Introduction
Stroke is the second leading cause of mortality and
the third leading cause of disability worldwide (1). In the past two decades, both the
morbidity and mortality rates of stroke have markedly increased in
China, which leads to substantial health care expenditures
(2). This is a result of limited
treatment options for stroke. Therefore, identifying novel drugs to
treat stroke and identifying its molecular mechanism is of great
significance.
Astrocytes are the most abundant glial cells in the
central nervous system. Astrocytes have been reported to serve
important roles in protecting neurons after stroke in multiple
ways, including antioxidant activities, ion homeostasis, energy
transfer and neurotransmitter transport (3). Furthermore, it has been reported that
impaired astrocytes in stroke exacerbate neuronal death (4). Therefore, alleviating astrocyte
injury is a promising direction to prevent excessive neuronal
damage following stroke.
Gap junctions are intercellular membrane channels
that directly connect the cytoplasm of adjacent cells (5). A gap junction channel contains two
hemichannels, each of which consists of six connexin (Cx) monomers
(5). Intercellular communication
via gap junctions serves a vital role in the development of
oxygen-glucose deprivation and re-oxygenation (OGD/R) injury, which
is a leading cause of stoke (6).
Cx43 is one of the most abundant gap junction proteins in
astrocytes (7). However, the
function of Cx43 in hypoxia/reperfusion (H/R) injury remains
debatable. For instance, Sun et al (8) identified that upregulation of Cx43
expression could inhibit oxidative damage and subsequently decrease
apoptotic cell death in cerebral ischemic injury. In addition, Cx43
knockout could increase infarct volumes in mice subjected to
transient middle cerebral artery occlusion (9). However, other studies have
demonstrated that Cx43 aggravates ischemia-induced cell damage by
spreading death signals to neighboring cells (10,11).
Therefore, the role of gap junctions and Cx43 in stroke development
remains unclear.
Propofol is widely used in anesthesia and intensive
care units for both short-term anesthesia and longer-term sedation
(12). Previous studies have found
that propofol exerts protective effects in models of cerebral H/R
injury (13–16). However, the underlying molecular
mechanisms of the neuroprotective effects of propofol against
cerebral ischemia remain unclear. The present study investigated
the effect of propofol on cerebral ischemic injury and its roles in
regulating gap junction function.
Materials and methods
Primary culture of astrocytes
The animal experiment protocol was reviewed and
approved by the Institutional Animal Care and Use Committee of Sun
Yat-sen University (Guangzhou, China). A total of 40 Sprague-Dawley
rats (age, 1–2 days; weight, 5–7 g; male) were obtained from Sun
Yat-sen University. The animals were housed at 22–24°C and 55±5%
relative humidity, with a 12-h light/dark cycle and free access to
food and water. For each primary culture, 6–10 rats were used to
isolate cortical astrocytes as previously described (17,18).
Specifically, the brain was excised from these rats and the
cerebral cortices were minced with forceps and harvested. The
cortical chunks were placed in Ca2+-free phosphate
buffered saline (PBS) containing 0.5% trypsin and 5 mM EDTA, and
digested at 37°C for 20 min. Subsequently, the dissociated cells
were centrifuged, resuspended in Dulbecco's modified Eagle's
medium/F12 (DMEM/F12) containing 10% fetal bovine serum (FBS), 100
U/ml penicillin and 100 µg/ml streptomycin (all cell culture
reagents were purchased from Gibco; Thermo Fisher Scientific,
Inc.), and seeded into poly-l-lysine-coated flasks at a density of
1×105 cells/cm2. The culture medium was
refreshed every 2 days. Upon reaching confluence (day 8–10 of
culture), the flasks were rotated at 200 rpm for 16 h at 37°C on an
orbital shaker to remove microglia and oligodendrocytes. The
astrocytes that attached to the bottom of the flasks were
trypsinized, resuspended and seeded into cell culture plates.
Immunocytochemistry
Astrocyte purity was assessed using
immunocytochemical staining of glial fibrillary acidic protein
(GFAP). Briefly, 1×105 astrocytes/cm2 were
seeded onto coverslips, grown for 48 h and fixed with 4% (w/v)
paraformaldehyde at room temperature for 30 min. The astrocytes
were then incubated in PBS containing 5% (w/v) sheep serum
(Sigma-Aldrich; Merck KGaA) and 0.3% (w/v) Triton X-100
(Sigma-Aldrich; Merck KGaA) at room temperature for 20 min. The
astrocytes were then transferred to 2% bovine serum albumin
(Sigma-Aldrich; Merck KGaA) containing anti-GFAP antibody (1:500;
cat. no. AB5804; Sigma-Aldrich; Merck KGaA) and incubated at 4°C
overnight. The next day, the coverslips were washed with PBS and
incubated with a Cy3-conjugated secondary antibody (1:500; cat. no.
A10520; Invitrogen; Thermo Fisher Scientific, Inc.) for 1 h at room
temperature. Nuclear counterstaining was performed using DAPI
(1:1,000; cat. no. D9542; Sigma-Aldrich; Merck KGaA) at room
temperature for 5 min. The coverslips were reviewed and scored by a
pathologist under a fluorescence microscope (magnification, ×40;
Olympus IX71; Olympus Corporation). Results are presented as the
number of cells marked with GFAP/the total number of nuclei
counterstained.
Drug treatment
Pure propofol (2,6-diisopropylphenol; Sigma-Aldrich;
Merck KGaA) was dissolved in intra-lipid (10% soybean oil, 2.25%
glycerol, 1.2% purified egg phosphatide; Clintec International
SARL) prior to use, at a final concentration of 10, 20 or 40 µM in
the culture medium. The control contained the same amount of
intra-lipid only. Carbenoxolone (CBX; Sigma-Aldrich; Merck KGaA)
was dissolved in PBS to give 50 mM stock solution. The final
concentration of CBX used was 50 µM, which was determined according
to a previous study (19). For the
experiments, propofol, intra-lipid and/or CBX were added to the
culture medium, and astrocytes were cultured for 1 h before OGD
exposure. The cultured primary astrocytes were randomly assigned to
different drug treatments.
OGD/R treatment of astrocytes
After the drug treatment, the astrocyte culture was
washed twice with glucose-free PBS, and glucose-free DMEM/F12 was
added. Subsequently, the culture plates were placed in an airtight
chamber pumped with a hypoxic gas mixture (95% N2, 5%
CO2; Advanced® Anoxomat; Advanced Instruments
Inc.) at 37°C and exposed to OGD for 6 h. Then, the medium was
changed to regular DMEM/F12 with 10% FBS and reoxygenated in a
water-saturated atmosphere of 5% CO2 and 95% air for 24
h in order to mimic a reperfusion period. Astrocytes that did not
undergo any treatment were defined as control cells. Astrocytes in
the sham cell group were exposed to 6 h OGD/24 h reoxygenation.
Astrocytes in the lipid group were pretreated with intra-lipid for
1 h before exposure to OGD/R.
Cell viability and lactate
dehydrogenase (LDH) secretion
Cell viability and LDH secretion assays were
performed to examine astrocyte damage as previously described
(20). Astrocyte viability was
assessed using a Cell Counting Kit-8 (CCK-8) assay, according to
the manufacturer's protocol (Dojindo Molecular Technologies, Inc.).
This kit was used to monitor the survival and proliferation of
cells. In the present study, astrocytes (1×104/well)
were plated in 96-well plates and incubated for 24 h to achieve
complete cell adhesion. After exposure to different experimental
conditions, as aforementioned, cell cultures were washed with
DMEM/F12 and 10 µl 10% CCK-8 solution was added into each well. The
cell were cultured for another 2 h. Cell viability was measured as
absorbance at an optical density of 450 nm using an Epoch™
microplate reader (BioTek Instruments, Inc.). Each assay was
performed in triplicate. Data were summarized as a percentage of
the control.
In addition, an LDH-Cytotoxicity assay kit (cat. no.
11644793001; Sigma-Aldrich; Merck KGaA) was used to assess LDH
activity, according to the manufacturer's protocol. The culture
media of astrocytes were collected after exposure to OGD/R and then
centrifuged at 100 × g for 5 min at room temperature to obtain the
supernatant. Subsequently, the astrocytes were lysed using 1%
Triton X-100 (Sigma Aldrich; Merck KGaA). Then, 50 µl culture
supernatant or cell lysates were incubated with LDH reaction
mixture at 37°C for 20 min. The intensity of color was measured at
490 nm using a spectrophotometer. Data were summarized as a
percentage of total LDH.
‘Parachute’ dye-coupling assay
Gap junction function was assessed, as previously
described (21,22). Briefly, cells were cultured to
confluence, and donor cells were labelled with 5 µmol/l
calcein-acetoxymethyl ester (Invitrogen; Thermo Fisher Scientific,
Inc.), which can permeate through gap junctions to adjacent cells.
After cells were incubated for 30 min at 37°C, the donor cells were
trypsinized and seeded onto the receiver cells at a 1:150
donor/receiver ratio. These two types of cells were allowed to
attach to form gap junctions at 37°C for 4 h, and were investigated
under a fluorescence microscope (magnification, ×40; Olympus IX71;
Olympus Corporation). The average number of receiver cells
containing dye per donor cell was evaluated and normalized to that
of control cultures as a measurement of gap junction intercellular
communication (GJIC) levels. The number of receiver cells for each
condition were counted by the same observer who was blinded to the
treatment conditions.
Flow cytometry apoptosis assay
Cell apoptosis was detected using an Alexa
Fluor® 488 Annexin V/propidium iodide (PI) staining kit
(Invitrogen; Thermo Fisher Scientific, Inc.). Cells from the
different experimental groups were harvested, washed in ice-cold
PBS, centrifuged at 100 × g for 5 min at 4°C, re-centrifuged using
the same conditions, and resuspended in 400 µl 1X Annexin-binding
buffer. Annexin V/PI staining solution was added to the mixture and
cells were incubated in the dark for 15 min. Afterwards, 400 µl 1X
Annexin-binding buffer was added to these mixtures and mixed
gently. The green fluorescence of Annexin V (FL1) vs. red
fluorescence of PI (FL2) was analyzed using a Beckman CytoFLEX flow
cytometer (Beckman Coulter, Inc.) at 530 and 575 nm, respectively.
At least 2×104 cells were examined per sample. Early
apoptotic cells bound to Annexin V, but not to PI, whereas late
apoptotic/necrotic cells displayed both types of binding, which was
analyzed using WinMDI 2.8 software (The Scripps Research
Institute).
Protein extraction and western
blotting
Astrocytes in different treatment groups were washed
three times with ice-cold PBS and lysed with RIPA lysis buffer
(Bio-Rad, Laboratories, Inc.) and protease inhibitors (1:200) on
ice for 15 min. Then, cells were sonicated on ice-cold water with
2–3 cycles of sonication for 15 and 45 sec of cooling time and then
centrifuged at 16,000 × g for 30 min at 4°C. Protein concentration
was determined by a bicinchoninic acid protein assay using a UV
spectrophotometer (DU 640; Beckman Coulter, Inc.). The protein
samples (25 µg) were separated by 10% SDS-PAGE, transferred onto
PVDF membranes and blocked with 5% fat-free milk for 1 h at room
temperature. Subsequently, the membranes were incubated at 4°C
overnight with an anti-Cx43 primary antibody (1:1,000; cat. no.
SAB4501175; Sigma-Aldrich; Merck KGaA). Following the primary
antibody incubation, the membrane was washed with TBS + 0.1%
Tween-20 (TBST) three times at room temperature and incubated for 1
h at room temperature with a secondary antibody (1:4,000; cat. no.
RABHRP1; Sigma-Aldrich; Merck KGaA). The membrane was then washed
with TBST five times at room temperature and visualized using an
ECL reagent (Beyotime Institute of Biotechnology) and an ECL
detection system (Beyotime Institute of Biotechnology). An
anti-GAPDH antibody (1:10,000; cat. no. G9545; Sigma-Aldrich; Merck
KGaA) was used to confirm equal loading. The protein bands were
analyzed using Quantity One 1-D v4.52 software and a GS-800
densitometer (Bio-Rad, Laboratories, Inc.).
Knockdown and overexpression of
Cx43
In order to knockdown Cx43 expression in astrocytes
(1×105 cells/cm2), a synthetic small
interfering RNA (siRNA; 100 pmol) from Guangzhou RiboBio Co., Ltd.
was transiently transfected into astrocytes for 48 h using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.). The target sequence of the siRNA was:
5′-GCTGGTTACTGGTGACAGA-3′. Non-targeting siRNA
(5′-UUCUCCGAACGUGUCACGU-3′; 100 pmol) was used as a negative
control. Overexpression of Cx43 was achieved by transient
transfection of 2 µg pcDNA3.1-Cx43 (Addgene, Inc.) using 3 µl
Lipofectamine 2000. An empty vector was transfected (2 µg) as the
negative control. Following 48 h of transfection at 37°C, the
knockdown and overexpression efficiency were assessed by western
blotting.
Statistical analysis
Data were statistically analyzed using SPSS 15.0
software (SPSS, Inc.) and are presented as the mean ± SEM. For each
treatment group, three repeats (n=3) were performed, and
experiments were performed according to our previous study
(23). Multiple comparisons among
groups were analyzed by one-way ANOVA with Tukey's post hoc test.
All analyses were plotted using SigmaPlot 13.0 (Systat Software,
Inc.). P<0.05 was considered to indicate a statistically
significant difference.
Results
OGD/R-induced cell injury is gap
junction-dependent
To study OGD/R-induced cell injury, cortical
astrocytes were isolated from rats and their purity was verified by
immunostaining. Immunofluorescence staining for GFAP was used to
label astrocytes, whereas DAPI was used to label cell nuclei
(Fig. 1A). The purity of isolated
astrocytes was >98% (data not shown). First, the gap junction
inhibitor CBX was used to study the function of gap junctions in
OGD/R-induced cell injury. Astrocytes were pretreated with 50 µM
CBX for 1 h, followed by hypoxia treatment for 6 h and reperfusion
for 24 h. The inhibitory effect of CBX on gap junction was verified
by a ‘parachute’ dye-coupling assay. The data demonstrated that 50
µM CBX pretreatment for 1 h could markedly decrease GJIC (Fig. 1B).
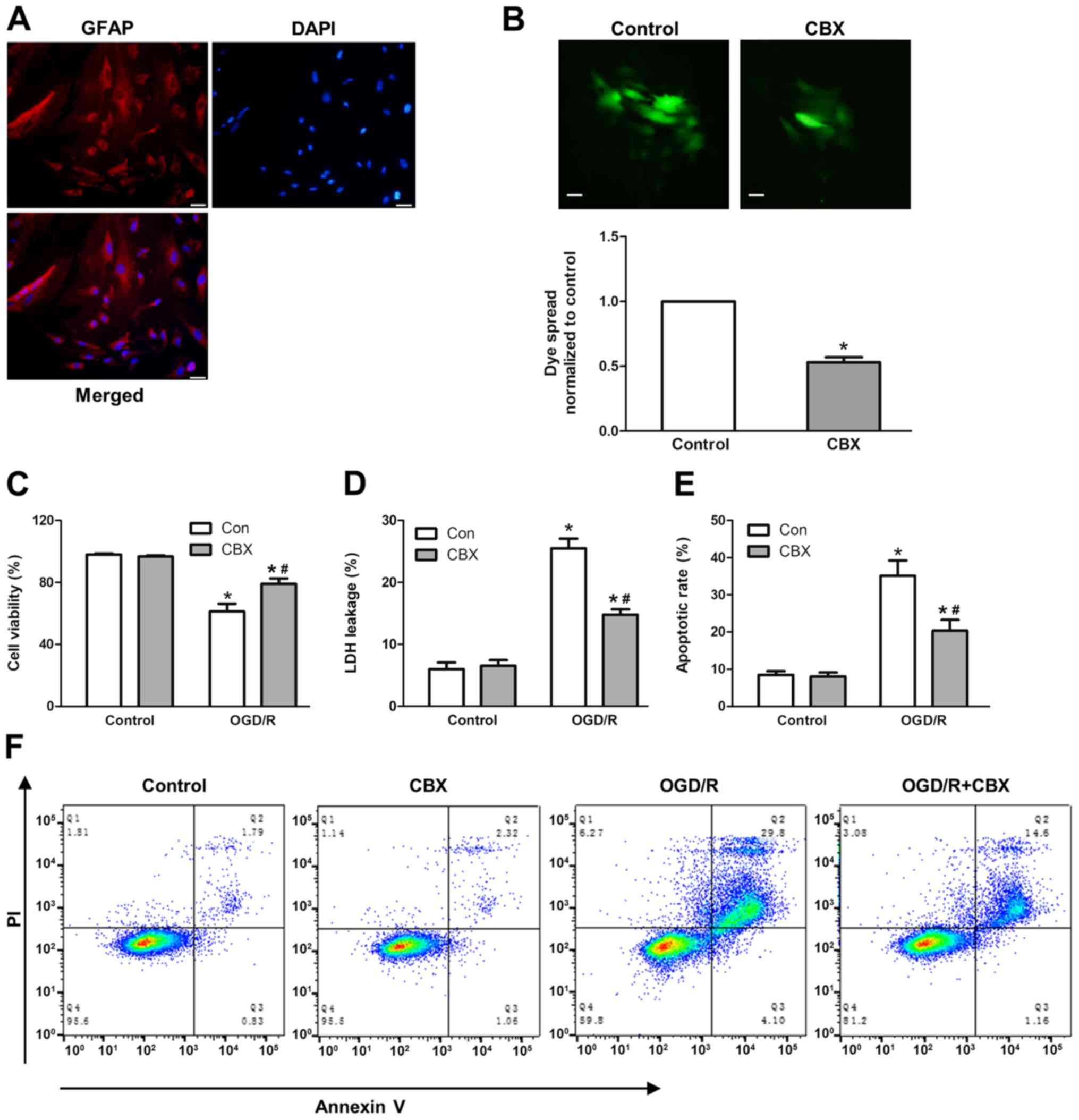 | Figure 1.Effect of gap junction function on
OGD/R-induced cell injury. (A) To confirm the establishment of a
primary culture of the astrocyte OGD injury model, primary cultured
astrocytes were labeled with anti-GFAP and counterstained with DAPI
nuclear stain. Scale bar, 20-µm. (B) Effect of GJIC inhibitor CBX
(50 µM) on dye coupling. Scale bar, 20-µm. (C) Astrocyte injury
following exposure to OGD/R in terms of cell viability, which was
assessed using a Cell Counting Kit-8 assay, and (D) LDH release
rate, assessed by the LDH-cytotoxicity assay. (E) Apoptotic rate
measured by flow cytometry. (F) Representative flow cytometry
images. The percentage of apoptotic cells includes cells that were
Annexin V+ and Annexin V+/PI+
after H/R. Values are presented as the mean ± SEM of three
independent experiments. *P<0.05 vs. control group without OGD/R
treatment; #P<0.05 vs. OGD/R-treated group without
CBX pretreatment. CBX, carbenoxolone; GFAP, glial fibrillary acidic
protein; GJIC, gap junction intercellular communication; H/R,
hypoxia/reoxygenation; LDH, lactate dehydrogenase; OGD/R,
oxygen-glucose deprivation and re-oxygenation; PI, propidium
iodide. |
Subsequently, to study the role of gap junctions in
OGD/R-induced cell injury, the cell viability, LDH leakage rate and
apoptotic rate were investigated using a WST-8 Cell Counting Kit,
LDH cytotoxicity assay kit and flow cytometry, respectively. It was
revealed that OGD/R treatment markedly decreased the viability of
astrocytes, and increased LDH leakage and cell apoptotic rates,
whereas CBX itself did not affect cell viability. In the
OGD/R-treated group that were pretreated with CBX, the cell
viability rate was increased by ~20%, the LDH leakage rate was
decreased by ~10%, and the apoptotic rate was decreased by ~15%
compared with those in the OGD/R-treated group without CBX
pretreatment (Fig. 1C-E).
Collectively, these data indicated that the gap junction promoted
OGD/R-induced cell injury in astrocytes.
Propofol protects astrocytes from
OGD/R-induced cell injury
To investigate the effect of propofol on
OGD/R-induced cell injury, astrocytes were pretreated with 10, 20
and 40 µM propofol for 1 h followed by OGD/R treatment. The cell
viability, LDH leakage rate and cell apoptotic rate were examined.
It was demonstrated that propofol (starting from 20 µM) increased
cell viability, and reduced LDH leakage and the cell apoptosis rate
compared with the sham group (OGD/R-treated without propofol
pretreatment) (Fig. 2A-D),
indicating that propofol protected astrocytes from OGD/R-induced
cell injury.
Propofol inhibits gap junction
function and decreases Cx43 expression in astrocytes
Since both propofol and gap junction inhibitor CBX
protected astrocytes from OGD/R-induced injury, to further study
whether propofol exerted protective effects by inhibiting gap
junction function, the present study tested the effect of propofol
on gap junction function by dye coupling between astrocytes.
Propofol (10, 20 and 40 µM) treatment for 1 h suppressed dye spread
in astrocytes in a dose-dependent manner when compared with control
group (Fig. 3A), indicating that
propofol inhibited gap junction function on astrocytes.
Gap junction function is mediated by the protein
levels of a variety of Cx proteins. Cx43 is one of the most
abundant Cx proteins in astrocytes (7). Furthermore, Cx43 has a
neuroprotective effect in cerebral ischemic injury (8). Therefore, it was investigated whether
propofol inhibited gap junction function via Cx43. Astrocytes were
treated with different concentrations of propofol (10, 20 and 40
µM) for 1 h and the protein levels of Cx43 were measured. Propofol
significantly decreased the protein levels of Cx43 in a
dose-dependent manner (Fig. 3B).
These data suggested that propofol inhibited gap junction function
by downregulating the expression levels of Cx43 in astrocytes.
Neuroprotective effect of propofol
against OGD/R is dependent on gap junctions and Cx43
To further validate the role of Cx43 in
propofol-mediated neuroprotective effects in injured astrocytes,
Cx43 was overexpressed or knocked down. As shown in Fig. 4A, the expression levels of Cx43
were upregulated in the Cx43 overexpression group (Cx43 OE),
whereas expression was markedly downregulated in the Cx43 knockdown
group (Cx43 KD) compared with the control group. First, the effect
of Cx43 OE and KD on gap junction function was investigated. As
expected, Cx43 OE significantly promoted gap junction function,
whereas Cx43 KD inhibited it, when measuring the dye coupling in
astrocytes (Fig. 4B).
Subsequently, the effects of propofol on Cx43 OE and
KD cells were studied. The cell viability of the Cx43 OE group was
significantly decreased after OGD/R injury compared with that in
the normal Cx43 group, whereas the apoptotic rate in the Cx43 OE
group was significantly increased. These data indicated that cells
with higher expression levels of Cx43 were more sensitive to OGD/R
injury. By contrast, the Cx43 KD group, with lower Cx43 expression,
exhibited higher cell viability rates and lower apoptotic rates
compared with the normal group after OGD/R injury, which further
demonstrated that Cx43 can aggravate OGD/R-induced cell injury
(Fig. 4C-E). Furthermore, propofol
(40 µM) diminished the OGD/R-induced cell injury in the normal and
Cx43 OE groups, but not in the Cx43 KD group. These data indicated
that Cx43 served an important role in propofol-suppressed OGD/R
cell injury. Overall, the data demonstrated that propofol exerted
neuroprotective effects against OGD/R cell injury by targeting Cx43
and inhibiting gap junction function.
Discussion
The present study revealed that GJIC serves an
important role in OGD/R-induced astrocyte injury. When GJIC was
inhibited by CBX, cell damage was alleviated. Furthermore,
propofol, at its clinically relevant concentrations, protected
astrocytes from OGD/R-induced cell death via the inhibition of GJIC
and Cx43 expression. This conclusion was supported by the following
findings: i) Propofol inhibited OGD/R-induced apoptosis in
astrocytes; ii) propofol markedly suppressed gap junction function
and expression levels of Cx43 in astrocytes (Fig. 3A); and iii) Cx43 OE increased
OGD/R-induced cell death and apoptosis, whereas propofol treatment
had the opposite effect. On the contrary, in Cx43 KD cells,
propofol exhibited only slight neuroprotective effects (Fig. 4).
Stroke is one of the leading causes of mortality and
disability worldwide (1). Current
treatments for stroke have limited benefits (2). In past decades, researchers have
mainly focused on neurons to treat hypoxic-ischemic injury
(24). Previously, astrocytes, the
most abundant cell type in the central nervous system, have been
reported to be another potential target for stroke therapy
(25). It has been reported that
impaired astrocytes in stroke could stimulate several important
apoptotic signaling pathways via gap junction communication, such
as cellular Ca2+ overload, to exacerbate neuronal death
(26). Accordingly, alleviating
astrocyte injury could be an important therapeutic direction to
protect neurons from stroke. In vitro OGD/R astrocyte models
have been widely used to mimic H/R injury that occurs in stroke.
The results of the present study demonstrated that OGD/R lead to
astrocyte injury. Compared with the control group, astrocytes
treated with OGD/R exhibited lower cell viability, a higher LDH
leakage rate and a higher apoptotic rate. This result was
consistent with previous studies (27,28),
which suggests that the OGD/R model used in the present study
successfully mimicked hypoxic-ischemic brain injury. Propofol is a
widely used intravenous sedative and anesthetic agent for both
short-term anesthesia and longer-term sedation (12). Propofol has been reported to exert
neuroprotective effects on cerebral ischemia-reperfusion injury
(20,29). In the present study, propofol, at
its clinically relevant concentrations (10–40 µM), dose-dependently
decreased OGD/R-induced cell injury via inhibition of gap junction
function. Gap junctions are important connections in direct
cell-to-cell communication that serve essential roles in the
pathogenesis of ischemic brain injury (30). Previous studies have demonstrated
that both beneficial and harmful substances may pass through gap
junctions to affect stroke in opposite ways (25,31–33).
The present results demonstrated that when GJIC was inhibited by
CBX, OGD/R-induced cell injury was decreased, including the
upregulation of cell viability, and downregulation of LDH leakage
and apoptotic rates. These results indicated that GJIC exacerbated
OGD/R-induced cell injury in primary cultured astrocytes, which is
consistent with a study by Orrenius et al (26), which reported that impaired
astrocytes in stroke could stimulate several important apoptotic
pathways via gap junction communication. Gap junction channels are
composed of Cx proteins, of which Cx43 is one of the most abundant
gap junction proteins in astrocytes (34). However, the role of Cx43 in stroke
remains controversial. It has been reported that Cx43 aggravates
ischemia-induced cell damage by spreading death signals to
neighboring cells (10,11,30).
The present study found that propofol decreased the protein
expression levels of Cx43, thereby protecting astrocytes from
OGD/R-induced cell injury.
In the future, it would be noteworthy to investigate
the mechanism by which propofol regulates the protein expression
levels of Cx43. One hypothesis is that propofol treatment may
trigger increased Cx43 degradation. Autophagy is a conserved
cellular process to degrade unnecessary proteins. It has been
reported that propofol treatment induces cellular protective
autophagy in COS-7 cells under hypoxic conditions (35), therefore it would be useful to
study whether propofol induces autophagy to degrade Cx43 in injured
astrocytes. Another possibility is that propofol treatment
suppresses the translation of Cx43. MicroRNAs are a class of
non-coding RNAs ~22 nucleotides in length that inhibit gene
expression; a series of studies have highlighted the roles of
propofol in regulating the amount of various microRNAs in
astrocytes (36–38). A recent study has found that
mirocRNA-206 targets the 3′untranslated regions of Cx43 in vascular
smooth muscle cells (39). So, it
would also be of note to investigate whether propofol downregulates
the expression levels of Cx43 via regulation of microRNAs in the
future.
In conclusion, the present study demonstrated that
propofol reduced OGD/R-induced cell injury in astrocytes by
inhibiting gap junction activity via downregulation of Cx43 protein
expression. Therefore, the present study provides novel insights
into the mechanism underlying the protective effect of propofol in
astrocytes following H/R injury.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YF, SZ and XW designed the experiments. YF, SZ, JW
and YZ conducted experiments and data analysis. YF and SZ wrote the
manuscript, and XW edited and approved the manuscript. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
The animal experiment protocol was reviewed and
approved by the Institutional Animal Care and Use Committee of Sun
Yat-sen University (Guangzhou, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Majid A: Neuroprotection in stroke: Past,
present, and future. ISRN Neurol. 2014:28962014. View Article : Google Scholar
|
|
2
|
Yang G, Wang Y, Zeng Y, Gao GF, Liang X,
Zhou M, Wan X, Yu S, Jiang Y, Naghavi M, et al: Rapid health
transition in China, 1990–2010: Findings from the global burden of
disease study 2010. Lancet. 381:1987–2015. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rossi DJ, Brady JD and Mohr C: Astrocyte
metabolism and signaling during brain ischemia. Nat Neurosci.
10:1377–1386. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nedergaard M and Dirnagl U: Role of glial
cells in cerebral ischemia. Glia. 50:281–286. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Harris AL: Connexin channel permeability
to cytoplasmic molecules. Prog Biophys Mol Biol. 94:120–143. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li X, Zhao H, Tan X, Kostrzewa RM, Du G,
Chen Y, Zhu J, Miao Z, Yu H, Kong J and Xu X: Inhibition of
connexin43 improves functional recovery after ischemic brain injury
in neonatal rats. Glia. 63:1553–1567. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Contreras JE, Sánchez HA, Véliz LP,
Bukauskas FF, Bennett MV and Sáez JC: Role of connexin-based gap
junction channels and hemichannels in ischemia-induced cell death
in nervous tissue. Brain Res Brain Res Rev. 47:290–303. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sun JB, Li Y, Cai YF, Huang Y, Liu S,
Yeung PK, Deng MZ, Sun GS, Zilundu PL, Hu QS, et al: Scutellarin
protects oxygen/glucose-deprived astrocytes and reduces focal
cerebral ischemic injury. Neural Regen Res. 13:1396–1407. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Siushansian R, Bechberger JF, Cechetto DF,
Hachinski VC and Naus CC: Connexin43 null mutation increases
infarct size after stroke. J Comp Neurol. 440:387–394. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Frantseva MV, Kokarovtseva L, Naus CG,
Carlen PL, MacFabe D and Perez Velazquez JL: Specific gap junctions
enhance the neuronal vulnerability to brain traumatic injury. J
Neurosci. 22:644–653. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wasielewski B, Jensen A, Roth-Härer A,
Dermietzel R and Meier C: Neuroglial activation and Cx43 expression
are reduced upon transplantation of human umbilical cord blood
cells after perinatal hypoxic-ischemic injury. Brain Res.
1487:39–53. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Deegan RJ: Propofol: A review of the
pharmacology and applications of an intravenous anesthetic agent.
Am J Med Sci. 304:45–49. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ergün R, Akdemir G, Sen S, Taşçi A and
Ergüngör F: Neuroprotective effects of propofol following global
cerebral ischemia in rats. Neurosurg Rev. 25:95–98. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bayona NA, Gelb AW, Jiang Z, Wilson JX,
Urquhart BL and Cechetto DF: Propofol neuroprotection in cerebral
ischemia and its effects on low-molecular-weight antioxidants and
skilled motor tasks. Anesthesiology. 100:1151–1159. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rossaint J, Rossaint R, Weis J, Fries M,
Rex S and Coburn M: Propofol: Neuroprotection in an in vitro model
of traumatic brain injury. Crit Care. 13:R612009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Huang Y, Zitta K, Bein B, Scholz J,
Steinfath M and Albrecht M: Effect of propofol on hypoxia
re-oxygenation induced neuronal cell damage in vitro*. Anaesthesia.
68:31–39. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yamamoto N, Sobue K, Miyachi T, Inagaki M,
Miura Y, Katsuya H and Asai K: Differential regulation of aquaporin
expression in astrocytes by protein kinase C. Brain Res Mol Brain
Res. 95:110–116. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schildge S, Bohrer C, Beck K and
Schachtrup C: Isolation and culture of mouse cortical astrocytes. J
Vis Exp. 500792013.PubMed/NCBI
|
|
19
|
Song MB, Yu XJ, Cui X, Zhu GX, Zhao G,
Chen JF and Huang L: Blockade of connexin 43 hemichannels reduces
neointima formation after vascular injury by inhibiting
proliferation and phenotypic modulation of smooth muscle cells. Exp
Biol Med (Maywood). 234:1192–1200. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sun B, Ou H, Ren F, Huan Y, Zhong T, Gao M
and Cai H: Propofol inhibited autophagy through
Ca(2+)/CaMKKβ/AMPK/mTOR pathway in OGD/R-induced neuron injury. Mol
Med. 24:582018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tao L and Harris AL: 2-aminoethoxydiphenyl
borate directly inhibits channels composed of connexin26 and/or
connexin32. Mol Pharmacol. 71:570–579. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Koreen IV, Elsayed WA, Liu YJ and Harris
AL: Tetracycline-regulated expression enables purification and
functional analysis of recombinant connexin channels from mammalian
cells. Biochem J. 383:111–119. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhao Y, Liu B, Wang Q, Yuan D, Yang Y,
Hong X, Wang X and Tao L: Propofol depresses the cytotoxicity of
X-ray irradiation through inhibition of gap junctions. Anesth
Analg. 112:1088–1095. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Busl KM and Greer DM: Hypoxic-ischemic
brain injury: Pathophysiology, neuropathology and mechanisms.
NeuroRehabilitation. 26:5–13. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu Z and Chopp M: Astrocytes, therapeutic
targets for neuroprotection and neurorestoration in ischemic
stroke. Prog Neurobiol. 144:103–120. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Orrenius S, Zhivotovsky B and Nicotera P:
Regulation of cell death: The calcium-apoptosis link. Nat Rev Mol
Cell Biol. 4:552–565. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li CY, Li X, Liu SF, Qu WS, Wang W and
Tian DS: Inhibition of mTOR pathway restrains astrocyte
proliferation, migration and production of inflammatory mediators
after oxygen-glucose deprivation and reoxygenation. Neurochem Int.
83-84:9–18. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang N, Zhang Y, Wu L, Wang Y, Cao Y, He
L, Li X and Zhao J: Puerarin protected the brain from cerebral
ischemia injury via astrocyte apoptosis inhibition.
Neuropharmacology. 79:282–289. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang H, Zheng S, Liu M, Jia C, Wang S,
Wang X, Xue S and Guo Y: The effect of propofol on mitochondrial
fission during oxygen-glucose deprivation and reperfusion injury in
rat hippocampal neurons. PLoS One. 11:e01650522016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Deng ZH, Liao J, Zhang JY, Liang C, Song
CH, Han M, Wang LH, Xue H, Zhang K, Zabeau L, et al: Inhibition of
the connexin 43 elevation may be involved in the neuroprotective
activity of leptin against brain ischemic injury. Cell Mol
Neurobiol. 34:871–879. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wallraff A, Köhling R, Heinemann U, Theis
M, Willecke K and Steinhäuser C: The impact of astrocytic gap
junctional coupling on potassium buffering in the hippocampus. J
Neurosci. 26:5438–5447. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nodin C, Nilsson M and Blomstrand F: Gap
junction blockage limits intercellular spreading of astrocytic
apoptosis induced by metabolic depression. J Neurochem.
94:1111–1123. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Perez Velazquez JL, Frantseva MV and Naus
CC: Gap junctions and neuronal injury: Protectants or executioners?
Neuroscientist. 9:5–9. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Rouach N, Avignone E, Meme W, Koulakoff A,
Venance L, Blomstrand F and Giaume C: Gap junctions and connexin
expression in the normal and pathological central nervous system.
Biol Cell. 94:457–475. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yoon JY, Baek CW, Kim EJ, Park BS, Yu SB,
Yoon JU and Kim EN: Propofol protects against
oxidative-stress-induced COS-7 cell apoptosis by inducing
autophagy. J Dent Anesth Pain Med. 17:37–46. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lu Y, Jian M, Xiong W and Han R: Effects
of propofol on miR-181a and Bcl-2 expression in glucose deprivation
cultured astrocytes. Zhonghua Yi Xue Za Zhi. 94:3020–3023. 2014.(In
Chinese). PubMed/NCBI
|
|
37
|
Sun WC, Liang ZD and Pei L:
Propofol-induced rno-miR-665 targets BCL2L1 and influences
apoptosis in rodent developing hippocampal astrocytes.
Neurotoxicology. 51:87–95. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sun W and Pei L: MicroRNA expression
profiling of propofol-treated developing rat hippocampal
astrocytes. DNA Cell Biol. 34:511–523. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li H, Xiang Y, Fan LJ, Zhang XY, Li JP, Yu
CX, Bao LY, Cao DS, Xing WB, Liao XH and Zhang TC: Myocardin
inhibited the gap protein connexin 43 via promoted miR-206 to
regulate vascular smooth muscle cell phenotypic switch. Gene.
616:22–30. 2017. View Article : Google Scholar : PubMed/NCBI
|

















