxicity (9,36).
Therefore, the present study investigated whether Dex exerted a
renoprotective effect on CP-induced AKI in a rat model by
inhibiting ERS-mediated apoptosis via the α2AR/PI3K/AKT pathway.
The present results suggested that Dex facilitated a significant
reduction in RI, body weight, and Scr and BUN levels, indicating a
significant renoprotective effect, which was further demonstrated
by H&E staining and apoptosis analysis. In addition, Dex
reduced the expression levels of ERS-dependent proteins and
increased the expression levels of p-PI3K and p-AKT. Furthermore,
the α2 receptor blocker Atip abolished the effects of Dex.
Therefore, the protective mechanism underlying Dex may be related
to ERS-mediated apoptosis via the α2AR/PI3K/AKT signaling pathway.
The present results provided a novel insight into the biological
benefits of Dex as a potential renoprotective agent.
As biomarkers of renal function, BUN and Scr are
often used to reflect the degree of renal injury (37). In the present study, BUN and Scr
levels in the CP group were significantly increased, and body
weight and RI were significantly changed, compared with in the Con
group. Furthermore, the results of histopathological examination
and TUNEL assay revealed that CP aggravated kidney damage compared
with the Con group. The present results were consistent with those
from previous studies (38). In
the present study, Dex administration significantly inhibited the
production of serum BUN and Scr and reversed the alterations in
body weight and RI. Furthermore, Atip abolished the effects of Dex.
The present study identified the beneficial effects of Dex on
prevention of renal tubular damage and dysfunction. However, the
mechanism underlying this protective effect requires further
investigation.
The ER is an intracellular organelle that serves an
important role in protein homeostasis (14). The ER can restore homeostasis via
the UPR. When the UPR fails to protect cells, ERS becomes one of
the main pathogenic factors that induces the apoptotic pathway and
results in an accumulation of misfolded proteins (6). As a major response to ERS, GRP78 is
released from transmembrane proteins and activates protein kinase
RNA-like ER kinase (PERK), inositol requiring enzyme 1 (IRE1) and
activating transcription factor 6 (ATF6), which induce the
pro-apoptotic transcription factor CHOP, eventually leading to
apoptosis (39). Caspase-12, a
marker of apoptosis, is localized on the ER and is specifically
activated via ERS, and not via membrane or mitochondrial signals.
It has been reported that ERS-mediated apoptosis is important in
CP-induced nephrotoxicity, suggesting that ER is a target of CP,
and that GRP78, caspase-12 and CHOP are important molecules
(9,40,41).
To investigate the mechanisms underlying the
protective effects of Dex against CP-induced tubular cell
apoptosis, the present study examined the protein expression levels
of GRP78, CHOP and caspase-12. It was revealed that GRP78, CHOP and
caspase-12 protein expression was increased following treatment
with CP, whereas Dex treatment significantly inhibited the
expression of these proteins. Therefore, the present results
suggested that Dex could suppress CP-induced ERS. Previous studies
have shown that the neuroprotective and cardioprotective effects of
Dex are mediated by inhibiting ERS-dependent apoptosis (27,42).
Furthermore, Liang et al (26) reported that Dex exerted an
anti-inflammatory effect via inhibition of the NF-κB signaling
pathway and had a role in the inhibition of apoptosis via
p53-mediated Bax induction during CP-induced AKI. The present study
also examined whether the α2AR served a role in the protective
effect of Dex. The present results suggested that the α2AR
antagonist Atip reversed the renoprotective effects of Dex,
indicating that Dex acted in an α2AR-dependent manner. Gu et
al (35) revealed that Dex
exerted a renoprotective effect against ischemia-reperfusion injury
via the α2AR-dependent pathway. Therefore, the present results
indicated that the potential mechanism underlying the effects of
Dex on the prevention of CP-induced AKI may be associated with the
suppression of ERS-mediated apoptosis via activation of α2AR.
The PI3K/AKT signaling pathway has a major role in
promoting cell survival by inhibiting the caspase-controlled
intrinsic apoptotic pathway (43).
A previous study has shown that ERS-induced apoptosis is closely
associated with the PI3K/AKT signaling pathway (44). In addition, CP aggravates renal
injury by inhibiting the PI3K/AKT signaling pathway in rat kidneys
(45), and a conventional dose of
CP is more lethal in PI3K-knockout mice (21). The present study identified a
significant increase in the expression levels of p-AKT following
treatment with Dex in CP-induced AKI. However, treatment with CP +
Dex + Atip had a similar effect to CP alone; the expression levels
of p-PI3K and p-AKT were significantly decreased in the CP + Dex +
Atip group, indicating that the effects of Dex were partially
blocked by the α2AR antagonist. Furthermore, a previous study
reported that AKT signaling is critical for recovery from renal
ischemia-reperfusion injury, and that Dex activates AKT via
α2AR-dependent and -independent PI3K coupling (38). Similarly, Dex exerts
neuroprotective (46),
cardioprotective (28) and
lung-protective effects (30,47)
via activation of the PI3K/AKT signaling pathway. Therefore, the
protective effects of Dex against CP-induced AKI may involve AKT
signaling.
There were some limitations to the present study.
The present results suggested that Dex alleviated CP-induced AKI by
attenuating ERS-induced apoptosis, at least in part, via the
α2AR/PI3K/AKT pathway. Since this study used an in vivo
model, the renoprotective effect of Dex may be associated with
other unidentified mechanisms, and whether Dex is able to directly
protect renal tubular epithelial cells remains unclear.
Furthermore, ERS is a complex process involving three transmembrane
proteins, PERK, ATF6 and IRE1, which are bound to GRP78 (48). The present study did not detect
changes in these transmembrane proteins; therefore, it is not clear
which membrane protein is involved in the renoprotective effects of
Dex. In addition, the activated status of caspase-12 is based on
the cleavage of procaspase-12 (30), and the present study did not detect
changes in the cleavage status of procaspase-12, which may not
accurately express changes in ERS. Furthermore, the sample sizes
were relatively small in the present study. Therefore, the present
results may have false-positive and false-negative errors. The
mechanism underlying the effects of Dex is complex and further
in-depth research is required to fully understand the effects of
Dex.
In conclusion, the present results suggested that
Dex may protect against CP-induced AKI, and the underlying
mechanism may be associated with the suppression of ERS-mediated
apoptosis via the α2AR/PI3K/AKT signaling pathway. These results
provided an insight into the novel biological benefits of Dex as a
renoprotective agent for patients with cancer. However, the
pathological process of CP-induced AKI involves a variety of
complex mechanisms, and whilst Dex has a protective effect, it
requires further study to be used as a therapeutic tool in clinical
settings.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during the present
study are included in this published manuscript.
Authors' contributions
HQJ and KSZ designed the experiments. YJC, KSZ and
XFW performed the experiments. KSZ, JMS, FFY and CL analyzed the
data and prepared the images. KSZ and YJC drafted the manuscript.
All authors reviewed the manuscript.
Ethics approval and consent to
participate
The present study was approved by The Animal Care
Committee of Hebei Medical University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Al-Jaghbeer M, Dealmeida D, Bilderback A,
Ambrosino R and Kellum JA: Clinical decision support for
in-hospital AKI. J Am SocNephrol. 29:654–660. 2018.
|
|
2
|
Zarbock A, Koyner JL, Hoste EAJ and Kellum
JA: Update on perioperative acute kidney injury. Anesth Analg.
127:1236–1245. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chertow GM, Burdick E, Honour M, Bonventre
JV and Bates DW: Acute kidney injury, mortality, length of stay,
and costs in hospitalized patients. J Am Soc Nephrol. 16:3365–3370.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gameiro J, Fonseca JA, Neves M, Jorge S
and Lopes JA: Acute kidney injury in major abdominal surgery:
Incidence, risk factors, pathogenesis and outcomes. Ann Intensive
Care. 8:222018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jin J, Wang Y, Shen Q, Gong J, Zhao L and
He Q: Acute kidney injury in cancer patients: A nationwide survey
in China. Sci Rep. 9:35402019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Izzedine H and Perazella MA: Anticancer
drug-induced acute kidney injury. Kidney Int Rep. 2:504–514. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dilruba S and Kalayda GV: Platinum-based
drugs: Past, present and future. Cancer Chemother Pharmacol.
77:1103–1124. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pabla N and Dong Z: Cisplatin
nephrotoxicity: Mechanisms and renoprotective strategies. Kidney
Int. 73:994–1007. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Manohar S and Leung N: Cisplatin
nephrotoxicity: A review of the literature. J Nephrol. 31:15–25.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Miao ZF, Liu XY, Wang ZN, Zhao TT, Xu YY,
Song YX, Huang JY, Xu H and Xu HM: Effect of neoadjuvant
chemotherapy in patients with gastric cancer: A PRISMA-compliant
systematic review and meta-analysis. BMC Cancer. 18:1182018.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lee EH, Kim HR, Baek SH, Kim KM, Chin JH,
Choi DK, Kim WJ and Choi IC: Risk factors of postoperative acute
kidney injury in patients undergoing esophageal cancer surgery. J
Cardiothorac Vasc Anesth. 28:936–942. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Salahudeen AK, Doshi SM, Pawar T, Nowshad
G, Lahoti A and Shah P: Incidence rate, clinical correlates, and
outcomes of AKI in patients admitted to a comprehensive cancer
center. Clin J Am Soc Nephrol. 8:347–354. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Oh GS, Kim HJ, Shen A, Lee SB, Khadka D,
Pandit A and So HS: Cisplatin-induced kidney dysfunction and
perspectives on improving treatment strategies. Electrolyte Blood
Press. 12:55–65. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yan M, Shu S, Guo C, Tang C and Dong Z:
Endoplasmic reticulum stress in ischemic and nephrotoxic acute
kidney injury. Ann Med. 50:381–390. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Volarevic V, Djokovic B, Jankovic MG,
Harrell CR, Fellabaum C, Djonov V and Arsenijevic N: Molecular
mechanisms of cisplatin-induced nephrotoxicity: A balance on the
knife edge between renoprotection and tumor toxicity. J Biomed Sci.
26:252019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bravo R, Parra V, Gatica D, Rodriguez AE,
Torrealba N, Paredes F, Wang ZV, Zorzano A, Hill JA, Jaimovich E,
et al: Endoplasmic reticulum and the unfolded protein response:
Dynamics and metabolic integration. Int Rev Cell Mol Biol.
301:215–290. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
García de la Cadena S and Massieu L:
Caspases and their role in inflammation and ischemic neuronal
death. Focus on caspase-12. Apoptosis. 21:763–777. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen B, Liu G, Zou P, Li X, Hao Q, Jiang
B, Yang X and Hu Z: Epigallocatechin-3-gallate protects against
cisplatin-induced nephrotoxicity by inhibiting endoplasmic
reticulum stress-induced apoptosis. Exp Biol Med (Maywood).
240:1513–1519. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gao Z, Liu G, Hu Z and Li X, Yang X, Jiang
B and Li X: Grape seed proanthocyanidin extract protects from
cisplatin-induced nephrotoxicity by inhibiting endoplasmic
reticulum stress-induced apoptosis. Mol Med Report. 9:801–807.
2014. View Article : Google Scholar
|
|
20
|
Gong X, Shao L, Fu YM and Zou Y: Effects
of olmesartan on endothelial progenitor cell mobilization and
function in carotid atherosclerosis. Med Sci Monit. 21:1189–1193.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kuwana H, Terada Y, Kobayashi T, Okado T,
Penninger JM, Irie-Sasaki J, Sasaki T and Sasaki S: The
phosphoinositide-3 kinase gamma-Akt pathway mediates renal tubular
injury in cisplatin nephrotoxicity. Kidney Int. 73:430–445. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen Y, Feng X, Hu X, Sha J, Li B, Zhang H
and Fan H: Dexmedetomidine ameliorates acute stress-induced kidney
injury by attenuating oxidative stress and apoptosis through
inhibition of the ROS/JNK signaling pathway. Oxid Med Cell Longev.
2018:40353102018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen Y, Luan L, Wang C, Song M, Zhao Y,
Yao Y, Yang H, Ma B and Fan H: Dexmedetomidine protects against
lipopolysaccharide-induced early acute kidney injury by inhibiting
the iNOS/NO signaling pathway in rats. Nitric Oxide. 85:1–9. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Si Y, Bao H, Han L, Chen L, Zeng L, Jing
L, Xing Y and Geng Y: Dexmedetomidine attenuation of renal
ischaemia-reperfusion injury requires sirtuin 3 activation. Br J
Anaesth. 121:1260–1271. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Qiu R, Yao W, Ji H, Yuan D, Gao X, Sha W,
Wang F, Huang P and Hei Z: Dexmedetomidine restores septic renal
function via promoting inflammation resolution in a rat sepsis
model. Life Sci. 204:1–8. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liang H, Liu HZ, Wang HB, Zhong JY, Yang
CX and Zhang B: Dexmedetomidine protects against cisplatin-induced
acute kidney injury in mice through regulating apoptosis and
inflammation. Inflamm Res. 66:399–411. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu C, Fu Q, Mu R, Wang F, Zhou C, Zhang
L, Yu B, Zhang Y, Fang T and Tian F: Dexmedetomidine alleviates
cerebral ischemia-reperfusion injury by inhibiting endoplasmic
reticulum stress dependent apoptosis through the
PERK-CHOP-Caspase-11 pathway. Brain Res. 1701:246–254. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cheng XY, Gu XY, Gao Q, Zong QF, Li XH and
Zhang Y: Effects of dexmedetomidine postconditioning on myocardial
ischemia and the role of the PI3K/Akt-dependent signaling pathway
in reperfusion injury. Mol Med Rep. 14:797–803. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
National Research Council: Guide for the
care and use of laboratory animals: Eighth edition. Washington, DC:
The National Academies Press; 2011
|
|
30
|
Zhang W, Zhang JQ, Meng FM and Xue FS:
Dexmedetomidine protects against lung ischemia-reperfusion injury
by the PI3K/Akt/HIF-1α signaling pathway. J Anesth. 30:826–833.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Alhoshani AR, Hafez MM, Husain S,
Al-Sheikh AM, Alotaibi MR, Al Rejaie SS, Alshammari MA, Almutairi
MM and Al-Shabanah OA: Protective effect of rutin supplementation
against cisplatin-induced Nephrotoxicity in rats. BMC Nephrol.
18:1942017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Perše M and Večerić-Haler Ž:
Cisplatin-induced rodent model of kidney injury: Characteristics
and challenges. Biomed Res Int. 2018:14628022018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Schröder M and Kaufman RJ: The mammalian
unfolded protein response. Annu Rev Biochem. 74:739–789. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Potočnjak I and Domitrović R: Carvacrol
attenuates acute kidney injury induced by cisplatin through
suppression of ERK and PI3K/Akt activation. Food Chem Toxicol.
98:251–261. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gu J, Sun P, Zhao H, Watts HR, Sanders RD,
Terrando N, Xia P, Maze M and Ma D: Dexmedetomidine provides
renoprotection against ischemia-reperfusion injury in mice. Crit
Care. 15:R1532011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mahgoub E, Kumaraswamy SM, Kader KH,
Venkataraman B, Ojha S, Adeghate E and Rajesh M: Genipin attenuates
cisplatin-induced nephrotoxicity by counteracting oxidative stress,
inflammation, and apoptosis. Biomed Pharmacother. 93:1083–1097.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang L, Gu Y, Li H, Cao H, Liu B, Zhang H
and Shao F: Daphnetin protects against cisplatin-induced
nephrotoxicity by inhibiting inflammatory and oxidative response.
Int Immunopharmacol. 65:402–407. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhang W, Hou J, Yan X, Leng J, Li R, Zhang
J, Xing J, Chen C, Wang Z and Li W: Platycodon grandiflorum
saponins ameliorate cisplatin-induced acute nephrotoxicity through
the NF-κB-mediated inflammation and PI3K/Akt/apoptosis signaling
pathways. Nutrients. 10(pii): E13282018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lin JH, Li H, Yasumura D, Cohen HR, Zhang
C, Panning B, Shokat KM, Lavail MM and Walter P: IRE1 signaling
affects cell fate during the unfolded protein response. Science.
318:944–949. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Soni KK, Kim HK, Choi BR, Karna KK, You
JH, Cha JS, Shin YS, Lee SW, Kim CY and Park JK: Dose-dependent
effects of cisplatin on the severity of testicular injury in
Sprague Dawley rats: Reactive oxygen species and endoplasmic
reticulum stress. Drug Des Devel Ther. 10:3959–3968. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Singh MP, Chauhan AK and Kang SC: Morin
hydrate ameliorates cisplatin-induced ER stress, inflammation and
autophagy in HEK-293 cells and mice kidney via PARP-1 regulation.
Int Immunopharmacol. 56:156–167. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Liu XR, Li T, Cao L, Yu YY, Chen LL, Fan
XH, Yang BB and Tan XQ: Dexmedetomidine attenuates H2O2-induced
neonatal rat cardiomyocytes apoptosis through mitochondria- and
ER-medicated oxidative stress pathways. Mol Med Report.
17:7258–7264. 2018.
|
|
43
|
Fruman DA, Meyers RE and Cantley LC:
Phosphoinositide kinases. Annu Rev Biochem. 67:481–507. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhou MF, Feng ZP, Ou YC, Peng JJ, Li K,
Gong HD, Qiu BH, Liu YW, Wang YJ and Qi ST: Endoplasmic reticulum
stress induces apoptosis of arginine vasopressin neurons in central
diabetes insipidus via PI3K/Akt pathway. CNS Neurosci Ther.
25:562–574. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kong D, Zhuo L, Gao C, Shi S, Wang N,
Huang Z, Li W and Hao L: Erythropoietin protects against
cisplatin-induced nephrotoxicity by attenuating endoplasmic
reticulum stress-induced apoptosis. J Nephrol. 26:219–227. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Shen M, Wang S, Wen X, Han XR, Wang YJ,
Zhou XM, Zhang MH, Wu DM, Lu J and Zheng YL: Dexmedetomidine exerts
neuroprotective effect via the activation of the PI3K/Akt/mTOR
signaling pathway in rats with traumatic brain injury. Biomed
Pharmacother. 95:885–893. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Oyadomari S and Mori M: Roles of
CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ.
11:381–389. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wu CT, Sheu ML, Tsai KS, Weng TI, Chiang
CK and Liu SH: The role of endoplasmic reticulum stress-related
unfolded protein response in the radiocontrast medium-induced renal
tubular cell injury. Toxicol Sci. 114:295–301. 2010. View Article : Google Scholar : PubMed/NCBI
|















