Introduction
Ischemia/reperfusion (IR) in the brain causes
selective neuronal death in vulnerable brain structure, such as the
hippocampus; especially, among neurons in the hippocampus,
pyramidal neurons of cornu ammonis 1 (CA1) are the most vulnerable
after IR (1,2). IR-induced death of CA1 pyramidal
neurons occurs a few days after IR, and this phenomenon is called
‘delayed neuronal death (DND)’ (1). Several factors, such as age and sex,
can affect the degree of neuronal damage in the brain (3–5).
Among them, brain and body temperature have been hypothesized to be
a major factor in neuronal survival and death after IR injury
(6–9).
Elevated temperature is closely related to
neurological deficits, enlargement of ischemic lesions,
microvascular injury and increase in neuronal damage in various
rodent models of brain ischemia (10–14).
Hyperthermia increases the metabolic rate (15), which is harmful to the ischemic
brain as there is an imbalance between energy supply and demand
during ischemia (7,16).
Metabolic adaptation, which is a switch between
oxidative and glycolytic metabolism, occurs during ischemia and
after reperfusion in brains (17).
Such metabolic changes increase the production and accumulation of
lactates in ischemic tissues (18,19).
Lactate trafficking between cells is facilitated mainly by
proton-dependent symporters known as monocarboxylate transporters
(MCTs). Currently, 14 isoforms of the MCT family are known, but
only three MCTs, MCT1, MCT2 and MCT4, have been elucidated as
lactate transporters in the central nervous system (20). Among them, MCT4 is known to be
expressed in neurons and/or astrocytes (21–24).
Our previous studies reported the effects of
hyperthermia on hippocampal neuronal damage after IR in gerbils
(8,9). In addition, a chronological change in
MCT4 protein expression in the gerbil hippocampus after IR was
revealed under normothermia (21,24).
However, to the best of our knowledge, there have been no studies
on changes of MCT protein expression levels in the hippocampus
under hyperthermia before and/or during ischemic insults.
Therefore, the present study compared MCT4 and TNF-α
immunoreactivities in IR-induced CA1 under hyperthermic condition
to that under normothermic condition.
Materials and methods
Experimental animals
Male Mongolian gerbils (Meriones unguiculatus;
weight, 66–74 g; age, 6 months) were obtained from the Experimental
Animal Center, Kangwon National University, Chuncheon, Republic of
Korea. Gerbils were housed under conventional housing conditions at
23±3°C with relative humidity of 55±5%, under a 12-h light/dark
cycle. Free access to food and water was allowed. Experimental
procedures for this study were approved by the Institutional Animal
Care and Use Committee at Kangwon National University (approval no.
KW-200113-1).
The process of handling and caring animals conformed
to the guidelines of the current international laws and policies
(25,26). The numbers of gerbils used in this
study and the suffering caused by the procedures used in all
experiments was minimized. Body weight and behavior of all animals
were monitored every other day. Humane endpoints were determined
when the animals showed weight loss >20%, dehydration, or loss
of ability to ambulate. No animals showed signs of humane endpoints
intended to be euthanized immediately. The method of euthanasia was
chemical and physical method. After each animal was profoundly
anesthetized using 60 mg/kg of pentobarbital sodium, and cardiac
perfusion was conducted. Confirmation of death was evaluated with
vital signs including heart beats, pupillary response, and
respiratory pattern (lack of cardiac activity for 5 min through
cardiac palpation, unresponsiveness to light with dilated pupils
using light into the eyes of the animal and lack of spontaneously
breathing pattern with shallow and irregular breathing
pattern).
Experimental groups and induction of
IR
Experimental animals (total n=109) were divided into
seven groups: i) Normal animals (normal group; n=5); ii) control
animals with normothermia (NT/control group; n=5); iii)
sham-operated animals with normothermia (NT/sham group; n=5); iv)
IR-operated animals with normothermia (NT/IR group; n=7 at each
time point); v) control animals with hyperthermia (HT/control
group; n=5); vi) sham-operated animals with hyperthermia (HT/sham
group; n=5); and vii) IR-operated animals with hyperthermia (HT/IR
group; n=7 at each time point). Since there were no significant
differences between the normal group and NT/control group in the
present study, data of the normal group are not shown.
The gerbils in each group were anesthetized with a
mixture of 2.5% isoflurane (Baxtor) in 30% oxygen and 70% nitrous
oxide using inhalation anesthesia equipment (Harvard Apparatus),
with a modification of methods of previous studies (27–29);
inhalation anesthetics for the gerbils maintained precise control
over the dosage of the anesthetic agent and enabled them to rapidly
recover (30). Hyperthermia was
induced by exposing the gerbils to a heating pad (homeothermic
monitoring system, Harvard Apparatus) connected to a rectal
thermistor under anesthesia until their rectal temperature was
elevated to 39.5±0.2°C, and the animals were maintained at this
temperature for 30 min before and during the surgery. For
normothermic condition, the rectal temperature was controlled at
37.0±0.5°C. For the induction of IR, as previously described
(9), both common carotid arteries
were isolated and occluded by using non-traumatic aneurysm clips
for 5 min. Then, the animals were maintained in thermal incubators
(temperature 23°C; humidity 60%) to maintain body temperature at a
normothermic level until they were euthanized (Fig. 1). The animals in the NT/sham and
HT/sham groups were exposed to the same surgical processes without
bilateral carotid artery occlusion.
Tissue processing for histology
The animals of the NT/IR and HT/IR groups were
sacrificed, and their brain sections containing the hippocampus
were prepared at designated times (3 h, 12 h, 1 day, 2 days, 3 days
and 5 days after IR). To reduce the number of animals, the brain
sections of the NT/control, NT/sham, HT/control and HT/sham groups
were obtained only at 5 days after sham operation. For preparation
of sections, as previously described (9,21),
the animals were perfused transcardially with solution of 4%
paraformaldehyde after checking vital signs to ensure that the
animals were profoundly anesthetized with 60 mg/kg pentobarbital
sodium (JW Pharmaceutical Co., Ltd.) (31). Their brains were removed and
post-fixed in the same fixative at room temperature for 6 h. The
brain tissues were cryoprotected with solution of 30% sucrose, and
the frozen tissues were serially sectioned into 30-µm coronal
sections.
Fluoro-Jade B (FJB) histofluorescence
staining
FJB is a fluorescent derivative used for
histological staining of degenerating neurons. In the present
study, FJB histofluorescence staining was conducted to examine
neuronal damage and death in the hippocampus after IR. As described
previously (9,21,32),
the tissues were immersed in a solution of 0.06% potassium
permanganate and stained with a solution of 0.0004% FJB (Histochem)
for 45 min at room temperature.
To analyze the numbers of damaged (dead) neurons,
the stained sections were observed with an epifluorescent
microscope at magnification, ×20 (Carl Zeiss AG). According to our
previously published method (21),
FJB-positive cells were examined with an epifluorescent microscope
(Carl Zeiss AG) equipped with 450–490 nm of blue excitation light
and a barrier filter. This microscope was equipped with a digital
camera connected to a PC monitor. Digital images of FJB-positive
cells in the same areas in the hippocampus were captured. These
captured cells were counted using an image analyzing software
(Optimas v6.5; CyberMetrics)
Immunohistochemistry
Immunohistochemical staining was performed to
examine changes in MCT4 expression in the hippocampus after IR
according to our previously published method (9,21,32).
Briefly, the sections (30-µm) were treated with a solution of 0.3%
hydrogen peroxide for 20 min at room temperature and followed by a
solution of 10% normal goat serum (cat. no. S-1000; Vector
Laboratories, Inc.) for 20 min at room temperature. These sections
were reacted with a 1:1,000 dilution of rabbit anti-TNF-α (cat. no.
ab66579, Abcam), a 1:200 dilution of rabbit anti-MCT4 (cat. no.
ab244385, Abcam) overnight at 4°C, and incubated in a 1:250
dilution of biotinylated goat anti-rabbit IgG (cat. no. BA-1000,
Vector Laboratories, Inc.) for 2 h at room temperature and treated
with a 1:200 dilution of streptavidin peroxidase complex (Vector
Laboratories, Inc.) for 1 h at room temperature. Finally, these
sections were visualized by reacting with a solution of
3,3′-diaminobenzidine tetrahydrochloride.
To quantitatively analyze TNF-α and MCT4
immunoreactivity, six sections with a 120-µm interval per animal
were selected. According to our previous method (9,32),
digital images of TNF-α and MCT4 immunoreactive structures were
captured in the hippocampus with Axio Imager 2 microscope (Carl
Zeiss AG) at magnification, ×20 equipped with a digital camera.
These images were calibrated into an array of 512 × 512 pixels. The
density of MCT4 immunoreactive structures was evaluated as relative
optical density (ROD) by using NIH ImageJ v1.59 software (National
Institutes of Health,). A ratio of ROD was calibrated as %, with
the NT/control group (100%).
Double immunofluorescence
staining
To examine cell types containing TNF-α and MCT4
immunoreactivity, double immunofluorescence staining was performed
according to our published protocol (33). In brief, rabbit anti-TNF-α (cat.
no. ab66579, dilution 1:500; Abcam), goat anti-Ionized calcium
binding adaptor molecule 1 (Iba1; cat. no. ab5076, dilution 1:400;
Abcam) for microglia and rabbit anti-MCT4 (cat. no. ab244385,
dilution 1:100; Abcam) and mouse anti-GFAP (cat. no. MAB360,
dilution 1:400; Abcam) for astrocytes were used. The sections were
incubated in the mixture of the antisera overnight at 4°C, and the
incubated sections were reacted in mixture of both donkey
anti-rabbit IgG, Alexa Fluor488 (cat. no. A32790, dilution 1:500;
Invitrogen; Thermo Fisher Scientific, Inc.) and goat anti-mouse
IgG, Alexa Fluor546 (cat. no. A-11030, dilution 1:500; Invitrogen;
Thermo Fisher Scientific, Inc.) and donkey anti-rabbit IgG, Alexa
Fluor546 (cat. no. A10040, dilution 1:500; Invitrogen; Thermo
Fisher Scientific, Inc.) and goat anti-mouse IgG, or Alexa Fluor488
(cat. no. A-11001, dilution 1:500; Invitrogen; Thermo Fisher
Scientific, Inc.) 2 h at room temperature. The immunoreaction was
examined under confocal microscope (LSM510 META NLO; Carl Zeiss AG)
at magnification, ×20 in the Korea Basic Science Institute
Chuncheon Center.
Statistical analysis
Data are expressed as the mean ± SEM. Differences in
the means among the groups were statistically analyzed by ANOVA
with a post hoc Bonferroni's multiple comparison tests with SPSS
v17.0 software (SPSS, Inc.). In order to elucidate ischemia-related
differences among experimental groups. In order to compare two
independent variables between normothermia and hyperthermia, and
their interaction, two-way ANOVA was used with the Bonferroni post
hoc. P<0.05 was considered to indicate a statically significant
difference.
Results
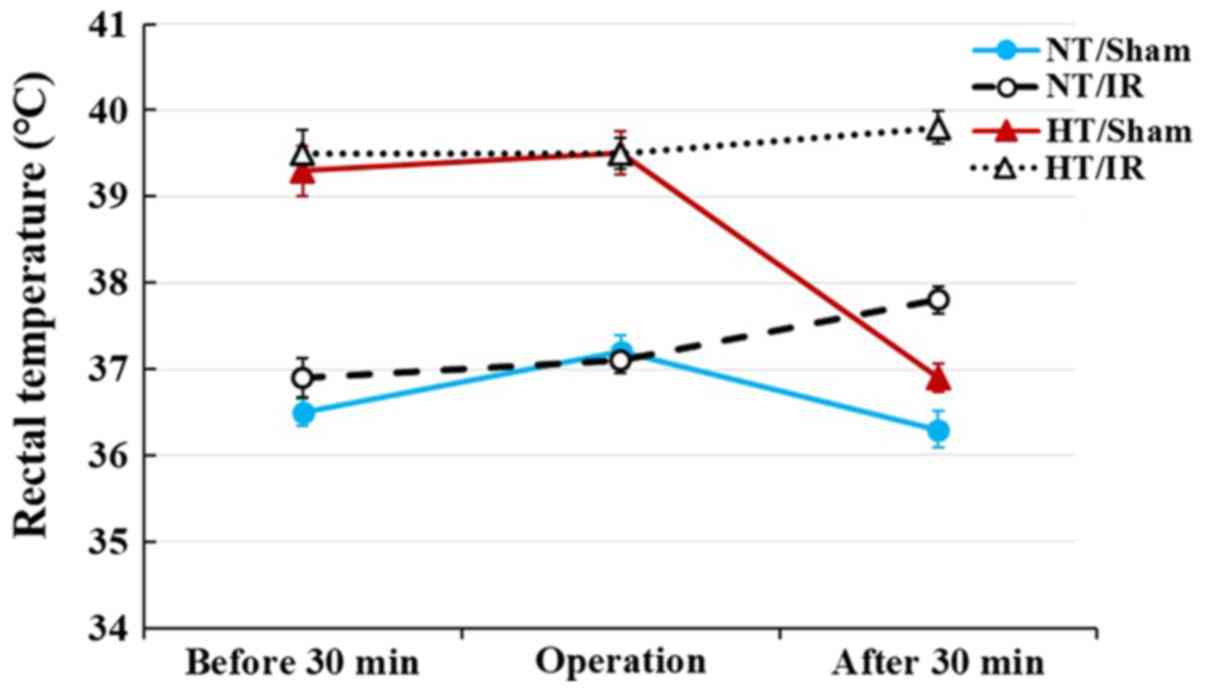
Change in body temperature
In the NT/sham group, rectal temperature was
36.5±0.2°C at 30 min before the sham operation, 37.2±0.2°C during
the sham operation and 36.3±0.2°C at 30 min after sham operation
(Fig. 1). In the NT/IR groups,
rectal temperature was 36.9±0.2°C at 30 min before the IR
operation, 37.1±0.1°C during the IR operation and 37.8±0.1°C at 30
min after the IR operation (Fig.
1).
In the HT/sham group, rectal temperature was
39.3±0.2°C at 30 min before the sham operation, maintained at
39.5±0.2°C during the sham operation and recovered to 36.9±0.1°C
(normal body temperature) at 30 min after the sham operation
(Fig. 1). In the HT/IR groups,
rectal temperature was 39.5±0.2°C at 30 min before the IR
operation, 39.5±0.1°C during the IR operation and 39.8±0.1°C at 30
mins after the IR operation (Fig.
1).
IR-induced neuronal death
To examine IR-induced neuronal damage/death in the
hippocampus, FJB (a marker for degenerating neurons)
histofluorescence staining was performed. In the NT/control and
NT/sham group, FJB-positive cells were not observed in CA1
(Fig. 2A, B and K). In the NT/IR
groups, FJB-positive cells were not observed 2 days after IR
(Fig. 2C and K). At 3 days after
IR, a few FJB-positive degenerating cells were observed in the
stratum pyramidale, in which pyramidal cells are located as
principal cells in the hippocampus (Fig. 2D and K). At 5 days after IR,
numerous FJB-positive degenerating cells were observed in the
stratum pyramidale, and the number of FJB-positive cells was
significantly increased (P<0.0001) compared with that at 3 days
after IR (Fig. 2E and K).
 | Figure 2.Fluoro-Jade B histofluorescence
staining in CA1 of the (A) NT/control, (B) NT/sham, NT/IR at (C) 2,
(D) 3 and (E) 5 days after IR, (F) HT/control, (G) HT/sham and
HT/IR groups at (H) 2, (I) 3 and (J) 5 days after IR. In the NT/IR
group, numerous FJB-positive cells were detected in the SP at 5
days after IR. However, in the HT/IR group, several FJB-positive
cells were found from 2 days after IR. Scale bar, 50 µm. (K)
Numbers of FJB-positive cells in CA1. *P<0.0001 vs. NT/sham or
HT/sham group; †P<0.0001, significantly different
from the corresponding NT group; #P<0.0001 vs.
pre-time point group. The bars indicate the means ± SEM. SO,
stratum oriens; SR, stratum radiatum; CA1, cornu ammonis 1; IR,
ischemia/reperfusion; NT, normothermia; HT, hyperthermia; SP,
stratum pyramidale. |
In the HT/control and HT/sham group, FJB-positive
cells were not detected in any layers of CA1 (Fig. 2F, G and K). In the HT/IR groups,
several FJB-positive cells began to be observed in the stratum
pyramidale at 2 days after IR (Fig. 2H
and K). At 3 days after IR, the number of FJB-positive
degenerating cells was increased to 112.1% compared to that at 2
days after IR (Fig. 2I and K),
and, at 5 days after IR, FJB-positive degenerating cells were
further increased (P=0.0035; 141.2% compared to that at 2 days
after IR) (Fig. 2J and K). In
addition, at this point in time, the number of FJB-positive
degenerating cells of the HT/IR group was significantly higher
(P=0.024, about 122.1% of the NT/IR group) than that in the NT/IR
group (Fig. 2J and K).
IR-induced change in TNF-α
immunoreactivity
In the NT/control and NT/sham groups, TNF-α
immunoreactivity was observed in pyramidal neurons (Fig. 3A and B). In the NT/IR group, TNF-α
immunoreactivity was significantly increased at 3 h (P<0.0001)
and 12 h (P<0.0001) after IR and peaked at 1 day (337.7% of the
NT/sham group) after IR compared with the NT/sham group (Fig. 3C, D, E and Q). Thereafter, TNF-α
immunoreactivity was gradually decreased at 2 days (P=0.0015) and 3
days (P<0.0001) after IR (Fig. 3F,
G and Q), and was barely observed at 5 days (p=0.0003) after IR
(Fig. 3H) compared with the
pre-time point group.
 | Figure 3.TNF-α immunohistochemistry in CA1 of
the (A) NT/control, (B) NT/sham, NT/IR at (C) 3 h, (D) 12 h, (E) 1
day, (F) 2 days, (G) 3 days and (H) 5 days after IR, (I)
HT/control, (J) HT/sham and HT/IR groups at (K) 3 h, (L) 12 h, (M)
1 day, (N) 2 days, (O) 3 days and (P) 5 days after IR. In the NT/IR
group, TNF-α immunoreactivity increased gradually in the SP, peaked
at 1 day post-IR and decreased thereafter. In the HT/control and
HT/sham groups, TNF-α immunoreactivity in the SP was much higher
(asterisks) than that in the NT/control and NT/sham groups. In the
HT/IR group, TNF-α immunoreactivity increased significantly from 3
h post-IR, peaked at 12 h post-IR, decreased from 1 day post-IR and
was barely observed at 3 and 5 days post-IR. Note that TNF-α
immunoreactivity was observed in non-pyramidal cells (arrows) in SO
and SR at 5 days after IR in both the NT/IR and HT/IR groups. Scale
bar, 50 µm. (Q) ROD of TNF-α immunoreactivity as % in CA1.
*P<0.0001, significantly different from the NT/sham or HT/sham
group; †P<0.0001, significantly different from the
corresponding NT group; #P<0.0001 vs. pre-time point
group. (R) Double immunofluorescence staining for TNF-α (green, a
and d), Iba-1 (red, b and e), and merged (c and f) images at 5 days
post-IR in the NT/IR (upper panels) and the HT/IR (lower panels)
groups. TNF-α immunoreactivity is merged with Iba-1 immunoreactive
microglia (arrows). Scale bar, 50 µm. CA1, cornu ammonis 1; IR,
ischemia/reperfusion; NT, normothermia; HT, hyperthermia; TNF,
Tumor necrosis factor; SP, stratum pyramidale; SO, strata oriens;
SR, stratum radiatum; ROD, Relative optical density; Iba, Ionized
calcium binding adaptor molecule 1. |
TNF-α immunoreactivity in the HT/control and HT/sham
groups was observed in pyramidal neurons, and TNF-α
immunoreactivity was significantly higher (P<0.0001) than that
in the NT/control and NT/sham group (255.6 and 265.7%,
respectively) (Fig. 3I, J and Q).
In the HT/IR group, TNF-α immunoreactivity was significantly
increased from 3 h (P<0.0001) (128.3% of the HT/sham group)
compared with the HT/sham group after IR and highest at 12 h
(P<0.0001) (221.4% of the HT/sham group) after IR (Fig. 3K, L and Q). Thereafter, TNF-α
immunoreactivity was significantly decreased at 1 day (P<0.0001)
(124.3% of the HT/sham group) after IR compared to that at 12 h
after ischemia and was barely observed at 2, 3 and 5 days after IR,
showing that TNF-α immunoreactivity at 5 days after IR was
increased in non-pyramidal cells of the strata oriens and radiatum
(Fig. 3M-Q).
Double immunofluorescence staining results showed
that, in the NT/IR and HT/IR groups, TNF-α immunoreactive
non-pyramidal cells at 5 days after IR were merged with Iba-1
immunoreactive microglia (Fig.
3R).
IR-induced change in MCT4
immunoreactivity
MCT4 immunoreactivity in the NT/control and NT/sham
groups was primarily observed in pyramidal neurons in CA1 (Fig. 4A and B). In the NT/IR group, MCT4
immunoreactivity in CA1 pyramidal neurons was significantly reduced
at 3 h (P<0.0001) and 12 h (P<0.0001) after IR, compared to
that in the NT/sham group (Fig. 4C, D
and Q). MCT4 immunoreactivity in the CA1 pyramidal neurons was
significantly increased at 1 day (P<0.0001) and 2 days
(P<0.0001) after IR compared to that in the NT/sham group
(Fig. 4E, F and Q). At 3 and 5
days after IR, MCT4 immunoreactivity in the CA1 pyramidal neurons
was barely observed compared with the NT/sham group, whereas MCT4
immunoreactivity was increased in non-pyramidal cells of the strata
oriens and radiatum at 5 days after IR (Fig. 4G and H).
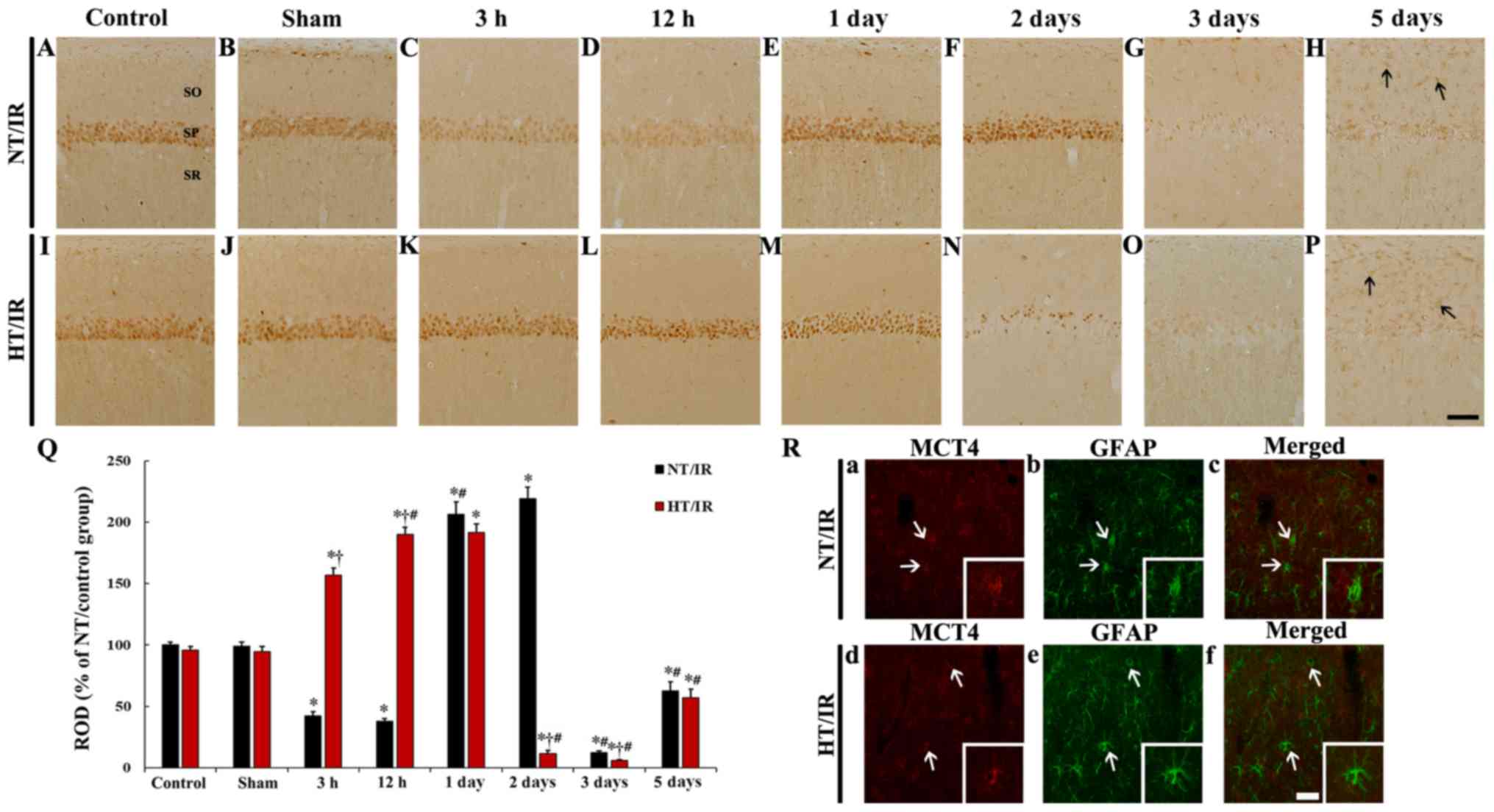 | Figure 4.MCT4 immunohistochemistry in CA1 of
the (A) NT/control, (B) NT/sham, NT/IR at (C) 3 h, (D) 12 h, (E) 1
day, (F) 2 days, (G) 3 days and (H) 5 days after IR, (I)
HT/control, (J) HT/sham and HT/IR groups at (K) 3 h, (L) 12 h, (M)
1 day, (N) 2 days, (O) 3 days and (P) 5 days after IR. In the NT/IR
group, MCT4 immunoreactivity in the SP was markedly reduced at 3
and 12 h post-IR, significantly increased at 1 and 2 days post-IR,
and barely observed at 3 and 5 days post-IR. In the HT/IR group,
MCT4 immunoreactivity in the SP was significantly increased from 3
h post-IR, increased until 1 day post-IR, dramatically decreased at
2 days post-IR, and barely observed at 3 and 5 days post-IR. Note
that MCT4 immunoreactivity was observed in non-pyramidal cells
(arrows) in SO and SR at 5 days post-IR in both NT/IR and HT/IR
groups. Scale bar, 50 µm. (Q) ROD of MCT4 immunoreactivity as % in
CA1. *P<0.0001, significantly different from the NT/sham or
HT/sham group; †P<0.0001, significantly different
from the corresponding NT group; #P<0.0001 vs.
pre-time point group. (R) Double immunofluorescence staining for
MCT4 (red, a and d), GFAP (green, b and e), and merged (c and f)
images at 5 days post-IR in the NT/IR (upper panels) and the HT/IR
(lower panels) groups. MCT4 immunoreactivity is merged with GFAP
immunoreactive astrocytes (arrows). Scale bar, 50 µm. CA1, cornu
ammonis 1; IR, ischemia/reperfusion; NT, normothermia; HT,
hyperthermia; MCT4, Monocarboxylate transporter 4; SP, stratum
pyramidale; SO, strata oriens; SR, stratum radiatum; ROD, Relative
optical density. |
In the HT/control and HT/sham group, no significant
difference in MCT4 immunoreactivity in CA1 pyramidal neurons was
shown, compared to that in the NT/control and NT/sham group
(Fig. 4I, J and Q). At 3 h after
IR, a significant increase (P<0.0001) (166.1% of the HT/sham
group) in MCT4 immunoreactivity was observed in the HT/IR group
when compared with that in the HT/sham group, and the corresponding
NT group (Fig. 4K and Q). MCT4
immunoreactivity in the CA1 pyramidal neurons in the HT/IR group
was increased until 1 day after IR (Fig. 4L, M and Q). At 2 days after IR,
only a few CA1 pyramidal neurons showed MCT4 immunoreactivity, and
there was a significant decrease compared with the corresponding NT
group (Fig. 4N). MCT4
immunoreactivity in the CA1 pyramidal neurons was barely observed
at 3 days after IR and was significantly decreased compared with
the corresponding NT group (Fig. 4O
and Q). At 5 days after IR MCT4 immunoreactivity in CA1
pyramidal neurons was not observed but was expressed in
non-pyramidal cells (Fig. 4P).
It was also found that MCT immunoreactivity in
non-pyramidal cells at 5 days after IR in the NT/IR and HT/IR
groups were identified as GFAP immunoreactive astrocytes (Fig. 4R).
Discussion
In the present study, body temperature of the NT/IR
and HT/IR group was slightly increased 30 min after IR, while in
the NT/sham and HT/sham group, body temperature was maintained
after sham operation. Similar to the present results, it has been
previously reported that cerebral ischemia raises body temperature
(38.5–39.5°C) until ~1 h after IR, and then gradually recovers to
normal body temperature in gerbils (34,35).
Therefore, the use of drugs to reduce body temperature immediately
after ischemia can effectively protect neurons (34,35).
However, it has been reported that hyperthermia during the acute
phase of cerebral ischemia within 24 h after IR exacerbates
ischemic brain damage and worsens outcomes in patients with
hyperthermia in acute ischemic stroke (36–38).
Our previous studies revealed that hyperthermia before and during
IR increased the extent and severity of IR-induced neuronal death
and IR-induced glial activation in the gerbil hippocampus (8,9). In
the present study, it was found that IR-induced death of CA1
pyramidal neurons (principal neurons) was markedly augmented and
occurred rapidly under hyperthermia when compared with that under
normothermia. This finding was coincident with the result of our
previous study (9).
There is extensive research on deleterious factors
of hyperthermia in cerebral ischemic conditions, showing that
induced hyperthermia can cause increases in oxidative stress and
DNA fragmentation finally exacerbating neuronal damage in the
hippocampus (39,40). In addition, it has been reported
that patients with hyperthermia have significantly higher plasma
levels of TNF-α and increased infarct volume compared with the
normothermic group, showing that there are significant correlations
between TNF-α level and infarct volume, and between body
temperature and infarct volume (37). Furthermore, brief hypoxia alone
significantly increases brain TNF-α expression, and hyperthermia at
39°C following hypoxia causes a more significant increase in TNF-α
expression in a rat model of perinatal inflammation (41). Similar to the results of the
previous studies, in the present study, TNF-α immunoreactivity in
CA1 pyramidal neurons located in the stratum pyramidale of the
NT/IR group was gradually increased from 3 h, peaked at 1 day and
significantly decreased at 3 days after IR. Notably, hyperthermia
without IR (HT/control and HT/sham group) significantly increased
TNF-α immunoreactivity in CA1 pyramidal neurons, and in the HT/IR
group, TNF-α immunoreactivity in the CA1 pyramidal neurons was much
higher, rapidly increased and peaked at 12 h after IR compared with
the NT/IR group. These results indicated that hyperthermia and IR
under hyperthermia increases TNF-α expression (inflammatory
response) in CA1 pyramidal neurons, showing that a significant
increase in TNF-α expression under hyperthermia may be closely
related to more severe ischemic damage to CA1 pyramidal
neurons.
It has been suggested that MCT4 expression is
closely related to TNF-α expression (42), indicating that the overexpression
of MCT4 accelerates glycolysis, increases lactate (end product of
glycolysis) and promotes pro-inflammatory cytokines in
arsenite-induced liver carcinogenesis. Based on this report, the
present study examined IR-induced changes in MCT4 immunoreactivity
and found that the IR-induced changes in MCT4 immunoreactivity in
CA1 pyramidal neurons under normothermia were significantly
different from those under hyperthermia.
It has been reported that MCT4 expression in the
brain is altered after ischemic insults. For instance, MCT4
expression was found to be increased in the ipsilateral hemisphere
at 1 h post-ischemia and then decreased at 24 h post-ischemia in a
mouse model of transient middle cerebral artery occlusion (MCAO),
which evoked focal brain ischemia (43). Another study showed that mRNA and
protein expression levels of MCT4 and lactate levels were
significantly increased in the rat brain after transient and
permanent MCAO (44). In our
previous and present studies, it was demonstrated that in CA1
region after IR under normothermia, MCT4 protein expression was
decreased early after IR and markedly increased 1 and 2 days after
IR (9). It has been reported that
MCT4 is a high-capacity lactate transporter in cells exhibiting
glycolytic activity and that MCT4 plays a crucial role in lactate
release from glycolytic cells (45,46).
In addition, in a gerbil model of transient forebrain ischemia, the
level of hippocampal lactate was significantly increased at 15 min
after IR, distinctively decreased from 15 min to 6 h and similar to
that in the control group at 2 days after IR (47). Collectively, it was indicated that
IR-induced a decrease in MCT4 expression in CA1 pyramidal neurons
soon after IR may be related to elevated usage of MCT4 to reduce
the IR-induced increase in lactate levels. However, increases in
MCT4 expression in CA1 pyramidal neurons at 1 and 2 days after IR
may be associated with compensatory mechanisms (24).
In the present study on MCT4 expression in CA1
neurons of the HT/IR group, MCT4 immunoreactivity was notably
increased from 3 h to 1 day after IR and markedly decreased at 2
days after IR, suggesting that the alteration pattern of MCT4
immunoreactivity was different from the NT/IR group. However, it is
difficult to hypothesis the reasons as to why MCT4 protein
expression in pyramidal neurons of CA1 induced by IR under
hyperthermia was significantly increased soon after IR; this
finding was different from that in pyramidal neurons of CA1 induced
by IR under normothermia. Lactate produced by glia at post-ischemia
is transported into neurons via MCTs for aerobic use, but
inhibition of lactate transport into neurons via acting on MCTs
(α-cyano-4-hydroxycinnamate, an MCT inhibitor) exacerbates neuronal
damage in the rat hippocampus after IR induced by cardiac arrest
(48). In addition, the alteration
of MCT4 expression harms cellular survival after hypoxic exposure
(23). Based on these studies and
the present findings, it was suggested that a marked increase in
MCT4 immunoreactivity in CA1 pyramidal neurons soon after IR under
hyperthermia may be associated with increased glycolytic activities
in the CA1 pyramidal neurons, and that the increase in MCT4
immunoreactivity at early points in time after IR may be related to
the marked reduction in MCT4 immunoreactivity, as well as the
advance of IR-induced death of CA1 pyramidal neurons from 2 days
after IR.
However, there is a limitation to the present study.
For instance, the present study examined IR-induced changes in
TNF-α and MCT4 immunoreactivity in the CA1 pyramidal neurons, but
did not investigate this using accurate quantitative experiments.
Therefore, to clearly elucidate the roles and the expression
changes in TNF-α and MCT4 following IR, quantitative analyses, such
as western blot analysis or reverse transcription-quantitative PCR,
should be performed in future studies.
In conclusion, IR under hyperthermia exacerbated the
death of CA1 pyramidal neurons, suggesting that TNF-α and MCT4
immunoreactivity in the CA1 pyramidal neurons was high soon after
IR under hyperthermia compared with that under normothermia. These
results indicated that acceleration of neuronal death following IR
under hyperthermia before and during IR may depend on the pattern
of TNF-α and MCT4 protein expression after IR under
hyperthermia.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Research
Foundation of Korea grant funded by the Korea government (MSIP;
Ministry of Science, ICT & Future Planning; grant no.
NRF-2017R1C1B5075773) and by the Basic Science Research Program
through the National Research Foundation of Korea funded by the
Ministry of Education (grant no. NRF-2018R1D1A1B07049453).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
YEP, TKL and BK performed the experiments and
measurements. JCL, JHC and JHP analyzed and interpreted the data.
TGO, JHA, MHW and CHL made substantial contributions to conception
and design of the research, and were involved in drafting, revising
the manuscript and interpreting all data. All authors read and
approved the manuscript, and agree to be accountable for all
aspects of the research in ensuring that the accuracy or integrity
of any part of the work are appropriately investigated and
resolved.
Ethics approval and consent to
participate
Experimental procedures for this study were approved
by the Institutional Animal Care and Use Committee at Kangwon
National University (approval no. KW-200113-1). The process of
handling and caring animals conformed to the guidelines of the
current international laws and policies (NIH Guide for the Care and
Use of Laboratory Animals, The National Academies Press, 8th
edition, 2011 and AVMA Guidelines for the Euthanasia of Animals,
2013 edition).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kirino T: Delayed neuronal death in the
gerbil hippocampus following ischemia. Brain Res. 239:57–69. 1982.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Onken M, Berger S and Kristian T: Simple
model of forebrain ischemia in mouse. J Neurosci Methods.
204:254–261. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lee CH, Yoo KY, Choi JH, Park OK, Hwang
IK, Kim SK, Kang IJ, Kim YM and Won MH: Neuronal damage is much
delayed and microgliosis is more severe in the aged hippocampus
induced by transient cerebral ischemia compared to the adult
hippocampus. J Neurol Sci. 294:1–6. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liu F and McCullough LD: Interactions
between age, sex, and hormones in experimental ischemic stroke.
Neurochem Int. 61:1255–1265. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Manwani B, Liu F, Scranton V, Hammond MD,
Sansing LH and McCullough LD: Differential effects of aging and sex
on stroke induced inflammation across the lifespan. Exp Neurol.
249:120–131. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Busto R, Dietrich WD, Globus MY and
Ginsberg MD: The importance of brain temperature in cerebral
ischemic injury. Stroke. 20:1113–1114. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Corbett D and Thornhill J: Temperature
modulation (hypothermic and hyperthermic conditions) and its
influence on histological and behavioral outcomes following
cerebral ischemia. Brain Pathol. 10:145–152. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kim DW, Cho JH, Cho GS, Kim IH, Park JH,
Ahn JH, Chen BH, Shin BN, Tae HJ, Hong S, et al: Hyperthermic
preconditioning severely accelerates neuronal damage in the gerbil
ischemic hippocampal dentate gyrus via decreasing SODs expressions.
J Neurol Sci. 358:266–275. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kim MJ, Cho JH, Cho JH, Park JH, Ahn JH,
Tae HJ, Cho GS, Yan BC, Hwang IK, Lee CH, et al: Impact of
hyperthermia before and during ischemia-reperfusion on neuronal
damage and gliosis in the gerbil hippocampus induced by transient
cerebral ischemia. J Neurol Sci. 348:101–110. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Barber PA, Hoyte L, Colbourne F and Buchan
AM: Temperature-regulated model of focal ischemia in the mouse: A
study with histopathological and behavioral outcomes. Stroke.
35:1720–1725. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Campos F, Blanco M, Barral D, Agulla J,
Ramos-Cabrer P and Castillo J: Influence of temperature on ischemic
brain: Basic and clinical principles. Neurochem Int. 60:495–505.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kim Y, Busto R, Dietrich WD, Kraydieh S
and Ginsberg MD: Delayed postischemic hyperthermia in awake rats
worsens the histopathological outcome of transient focal cerebral
ischemia. Stroke. 27:2274–2280, discussion 2281. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Satoh K, Niwa M, Binh NH, Nakashima M,
Kobayashi K, Takamatsu M and Hara A: Increase of galectin-3
expression in microglia by hyperthermia in delayed neuronal death
of hippocampal CA1 following transient forebrain ischemia. Neurosci
Lett. 504:199–203. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang CX, Stroink A, Casto JM and Kattner
K: Hyperthermia exacerbates ischaemic brain injury. Int J Stroke.
4:274–284. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Saxton C: Effects of severe heat stress on
respiration and metabolic rate in resting man. Aviat Space Environ
Med. 52:281–286. 1981.PubMed/NCBI
|
|
16
|
Wood SC and Gonzales R: Hypothermia in
hypoxic animals: Mechanisms, mediators, and functional
significance. Comp Biochem Physiol B Biochem Mol Biol. 113:37–43.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Arnberg F, Grafström J, Lundberg J,
Nikkhou-Aski S, Little P, Damberg P, Mitsios N, Mulder J, Lu L,
Söderman M, et al: Imaging of a clinically relevant stroke model:
Glucose hypermetabolism revisited. Stroke. 46:835–842. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Karaszewski B, Wardlaw JM, Marshall I,
Cvoro V, Wartolowska K, Haga K, Armitage PA, Bastin ME and Dennis
MS: Early brain temperature elevation and anaerobic metabolism in
human acute ischaemic stroke. Brain. 132:955–964. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Schurr A: Lactate, glucose and energy
metabolism in the ischemic brain (Review). Int J Mol Med.
10:131–136. 2002.(Review). PubMed/NCBI
|
|
20
|
Pierre K and Pellerin L: Monocarboxylate
transporters in the central nervous system: Distribution,
regulation and function. J Neurochem. 94:1–14. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hong S, Ahn JY, Cho GS, Kim IH, Cho JH,
Ahn JH, Park JH, Won MH, Chen BH, Shin BN, et al: Monocarboxylate
transporter 4 plays a significant role in the neuroprotective
mechanism of ischemic preconditioning in transient cerebral
ischemia. Neural Regen Res. 10:1604–1611. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pellerin L, Bergersen LH, Halestrap AP and
Pierre K: Cellular and subcellular distribution of monocarboxylate
transporters in cultured brain cells and in the adult brain. J
Neurosci Res. 79:55–64. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rosafio K and Pellerin L: Oxygen tension
controls the expression of the monocarboxylate transporter MCT4 in
cultured mouse cortical astrocytes via a hypoxia-inducible
factor-1α-mediated transcriptional regulation. Glia. 62:477–490.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yoo DY, Park JH, Lee KY, Kwon HJ, Jung HY,
Kim JW, Kim DW, Choi JH, Moon SM, Yoon YS, et al: Temporal and
spatial changes of monocarboxylate transporter 4 expression in the
hippocampal CA1 region following transient forebrain ischemia in
the Mongolian gerbil. Mol Med Rep. 15:4225–4230. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Council NR: Guide for the Care and Use of
Laboratory Animals. National Academies Press (Washington, DC).
2010.
|
|
26
|
Leary SL, Underwood W, Anthony R, Cartner
S, Corey D, Grandin T, Greenacre C, Gwaltney-Brant S, McCrackin M
and Meyer R: AVMA guidelines for the euthanasia of animals. 1st
edition. American Veterinary Medical Association. Schaumburg. (IL).
2013.
|
|
27
|
Kaufman GD, Shinder ME and Perachio AA:
Correlation of Fos expression and circling asymmetry during gerbil
vestibular compensation. Brain Res. 817:246–255. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Du X, Wang D, Li Y, Huo X, Li C, Lu J,
Wang Y, Guo M and Chen Z: Newly breeding an inbred strain of
ischemia-prone Mongolian gerbils and its reproduction and genetic
characteristics. Exp Anim. 67:83–90. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhu X, Yan B, Tang C, Qiu G, Wu Y, Wang J
and Bo P: Neuroprotective effect of Paeoniae Radix Rubra on
hippocampal CA1 region of mice induced by transient focal cerebral
ischemia via anti-gliosis and anti-oxidant activity. Chin Herb Med.
11:86–91. 2019. View Article : Google Scholar
|
|
30
|
Diven K: Inhalation anesthetics in
rodents. Lab Anim (NY). 32:44–47. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Carpenter JW: Exotic Animal
Formulary-eBook. 4th edition. Elsevier Health Sciences (Amsterdam,
Netherlands). 2012.
|
|
32
|
Ahn JH, Kim DW, Park JH, Lee TK, Lee HA,
Won MH and Lee CH: Expression changes of CX3CL1 and CX3CR1 proteins
in the hippocampal CA1 field of the gerbil following transient
global cerebral ischemia. Int J Mol Med. 44:939–948.
2019.PubMed/NCBI
|
|
33
|
Park JH, Shin BN, Ahn JH, Cho JH, Kim IH,
Kim DW, Won MH, Hong S, Cho JH and Lee CH: Ischemia-Induced Changes
of PRAS40 and p-PRAS40 Immunoreactivities in the gerbil hippocampal
CA1 region after transient cerebral ischemia. Cell Mol Neurobiol.
36:821–828. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Won MH, Lee JC, Kim YH, Song DK, Suh HW,
Oh YS, Kim JH, Shin TK, Lee YJ and Wie MB: Postischemic hypothermia
induced by eugenol protects hippocampal neurons from global
ischemia in gerbils. Neurosci Lett. 254:101–104. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yang GE, Tae H-J, Lee T-K, Park YE, Cho
JH, Kim DW, Park JH, Ahn JH, Ryoo S, Kim Y-M, et al: Risperidone
treatment after transient ischemia induces hypothermia and provides
neuroprotection in the gerbil hippocampus by decreasing oxidative
stress. Int J Mol Sci. 20:46212019. View Article : Google Scholar
|
|
36
|
Zaremba J: Hyperthermia in ischemic
stroke. Med Sci Monit. 10:RA148–RA153. 2004.PubMed/NCBI
|
|
37
|
Leira R, Rodríguez-Yáñez M, Castellanos M,
Blanco M, Nombela F, Sobrino T, Lizasoain I, Dávalos A and Castillo
J: Hyperthermia is a surrogate marker of inflammation-mediated
cause of brain damage in acute ischaemic stroke. J Intern Med.
260:343–349. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
de Jonge JC, Wallet J and van der Worp HB:
Fever worsens outcomes in animal models of ischaemic stroke: A
systematic review and meta-analysis. Eur Stroke J. 4:29–38. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hara A, Niwa M, Iwai T, Yano H, Bunai Y,
Uematsu T, Yoshimi N and Mori H: Increase of fragmented DNA
transport in apical dendrites of gerbil CA1 pyramidal neurons
following transient forebrain ischemia by mild hypothermia.
Neurosci Lett. 280:73–77. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kil HY, Zhang J and Piantadosi CA: Brain
temperature alters hydroxyl radical production during cerebral
ischemia/reperfusion in rats. J Cereb Blood Flow Metab. 16:100–106.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang W, Dow KE and Flavin MP: Hyperthermia
amplifies brain cytokine and reactive oxygen species response in a
model of perinatal inflammation. Neurosci Lett. 445:233–235. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Luo F, Zou Z, Liu X, Ling M, Wang Q, Wang
Q, Lu L, Shi L, Liu Y, Liu Q, et al: Enhanced glycolysis, regulated
by HIF-1α via MCT-4, promotes inflammation in arsenite-induced
carcinogenesis. Carcinogenesis. 38:615–626. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Rosafio K, Castillo X, Hirt L and Pellerin
L: Cell-specific modulation of monocarboxylate transporter
expression contributes to the metabolic reprograming taking place
following cerebral ischemia. Neuroscience. 317:108–120. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Geng X, Sy CA, Kwiecien TD, Ji X, Peng C,
Rastogi R, Cai L, Du H, Brogan D, Singh S, et al: Reduced cerebral
monocarboxylate transporters and lactate levels by ethanol and
normobaric oxygen therapy in severe transient and permanent
ischemic stroke. Brain Res. 1603:65–75. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Dimmer KS, Friedrich B, Lang F, Deitmer JW
and Bröer S: The low-affinity monocarboxylate transporter MCT4 is
adapted to the export of lactate in highly glycolytic cells.
Biochem J. 350:219–227. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Halestrap AP and Meredith D: The SLC16
gene family-from monocarboxylate transporters (MCTs) to aromatic
amino acid transporters and beyond. Pflugers Arch. 447:619–628.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kim W, Kwon HJ, Jung HY, Yoo DY, Kim DW
and Hwang IK: Phosphoglycerate mutase 1 reduces neuronal damage in
the hippocampus following ischemia/reperfusion through the
facilitation of energy utilization. Neurochem Int. 133:1046312020.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Schurr A, Payne RS, Miller JJ, Tseng MT
and Rigor BM: Blockade of lactate transport exacerbates delayed
neuronal damage in a rat model of cerebral ischemia. Brain Res.
895:268–272. 2001. View Article : Google Scholar : PubMed/NCBI
|