Introduction
Ginseng (Panax ginseng), which is documented
in Shennong's Classic of Materia Medica as having
intellect-enhancing effects, has been widely used in China for
millennia (1). Ginsenoside Rg1, a
primary ginseng components, has multiple neuroprotective effects
against Alzheimer's disease (AD), including the improvement of
memory impairment (2), inhibition
of neuronal apoptosis (3),
amelioration of oxidative stress (4) and attenuation of mitochondrial
dysfunction (5). The accumulation
of β-amyloid peptides (Aβ) is the most prominent pathological
feature of AD and significantly influences the pathogenesis of the
disease (6). The aggregation and
accumulation of Aβ in the brain may lead to neuroinflammatory
responses (7), neurofibrillary
tangle formation (8), synaptic
loss (9), oxidative stress
(10), neuronal apoptosis
(11), cholinergic dysfunction
(12) and tau phosphorylation
(13). It has been reported that
ginsenoside Rg1 has Aβ-scavenging effects, for example, ginsenoside
Rg1 inhibits the transcription and translation of β-amyloid
cleavage enzyme 1 (Bace1), a target gene of peroxisome
proliferator-activated receptor γ (PPARγ), by enhancing the binding
of PPARγ to the Bace1 promoter, thereby decreasing BACE1
activity, which ultimately attenuates Aβ production (14). Additionally, ginsenoside Rg1
upregulates PPARγ expression in the rat hippocampus AD model,
thereby increasing the expression of insulin-degrading enzyme
(Ide) (another PPARγ target gene) and promoting the
clearance of Aβ1-42 from the hippocampus (15). These studies suggest that PPARγ is
involved in the reduction of Aβ levels caused by ginsenoside Rg1;
however, the mechanisms by which ginsenoside Rg1 affects PPARγ
remain unclear.
Cyclin-dependent kinase 5 (CDK5), a cyclin-dependent
kinase, is a proline-directed serine/threonine kinase predominantly
activated in post-mitotic cells and has various activities
including cytoskeletal dynamics, signaling cascades, gene
expression, cell survival, neurodevelopment and brain function
(16–19). Phosphorylation is an important
post-translational modification of PPARγ. It has been demonstrated
that in the adipose tissue, CDK5 induces PPARγ phosphorylation at
serine 273 (Ser273), which is located in the hinge region between
the DNA- and the ligand-binding domains in vivo and in
vitro (20). Furthermore, our
previous study demonstrated that in rat primary hippocampal neurons
CDK5 regulates the expression of IDE and BACE1 by
mediating the phosphorylation of PPARγ, resulting in decreased Aβ
clearance and increased Aβ production (21). The present study aimed to
investigate whether ginsenoside Rg1 inhibits the phosphorylation of
PPARγ through the downregulation of the CDK5 pathway. The findings
of this study will deepen the understanding of the neuroprotective
properties of ginsenoside Rg1 and its potential use in the
treatment of AD.
Materials and methods
Reagents
Ginsenoside Rg1 (molecular formula: C42H72O14;
molecular weight: 801.01; HPLC purity: 98%) was purchased from
Baoji Herbest Bio-Tech Co., Ltd. Aβ1-42 and roscovitine
were purchased from Sigma Aldrich; Merck KGaA. Rabbit anti-rat IDE
(cat. no. ab133561), BACE1 (cat. no. ab10716) and amyloid precursor
protein (APP; cat. no. ab15272) polyclonal antibodies were
purchased from Abcam. Rabbit anti-rat p-PPARγ-Ser273 polyclonal
antibody (cat. no. bs-4888R) was purchased from BIOSS Antibodies.
Rabbit anti-rat CDK5 (cat. no. WL01673), PPARγ (cat. no. WL0269)
and Aβ1-42 (cat. no. WL01427) polyclonal antibodies,
anti-β-actin antibody (cat. no. WL01845), goat anti-rabbit
secondary horseradish peroxidase-conjugated antibody (cat. no.
WLA023), TUNEL assay kit, total protein extraction kit,
bicinchoninic acid (BCA) protein assay kit and enhanced
chemiluminescence (ECL) reagent were purchased from Wanleibio Co.,
Ltd. Dulbecco's modified Eagle's medium (DMEM) was purchased from
Gibco; Thermo Fisher Scientific, Inc. Fetal bovine serum (FBS) was
purchased from Biological Industries. Trypsin and
4′,6-diamidino-2-phenylindole (DAPI) were purchased from Beyotime
Institute of Biotechnology. TRIzol® and 2X Power Taq PCR
MasterMix were purchased from BioTeke Corporation. SYBR Green
master mix was purchased from Beijing Solarbio Science &
Technology Co., Ltd.
Isolation and culture of rat
hippocampal neurons
Rat hippocampal neurons were isolated and cultured
using the methods previously described (22). In total, 150 2-day-old Sprague
Dawley rats (weight, 8±2 g; males, 75; females, 75) were used in
the study. These rats were obtained from the Experimental Animal
Center of Xi'an Jiaotong University Health Science Center [License
no. SCXK (Shaan) 2018-001], which were housed in a
specific-pathogen free facility maintained at 23°C with a 12-h
light-dark cycle and 24-h atmosphere purification, and were allowed
free access to breastmilk from their mother. In brief, brain
tissues were isolated from these rats under aseptic conditions. The
hippocampal tissues were then dissected on an ultra-clean bench,
cut into pieces, and digested by trypsin. Subsequently, DMEM
containing 10% FBS and penicillin/streptomycin was added for
trypsin neutralization. Cell suspension was repeatedly digested
five-six times after centrifugation (168 × g, 7 min, 25°C). After
filtration and centrifugation, the hippocampal neurons were placed
in a poly-lysine-coated 6-well plate at a density of
5×105 cells/ml and cultured for 8 h at 37°C with 5%
CO2 and saturated humidity. Subsequently, the culture
medium was changed to neurobasal medium containing 2% B27, 0.5 mM
glutamine, 100 U/ml penicillin, and 100 U/ml streptomycin and the
cells were cultured for 48 h. Cytarabine was then added to the
medium (final concentration, 10 µM) to inhibit the growth of glial
cells. The media were changed once every 3 days until maturation
and network formation of the hippocampal neurons in ~15 days. All
experimental procedures in this study were approved by the Ethics
Committee of The Second Affiliated Hospital of Xi'an Jiaotong
University (Shaanxi, China).
Drug treatment
The cultured neurons were divided into the following
three groups: Control, model and ginsenoside Rg1 (Rg1) groups. The
control group was used as the vehicle treated group, in which no
drugs were added to the culture medium; in the model group,
cultured neurons were treated with 8 µM Aβ1-42 (23) for 24 h at 37°C; in the Rg1 group,
the cultured neurons were exposed to 60 µM ginsenoside Rg1
(24) for 1 h at 37°C and then to
8 µM Aβ1-42 for 24 h at 37°C. To confirm whether
ginsenoside Rg1 regulates PPARγ phosphorylation by acting on CDK5,
the effects of ginsenoside Rg1 on cultured neurons that were
treated with Aβ1-42 after CDK5 expression was inhibited
using the CDK5 inhibitor roscovitine were investigated. Neurons
were divided into the three following groups: Model, roscovitine
and roscovitine+Rg1 groups. In the model group, the cultured
neurons were treated with 8 µM Aβ1-42 for 24 h at 37°C;
in the roscovitine group, the cultured neurons were first exposed
to 25 µM roscovitine (25) for 1 h
at 37°C and then to 8 µM Aβ1-42 for 24 h at 37°C; in the
roscovitine+Rg1 group, cultured neurons were first treated with 25
µM roscovitine for 0.5 h at 37°C followed by 60 µM ginsenoside Rg1
for 1 h at 37°C and subsequently 8 µM Aβ1-42 for 24 h at
37°C.
TUNEL staining
TUNEL staining was performed using an assay kit
according to the manufacturer's instructions. After the slides were
fixed with 4% paraformaldehyde at 25°C for 10 min, the cells were
permeabilized with 50 µl of 0.1% Triton X-100 for 15 min at 25°C.
After being washed with phosphate-buffered saline (PBS), the cells
were incubated with the TUNEL reaction mixture (formulated by
mixing the enzyme and label solutions at a ratio of 1:9) in a wet
chamber in the dark at 37°C for 60 min. Subsequently, the cells
were washed with PBS and counterstained with DAPI in the dark at
25°C for 5 min. After washing with PBS again, the slides were
mounted using mounting medium with anti-fluorescent quenchers. The
neurons were counted in a blind manner by two pathologists under a
BX 53 fluorescence microscope (Olympus Corporation) at ×400
magnification.
The average number of neurons from four random
fields of view was used as the final result for each pathologist.
The results from the two pathologists were averaged and used to
calculate the percentage of TUNEL-positive neurons.
Western blotting
Total proteins of the neurons were extracted using
the total protein extraction kit according to the manufacturer's
instructions and the protein concentration was measured using the
BCA protein assay method. After denaturation at 95°C for 5 min, 20
µl protein sample (including 40 µg protein) was added into each
electrophoretic lane, separated in an 8% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis and transferred to a
nitrocellulose (NC) membrane. The membrane was blocked with 5%
non-fat powdered milk at 37°C for 1 h and was then incubated with
rabbit anti-rat CDK5 (1:500), p-PPARγ-Ser273 (1:500), PPARγ
(1:400), IDE (1:900), BACE1 (1:600), APP (1:800), and
Aβ1-42 (1:500) polyclonal antibodies overnight at 4°C.
After being washed with Tris-buffered saline buffer with 0.05%
Tween 20, the NC membrane was incubated with the secondary
horseradish peroxidase-conjugated antibody (goat anti-rabbit,
1:1,000) at 37°C for 45 min. Subsequently, the NC membrane was
developed using ECL reagent and the blots were scanned. The optical
densities of the target bands were analyzed using Gel-Pro Analyzer
software (version 4.0, Media Cybernetics, Inc.).
Reverse transcription-quantitative
(RT-q)PCR
Total RNA from the neurons of the three groups was
extracted using TRIzol and the concentrations were measured using a
UV spectrophotometer. Reverse transcription was performed according
to the manufacturer's instructions. In brief, each RNA sample was
added into a nuclease-free centrifuge tube in an ice bath based on
the concentration of the extracted RNA sample (consistent RNA
concentrations during sample loading), followed by the addition of
1 µl of oligo (dT)15, 1 µl of random primers, and a
sufficient volume of double-distilled water to reach a total volume
of 12.5 µl. The mixture was incubated at 70°C for 5 min and was
then rapidly cooled on ice for 2 min. After centrifugation (671 ×
g, 1 min, 4°C), the reaction mixture was mixed with 2 µl of
deoxynucleoside triphosphate (2.5 mM each), 4 µl of 5X buffer, 0.5
µl of RNase inhibitor and 1 µl of Moloney-murine leukemia virus
(200 U), and was then sequentially subjected to the following
conditions: 25°C for 10 min, 42°C for 50 min and 80°C for 10 min to
terminate the reaction. The resultant cDNA was stored at −20°C for
further use. Primers (Table I) of
rat Cdk5, Pparγ, Ide, Bace1 and App genes were
designed using Primer Premier 5.0 (Premier Biosoft International).
All primers were synthesized by Sangon Biotech Co., Ltd.
Fluorescence-based RT-qPCR was performed using a 2X Power Taq PCR
Master Mix kit with a Exicycler™ 96 real-time PCR instrument
(Bioneer Corp.). The PCR reaction was performed according to the
manufacturer's instructions in a 20 µl mixture including 1 µl of
cDNA, 0.5 µl of the forward primer (10 µM), 0.5 µl of the reverse
primer (10 µM), 10 µl of the SYBR Green Master Mix and sufficient
double-distilled water. Reaction conditions were as follows:
Initial denaturation at 94°C for 5 min, followed by denaturation at
94°C for 10 sec, annealing at 60°C for 20 sec and extension at 72°C
for 30 sec for 40 cycles. Relative mRNA expression levels were
calculated using the 2−ΔΔCq method (26). β-actin was used as the internal
control.
 | Table I.Primers used for reverse
transcription-quantitative PCR. |
Table I.
Primers used for reverse
transcription-quantitative PCR.
| Gene | Primer sequence
(5′→3′) | Primer length | Temperature,
°C | PCR product length,
bp |
|---|
| Cyclin-dependent
kinase 5 | F:
GGACACCGACTGAGGAAC | 18 | 52.0 | 103 |
|
| R:
TTGGGCACGACATTCAC | 17 | 52.5 |
|
| Peroxisome
proliferator-activated | F:
TACCACGGTTGATTTCTC | 18 | 47.7 | 155 |
| receptor γ | R:
AATAATAAGGCGGGGACG | 18 | 55.3 |
|
| Insulin-degrading
enzyme | F:
TCCCGTGAAGCGACTGT | 17 | 54.3 | 180 |
|
| R:
GACTTGTCCGTGGTGGG | 17 | 53.6 |
|
| β-amyloid cleavage
enzyme 1 | F:
TCCGCATCACCATCCTT | 17 | 54.0 | 123 |
|
| R:
TGACCGCTCCCATAACG | 17 | 55.1 |
|
| Amyloid precursor
protein | F:
ACTCTGTGCCAGCCAATA | 18 | 51.2 | 158 |
|
| R:
TGAATCATGTCCGAACTCC | 19 | 53.0 |
|
| β-actin | F:
GGAGATTACTGCCCTGGCTCCTAGC | 25 | 60.1 | 155 |
|
| R:
GGCCGGACTCATCGTACTCCTGCTT | 25 | 62.0 |
|
Data analysis
All data are expressed as the mean ± SEM. One-way
analysis of variance (ANOVA) followed by a Least Significant
Difference post hoc test was performed for multiple comparisons.
Statistical analyses were conducted using SPSS (version 16.0, SPSS,
Inc.). P<0.05 was considered to indicate a statistically
significant difference.
Results
Ginsenoside Rg1 inhibits PPARγ
phosphorylation in the AD model
In primary cultured rat hippocampal neurons
Aβ1-42 treatment significantly enhanced PPARγ
phosphorylation at Ser273, increased the p-PPARγ/PPARγ ratio and
decreased PPARγ protein and mRNA expression levels compared with
those in the control group (P<0.05; Fig. 1). These results suggested the
presence of PPARγ phosphorylation in the AD neuron model induced by
Aβ1-42. In addition, pretreatment with ginsenoside Rg1
significantly attenuated the aforementioned
Aβ1-42-induced effects in these neurons (P<0.05;
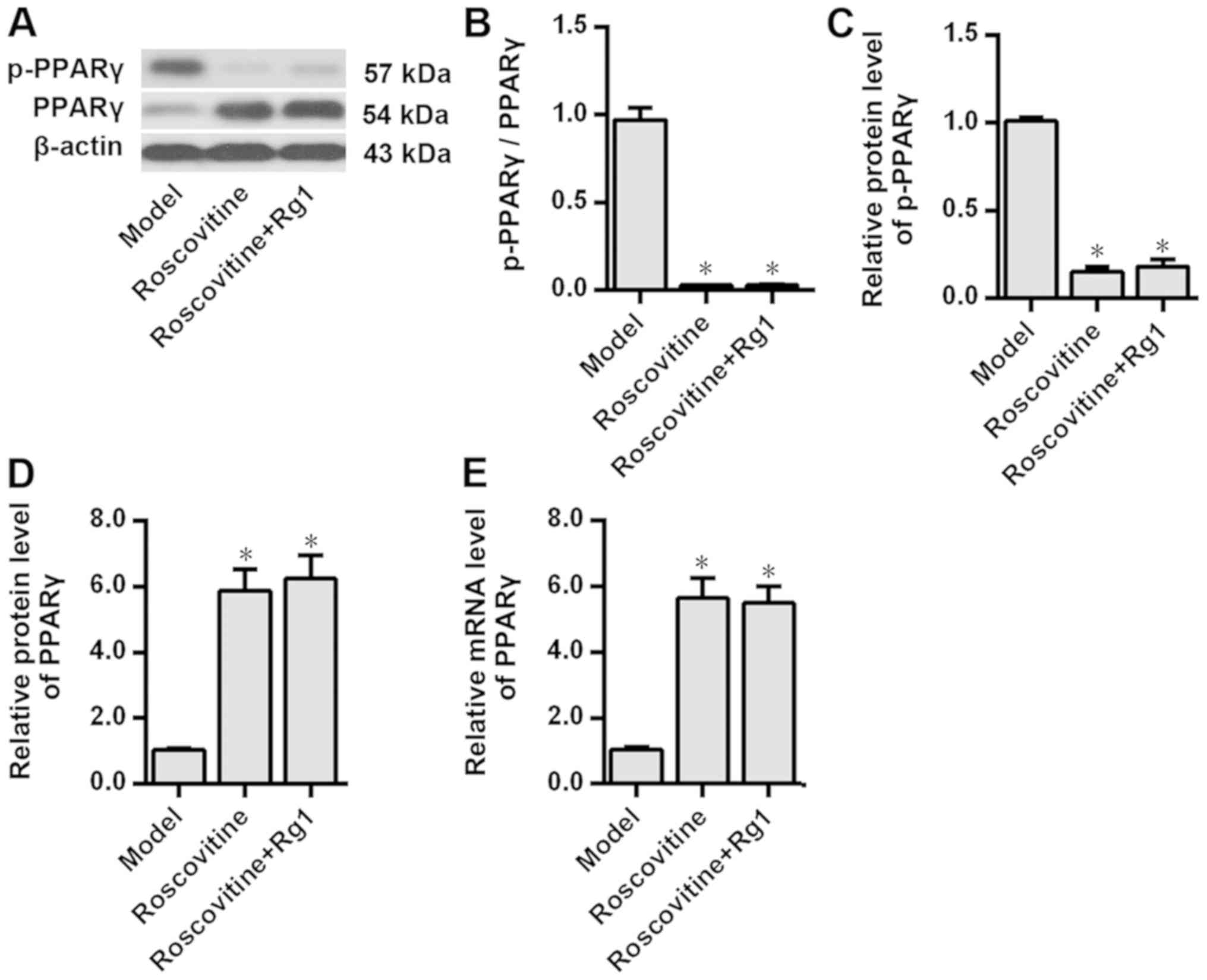
Fig. 1). Notably, after inhibiting
CDK5 expression using roscovitine, the results demonstrated that
compared with those in the model group PPARγ phosphorylation was
significantly inhibited and PPARγ expression levels were
significantly increased in the Aβ1-42-treated neurons
(P<0.05; Fig. 2). Additionally,
PPARγ phosphorylation and the expression levels of PPARγ protein in
the Aβ1-42-treated neurons were not further affected by
ginsenoside Rg1 treatment after CDK5 inhibition (P>0.05;
Fig. 2), suggesting that CDK5 may
be involved in the inhibition of PPARγ phosphorylation induced by
ginsenoside Rg1.
Ginsenoside Rg1 decreases CDK5
expression levels in the AD model
The effects of ginsenoside Rg1 on CDK5 expression in
the neuron model of AD were investigated. Western blotting and
RT-qPCR analyses demonstrated that in the primary cultured rat
hippocampal neurons Aβ1-42 treatment significantly
increased the protein and mRNA expression levels of CDK5 compared
with those in the control group (P<0.05); but, ginsenoside Rg1
pretreatment significantly attenuated the Aβ1-42-induced
increase in the protein and mRNA expression levels of CDK5
(P<0.05; Fig. 3).
Ginsenoside Rg1 regulates the
expression of PPARγ target genes and decreases intracellular
Aβ1-42 levels in the AD model
The effects of ginsenoside Rg1 on the expression of
PPARγ target genes and intracellular Aβ1-42 level when
CDK5 was inhibited were examined. The results demonstrated that
compared with the rat hippocampal neurons in the control group,
those in the model group exhibited significantly decreased IDE
protein and mRNA expression levels (P<0.05), but significantly
increased BACE1, APP and enhanced intracellular Aβ1-42
levels (P<0.05; Fig. 4).
Additionally, pretreatment with ginsenoside Rg1 significantly
attenuated Aβ1-42-induced effects in these neurons
(P<0.05; Fig. 4). Additionally,
compared with those in the model group inhibition of CDK5
expression using roscovitine significantly increased the expression
levels of IDE and reduced the expression levels of BACE1, APP and
Aβ1-42 in rat hippocampal neurons treated with
Aβ1-42 (P<0.05; Fig.
5). In addition, no significant differences were observed in
the expression levels of IDE, BACE1, APP and Aβ1-42
after ginsenoside Rg1 treatment following CDK5 inhibition with
roscovitine (P>0.05; Fig.
5).
 | Figure 4.Effects of ginsenoside Rg1 on the
expression levels of PPARγ target genes and intracellular
Aβ1-42 levels in the Alzheimer's disease neuron model.
Protein expression levels of (A) IDE, (D) BACE1, (G) APP and (J)
Aβ1-42 were assessed by western blotting analysis. IDE
(B) protein and (C) mRNA expression levels, BACE1 (E) protein and
(F) mRNA expression levels, APP (H) protein and (I) mRNA expression
levels, and (K) Aβ1-42 protein expression levels were
compared among the three groups. n=6. *P<0.05 vs. control group;
#P<0.05 vs. model group. PPARγ, peroxisome
proliferator-activated receptor γ; IDE, insulin-degrading enzyme;
BACE1, β-amyloid cleavage enzyme 1; APP, amyloid precursor protein;
Aβ, β-amyloid peptides. |
Ginsenoside Rg1 attenuates
Aβ1-42-induced apoptosis in rat hippocampal neurons
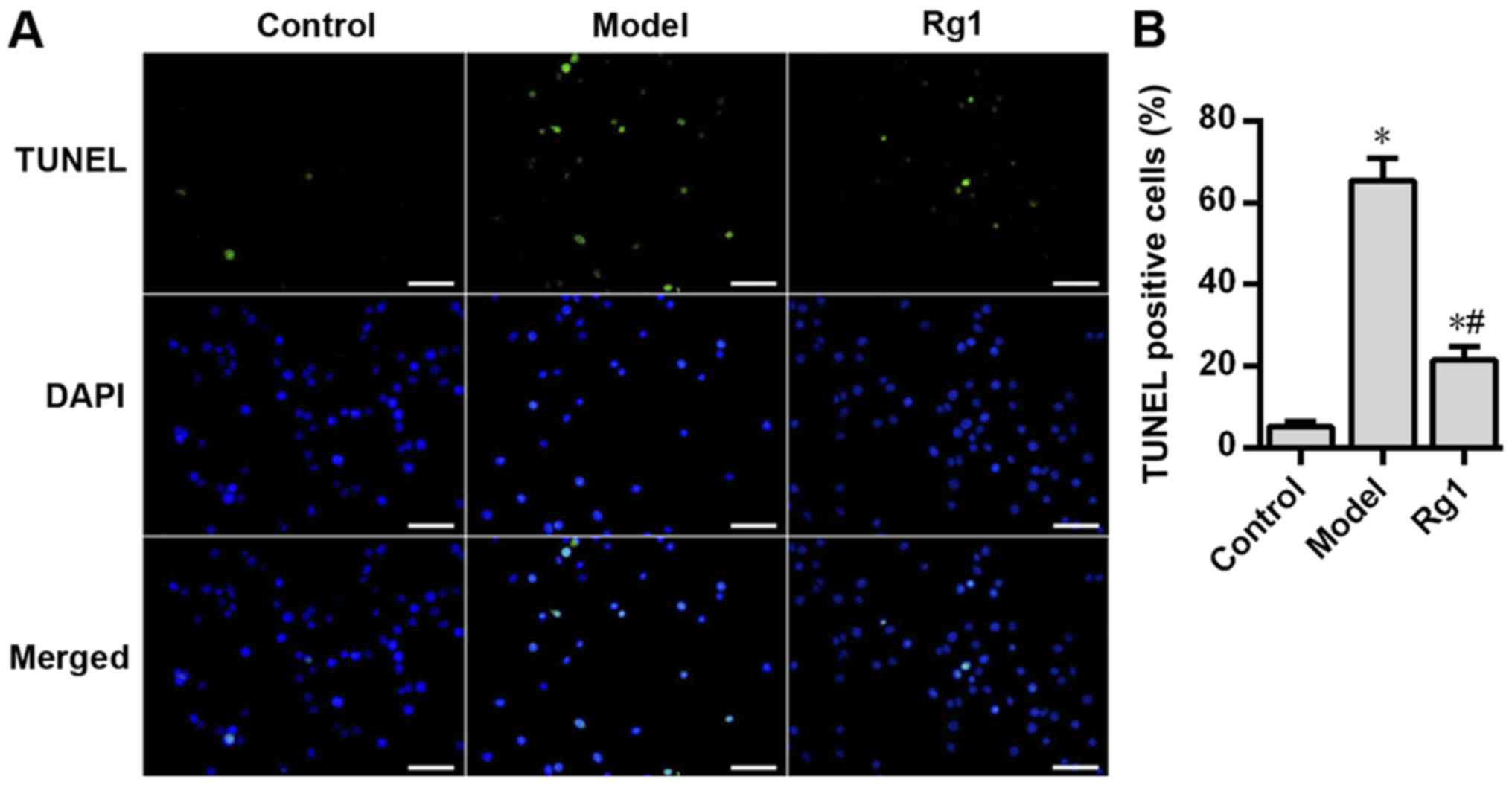
In the present study, TUNEL staining was performed
to determine the effects of Aβ1-42 and ginsenoside Rg1
on the apoptosis of rat hippocampal neurons. Compared with that in
the control group, Aβ1-42 treatment significantly
increased neuronal apoptosis in the model group (P<0.05;
Fig. 6), but pretreatment with
ginsenoside Rg1 significantly decreased Aβ1-42-induced
neuronal apoptosis (P<0.05; Fig.
6). In addition, the neuronal apoptosis rate in the roscovitine
group was significantly lower compared with that in the model group
(P<0.05; Fig. 7). Additionally,
no significant difference was observed in the in the neuronal
apoptosis rate between the roscovitine group and the
roscovitine+Rg1 group (P>0.05; Fig.
7).
Discussion
This study investigated whether ginsenoside Rg1
inhibits the phosphorylation of PPARγ through the downregulation of
the CDK5 pathway, thereby affecting the expression of PPARγ target
genes Bace1 and Ide and reducing Aβ levels (Fig. 8). PPARs are ligand-activated
nuclear transcription factors, which regulate the transcription of
target genes by binding to the peroxisome proliferator response
element (PPRE) located on the promoters of these genes (27). Different types of fatty acids
(FAs), including docosahexaenoic acid, activate PPARs in adipose
tissues (28,29). FA binding to PPARs control the
transcription of specific genes including those encoding for
various metabolic and cellular processes such as FA β-oxidation and
adipogenesis, making them key mediators of lipid homeostasis
(29). PPARγ is one of PPARs
superfamily members (along with PPARα, PPARβ/δ). A previous study
has reported that PPARγ inhibits BACE1 expression by binding to the
PPRE on the Bace1 gene promoter in N2a/APP695 cells
(30), which is an essential
enzyme in the generation of Aβ as it hydrolyzes APP to form Aβ
(31). Additionally, PPARγ also
bind to the PPRE in the Ide promoter, thereby regulating Ide
gene transcription and promoting IDE protein expression (32), which has been demonstrated to
degrade Aβ (33). These findings
indicate that PPARγ serves a key role in the inhibition of Aβ
generation and the promotion of Aβ degradation. Moreover, PPARγ has
anti-inflammatory effects through inhibiting the generation of
certain proinflammatory cytokines (such as tumor necrosis factor,
interleukin-1β and interleukin-6), the production of nitric oxide,
and the expression of matrix metalloproteinase 9 and macrophage
scavenger receptor 1 (34).
Studies have reported that these molecules are closely associated
with the onset of AD (35–40). Furthermore, oxidative stress
usually occurs during early stages of AD and elevates with
increased AD severity (41); PPARγ
inhibits Aβ-induced oxidative stress (42). Therefore, PPARγ agonists may
provide neuroprotective effects against AD.
Several clinical and experimental studies have
investigated the role of thiazolidinediones (TZDs), PPARγ agonists,
in AD, for example, pioglitazone reduces brain Aβ levels (43) and improves learning and memory in
APP/PS1 mice (44), and decreases
tau phosphorylation (45) and
neuronal apoptosis (46) in an AD
cell model; rosiglitazone decreases Aβ1-40 and Aβ1-42
levels, reduces tau phosphorylation, alleviates memory impairment
(47), and inhibits Aβ-induced
oxidative stress (48),
inflammatory responses (49) and
mitochondrial dysfunction (50) in
both in vitro and in vivo systems. Oral
administration of rosiglitazone improves cognitive function in
patients with mild-to-moderate AD (51). Troglitazone and ciglitazone prevent
Aβ-induced microglial- and monocyte-mediated neurotoxicity and
inhibit Aβ-induced increased expression of interleukin 6, tumor
necrosis factor α, and cyclooxygenase-2 (52). Recently, a study demonstrated that
pioglitazone inhibits the phosphorylation of PPARγ at Ser273 in
vitro by inhibiting CDK5 expression, which in turn affected the
expression of PPARγ target genes Ide and Bace1,
thereby promoting Aβ degradation and reducing Aβ production. This
reduced Aβ levels in the brain, thereby exerting neuroprotective
effects in an AD model (53).
These findings indicate that TZDs have neuroprotective effects
against AD. However, as a long-term medication is required for AD,
which is a chronic disease with complex pathogenic mechanisms and a
long disease course, the side effects of TZDs will substantially
limit their application in AD treatment. Studies have demonstrated
that rosiglitazone may lead to an increased risk of fractures
(54), heart failure (55,56)
and increased incidence of stroke (56); pioglitazone may lead to fractures
and bladder cancer (54,57); and troglitazone may induce severe
hepatotoxicity (58). Therefore,
there is a need for TZD replacement with safer and effective drugs
presenting mild side effects and PPARγ-agonistic effects in the
study of AD therapies.
Ginseng is a natural herbal remedy that has been
used in China over several millennia (1). Ginseng has multi-target therapeutic
and pharmacological effects in central nervous system, which have
been well-demonstrated in the clinical practice (59). Ginsenosides, which mainly include
Rb1, Rb2, Rb3, Rc, Rd, Re, Rg1 and Rg2, are the main components of
ginseng (60). In particular,
ginsenoside Rg1 is one of the most studied and representative
ginsenoside components in the field of AD treatment (61). Previous studies have observed that
ginsenoside Rg1 may potentially activate PPARγ and facilitate Aβ
removal by enhancing the binding of PPARγ to target genes or by
upregulating PPARγ expression (14,15).
Accordingly, the present study further elucidated the mechanisms by
which ginsenoside Rg1 affects PPARγ. The results of the present
study suggested that ginsenoside Rg1 inhibits PPARγ phosphorylation
by downregulating the expression of CDK5, thereby affecting the
expression of PPARγ target genes.
Extracellular and intracellular Aβ serve essential
roles in the onset of AD (62).
BACE1-mediated APP proteolysis produces a soluble β-fragment of the
amyloid precursor protein and a C-terminal fragment containing 99
amino acids (C99). Subsequently, C99 is enzymatically cleaved by
γ-secretase to generate APP intracellular domain and Aβ; the latter
is released into the extracellular matrix (63,64)
(Fig. 8). APP proteolysis also
occurs in the endoplasmic reticulum and golgi apparatus (64,65),
producing intracellular Aβ. Gouras et al (66) reported that intraneuronal Aβ
immunoreactivity appeared to precede the deposition of both
neurofibrillary tangles and senile plaques, indicating that
intraneuronal accumulation of Aβ is an early event in the onset of
AD. The neuron model of AD in this study was established with the
addition of exogenous Aβ, so exogenous and neuro-secreted Aβ could
not be distinguished, and this study only observed the effect of
ginsenoside Rg1 on the intracellular Aβ and did not investigate the
changes in extracellular Aβ levels.
This study demonstrated that Rg1 inhibited CDK5
expression. However, the mechanism remains unclear. The results of
the present study suggested that ginsenoside Rg1 may affect CDK5
directly, or through other mechanisms; this needs to be
investigated. The results of the present also demonstrated that
exogenously added Aβ increased CDK5 expression levels, aggravated
PPARγ phosphorylation, decreased PPARγ expression levels, and
affected the expression levels of the downstream PPARγ target genes
Ide and Bace1. The exact mechanisms of these phenomena
remain unclear and are subjects for future studies. Previous
studies have observed a negative correlation between IDE expression
and Aβ levels in AD brains (67),
and a decrease in IDE expression levels were also observed in an AD
rat model established through the injection of Aβ1-42 in
the hippocampus (15). These
findings are consistent with those of the present study, which
demonstrated decreased IDE expression levels in Aβ-treated primary
cultured hippocampal neurons. After treatment with ginsenoside Rg1,
IDE expression levels increased and Aβ levels decreased. Several
possible reasons may account for these findings: i) The consumption
of IDE through its effects on degradation of Aβ exceeds the
compensatory generation of IDE; ii) decreased IDE expression may
consequently promote Aβ generation; and iii) mutual interactions
may exist between IDE and Aβ, i.e., generated Aβ affects IDE
expression, whereas decreased IDE expression promotes an increase
in Aβ levels. Accordingly, further studies are required to verify
these explanations. This study demonstrated that CDK5 mediates the
ginsenoside Rg1 reduction of Aβ levels by phosphorylating PPARγ. In
addition to CDK5, other CDKs such as CDK7 and CDK9 also take part
in PPARγ phosphorylation. A previous study has reported that CDK7
phosphorylates PPARγ at Ser112 to inhibit the activity of PPARγ
(68). However, CDK9 increases
PPARγ activity after phosphorylating PPARγ at Ser112 (69). Thus, further investigation is
necessary into whether the phosphorylation of PPARγ by CDK7 and
CDK9 is involved in the ginsenoside Rg1 reduction of Aβ levels.
Additionally, the results of the present study demonstrated that
the protein and mRNA expression levels of PPARγ in the Rg1 group
were higher compared with those in the control group. It was
speculated this is due to the high concentrations of ginsenoside
Rg1 used in the study, which were able to stimulate a significant
response compared with the control.
There are several limitations to this study. In the
study, no experiments such as chromatin immunoprecipitation
RT-qPCR, were performed to confirm whether CDK5 directly regulates
PPARγ. However, our previous study reported that CDK5 regulates
PPARγ (21), and performed
co-immunoprecipitation experiments to confirm that Aβ promotes the
binding of CDK5 to PPARγ (53). In
addition, our previous study demonstrated that the PPARγ agonist
pioglitazone inhibits PPARγ phosphorylation by inhibiting CDK5
expression, thereby promoting Aβ degradation and reducing Aβ
production (53). Therefore, no
PPARγ agonist was used for comparison with ginsenoside Rg1 in this
study. Since a CDK5 inhibitor was used in the study, no in
vivo studies were conducted and only in vitro
experiments were performed; therefore, the effects of ginsenoside
Rg1 in vivo are unknown. Furthermore, since there are no
available drugs that target CDK5, no positive-control drug was used
in the study. A set of CDK5 overexpressing cells would allow for
testing of the results of this study; unfortunately, CDK5
overexpression experiments were not be performed due to limitations
on time and funding.
In conclusion, the results of the present study
suggested that ginsenoside Rg1 inhibits PPARγ phosphorylation
possibly through the downregulation of CDK5 expression, thereby
affecting the expression of PPARγ target genes (Ide and
Bace1) and decreasing Aβ levels through the promotion of Aβ
degradation and reduction of Aβ synthesis, which ultimately
provides neuroprotective effects against AD.
Acknowledgements
Not applicable.
Funding
This study was supported by the Chinese Postdoctoral
Science Foundation (grant no. 2017M623191), the Natural Science
Foundation of Shaanxi Province (grant no. 2017JQ8039) and the
Foundation of The Second Affiliated Hospital of the Xi'an Jiaotong
University [grant no. YJ(ZD)201517].
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
QQ, XL and JF designed the study and wrote the
manuscript. QQ, JH, ML and BZ performed the experiments. QQ and JH
collected and analyzed the data. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
All experimental procedures in the present study
were approved by the Ethics Committee of The Second Affiliated
Hospital of Xi'an Jiaotong University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Chen CF, Chiou WF and Zhang JT: Comparison
of the pharmacological effects of Panax ginseng and Panax
quinquefolium. Acta Pharmacol Sin. 29:3277–1108. 2008. View Article : Google Scholar
|
|
2
|
Nie L, Xia J, Li H, Zhang Z, Yang Y, Huang
X, He Z, Liu J and Yang X: Ginsenoside Rg1 ameliorates behavioral
abnormalities and modulates the hippocampal proteomic change in
triple transgenic mice of Alzheimer's disease. Oxid Med Cell
Longev. 2017:64735062017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mu JS, Lin H, Ye JX, Lin M and Cui XP: Rg1
exhibits neuroprotective effects by inhibiting the endoplasmic
reticulum stress-mediated c-Jun N-terminal protein kinase apoptotic
pathway in a rat model of Alzheimer's disease. Mol Med Rep.
12:3862–3868. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liu QA, Kou JP and Yu BY: Ginsenoside Rg1
protects against hydrogen peroxide-induced cell death in PC12 cells
via inhibiting NF-κB activation. Neurochem Int. 58:119–125. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Huang T, Fang F, Chen L, Zhu Y, Zhang J,
Chen X and Yan SS: Ginsenoside Rg1 attenuates oligomeric
Aβ(1–42)-induced mitochondrial dysfunction. Curr Alzheimer Res.
9:388–395. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tanzi RE, Moir RD and Wagner SL: Clearance
of Alzheimer's Abeta peptide: The many roads to perdition. Neuron.
43:605–608. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Álvarez-Arellano L, Pedraza-Escalona M,
Blanco-Ayala T, Camacho-Concha N, Cortés-Mendoza J, Pérez-Martínez
L and Pedraza-Alva G: Autophagy impairment by caspase-1-dependent
inflammation mediates memory loss in response to β-amyloid peptide
accumulation. J Neurosci Res. 96:234–246. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Seino Y, Kawarabayashi T, Wakasaya Y,
Watanabe M, Takamura A, Yamamoto-Watanabe Y, Kurata T, Abe K, Ikeda
M, Westaway D, et al: Amyloid β accelerates phosphorylation of tau
and neurofibrillary tangle formation in an amyloid precursor
protein and tau double-transgenic mouse model. J Neurosci Res.
88:3547–3554. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Vargas LM, Leal N, Estrada LD, González A,
Serrano F, Araya K, Gysling K, Inestrosa NC, Pasquale EB and
Alvarez AR: EphA4 activation of c-Abl mediates synaptic loss and
LTP blockade caused by amyloid-β oligomers. PLoS One. 9:e923092014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ibi D, Tsuchihashi A, Nomura T and
Hiramatsu M: Involvement of GAT2/BGT-1 in the preventive effects of
betaine on cognitive impairment and brain oxidative stress in
amyloid β peptide-injected mice. Eur J Pharmacol. 842:57–63. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hooshmandi E, Ghasemi R, Iloun P and
Moosavi M: The neuroprotective effect of agmatine against amyloid
β-induced apoptosis in primary cultured hippocampal cells involving
ERK, Akt/GSK-3β, and TNF-α. Mol Biol Rep. 46:489–496. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Schirinzi T, Di Lorenzo F, Sancesario GM,
Di Lazzaro G, Ponzo V, Pisani A, Mercuri NB, Koch G and Martorana
A: Amyloid-mediated cholinergic dysfunction in motor impairment
related to Alzheimer's disease. J Alzheimers Dis. 64:525–532. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ali T, Yoon GH, Shah SA, Lee HY and Kim
MO: Osmotin attenuates amyloid beta-induced memory impairment, tau
phosphorylation and neurodegeneration in the mouse hippocampus. Sci
Rep. 5:117082015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen LM, Lin ZY, Zhu YG, Lin N, Zhang J,
Pan XD and Chen XC: Ginsenoside Rg1 attenuates β-amyloid generation
via suppressing PPARγ-regulated BACE1 activity in N2a-APP695 cells.
Eur J Pharmacol. 675:15–21. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Quan Q, Wang J, Li X and Wang Y:
Ginsenoside Rg1 decreases Aβ1-42 level by upregulating
PPARγ and IDE expression in the hippocampus of a rat model of
Alzheimer's disease. PLoS One. 8:e591552013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu C, Zhai X, Zhao B, Wang Y and Xu Z:
Cyclin I-like (CCNI2) is a Cyclin-dependent kinase 5 (CDK5)
activator and is involved in cell cycle regulation. Sci Rep.
7:409792017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Su SC and Tsai LH: Cyclin-dependent
kinases in brain development and disease. Annu Rev Cell Dev Biol.
27:465–491. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lopes JP and Agostinho P: Cdk5:
Multitasking between physiological and pathological conditions.
Prog Neurobiol. 94:49–63. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wilkaniec A, Czapski GA and Adamczyk A:
Cdk5 at crossroads of protein oligomerization in neurodegenerative
diseases: Facts and hypotheses. J Neurochem. 136:222–233. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Choi JH, Banks AS, Estall JL, Kajimura S,
Boström P, Laznik D, Ruas JL, Chalmers MJ, Kamenecka TM, Blüher M,
et al: Anti-diabetic drugs inhibit obesity-linked phosphorylation
of PPARγ by Cdk5. Nature. 466:451–456. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Quan Q, Qian Y, Li X and Li M: CDK5
participates in amyloid-β production by regulating PPARγ
phosphorylation in primary rat hippocampal neurons. J Alzheimers
Dis. 71:443–460. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Vadukul DM, Gbajumo O, Marshall KE and
Serpell LC: Amyloidogenicity and toxicity of the reverse and
scrambled variants of amyloid-β 1-42. FEBS Lett. 591:822–830. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang EJ, Ahn S, Ryu J, Choi MS, Choi S,
Chong YH, Hyun JW, Chang MJ and Kim HS: Phloroglucinol attenuates
the cognitive deficits of the 5XFAD mouse model of Alzheimer's
sisease. PLoS One. 10:e01356862015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li Y, Guan Y, Wang Y, Yu CL, Zhai FG and
Guan LX: Neuroprotective effect of the ginsenoside Rg1 on cerebral
ischemic injury in vivo and in vitro is mediated by PPARγ-regulated
antioxidative and anti-inflammatory pathways. Evid Based Complement
Alternat Med. 2017:78420822017.PubMed/NCBI
|
|
25
|
Manser C, Vagnoni A, Guillot F, Davies J
and Miller CC: Cdk5/p35 phosphorylates lemur tyrosine kinase-2 to
regulate protein phosphatase-1C phosphorylation and activity. J
Neurochem. 121:343–348. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mandrekar-Colucci S, Karlo JC and Landreth
GE: Mechanisms underlying the rapid peroxisome
proliferator-activated receptor-γ-mediated amyloid clearance and
reversal of cognitive deficits in a murine model of Alzheimer's
disease. J Neurosci. 32:10117–10128. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Houseknecht KL, Cole BM and Steele PJ:
Peroxisome proliferator-activated receptor gamma (PPARgamma) and
its ligands: A review. Domest Anim Endocrinol. 22:1–23. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Echeverría F, Valenzuela R, Catalina
Hernandez-Rodas M and Valenzuela A: Docosahexaenoic acid (DHA), a
fundamental fatty acid for the brain: New dietary sources.
Prostaglandins Leukot Essent Fatty Acids. 124:1–10. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Echeverría F, Ortiz M, Valenzuela R and
Videla LA: Long-chain polyunsaturated fatty acids regulation of
PPARs, signaling: Relationship to tissue development and aging.
Prostaglandins Leukot Essent Fatty Acids. 114:28–34. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lin N, Chen LM, Pan XD, Zhu YG, Zhang J,
Shi YQ and Chen XC: Tripchlorolide attenuates β-amyloid generation
via suppressing PPARγ-regulated BACE1 activity in N2a/APP695 cells.
Mol Neurobiol. 53:6397–6406. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sadleir KR, Eimer WA, Cole SL and Vassar
R: Aβ reduction in BACE1 heterozygous null 5XFAD mice is associated
with transgenic APP level. Mol Neurodegener. 10:12015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Du J, Zhang L, Liu SB, Zhang C, Huang XQ,
Li J, Zhao NM and Wang Z: PPARgamma transcriptionally regulates the
expression of insulin-degrading enzyme in primary neurons. Biochem
Biophys Res Commun. 383:485–490. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Vingtdeux V, Chandakkar P, Zhao H, Blanc
L, Ruiz S and Marambaud P: CALHM1 ion channel elicits amyloid-β
clearance by insulin-degrading enzyme in cell lines and in vivo in
the mouse brain. J Cell Sci. 128:2330–2338. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Delerive P, Fruchart JC and Staels B:
Peroxisome proliferator-activated receptors in inflammation
control. J Endocrinol. 169:453–459. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Decourt B, Lahiri DK and Sabbagh MN:
Targeting tumor necrosis factor alpha for Alzheimer's disease. Curr
Alzheimer Res. 14:412–425. 2017.PubMed/NCBI
|
|
36
|
Mendiola AS and Cardona AE: The IL-1β
phenomena in neuroinflammatory diseases. J Neural Transm (Vienna).
125:781–795. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Haddick PC, Larson JL, Rathore N, Bhangale
TR, Phung QT, Srinivasan K, Hansen DV, Lill JR; Alzheimer's Disease
Genetic Consortium (ADGC); Alzheimer's Disease Neuroimaging
Initiative (ADNI), ; et al: A common variant of IL-6R is associated
with elevated IL-6 pathway activity in Alzheimer's disease brains.
J Alzheimers Dis. 56:1037–1054. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cifuentes D, Poittevin M, Bonnin P, Ngkelo
A, Kubis N, Merkulova-Rainon T and Lévy BI: Inactivation of nitric
oxide synthesis exacerbates the development of Alzheimer disease
pathology in APPPS1 mice (Amyloid Precursor Protein/Presenilin-1).
Hypertension. 70:613–623. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Moussa C, Hebron M, Huang X, Ahn J,
Rissman RA, Aisen PS and Turner RS: Resveratrol regulates
Neuro-inflammation and induces adaptive immunity in Alzheimer's
disease. J Neuroinflammation. 14:12017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Spitzer P, Weinbeer J, Herrmann M,
Oberstein TJ, Condic M, Lewczuk P, Kornhuber J and Maler JM:
Analysis of surface levels of IL-1 receptors and macrophage
scavenger receptor I in peripheral immune cells of patients with
Alzheimer's disease. J Geriatr Psychiatry Neurol. 32:211–220. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
de la Monte SM and Wands JR: Molecular
indices of oxidative stress and mitochondrial dysfunction occur
early and often progress with severity of Alzheimer's disease. J
Alzheimers Dis. 9:167–181. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhang ZX, Li YB and Zhao RP:
Epigallocatechin gallate attenuates β-Amyloid generation and
oxidative stress involvement of PPARγ in N2a/APP695 cells.
Neurochem Res. 42:468–480. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Silva-Abreu M, Calpena AC, Andrés-Benito
P, Aso E, Romero IA, Roig-Carles D, Gromnicova R, Espina M, Ferrer
I, García ML and Male D: PPARγ agonist-loaded PLGA-PEG nanocarriers
as a potential treatment for Alzheimer's disease: In vitro and in
vivo studies. Int J Nanomedicine. 13:5577–5590. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Searcy JL, Phelps JT, Pancani T, Kadish I,
Popovic J, Anderson KL, Beckett TL, Murphy MP, Chen KC, Blalock EM,
et al: Long-term pioglitazone treatment improves learning and
attenuates pathological markers in a mouse model of Alzheimer's
disease. J Alzheimers Dis. 30:943–961. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hamano T, Shirafuji N, Makino C, Yen SH,
Kanaan NM, Ueno A, Suzuki J, Ikawa M, Matsunaga A, Yamamura O, et
al: Pioglitazone prevents tau oligomerization. Biochem Biophys Res
Commun. 478:1035–1042. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Dehghani L, Meamar R, Askari G, Khorvash
F, Shaygannejad V, Pour AF and Javanmard SH: The effect of
pioglitazone on the Alzheimer's disease-induced apoptosis in human
umbilical vein endothelial cells. Int J Prev Med. 4 (Suppl
2):S205–S210. 2013.PubMed/NCBI
|
|
47
|
Escribano L, Simón AM, Gimeno E,
Cuadrado-Tejedor M, de Maturana RL, García-Osta A, Ricobaraza A,
Pérez-Mediavilla A, Del Río J and Frechilla D: Rosiglitazone
rescues memory impairment in Alzheimer's transgenic mice:
Mechanisms involving a reduced amyloid and tau pathology.
Neuropsychopharmacology. 35:1593–1604. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Chiang MC, Nicol CJ, Cheng YC, Lin KH, Yen
CH and Lin CH: Rosiglitazone activation of PPARγ-dependent pathways
is neuroprotective in human neural stem cells against
amyloid-beta-induced mitochondrial dysfunction and oxidative
stress. Neurobiol Aging. 40:181–190. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Toledo EM and Inestrosa NC: Activation of
Wnt signaling by lithium and rosiglitazone reduced spatial memory
impairment and neurodegeneration in brains of an APPswe/PSEN1ΔE9
mouse model of Alzheimer's disease. Mol Psychiatry. 15:272–285,
228. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Strum JC, Shehee R, Virley D, Richardson
J, Mattie M, Selley P, Ghosh S, Nock C, Saunders A and Roses A:
Rosiglitazone induces mitochondrial biogenesis in mouse brain. J
Alzheimers Dis. 11:45–51. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Risner ME, Saunders AM, Altman JF, Ormandy
GC, Craft S, Foley IM, Zvartau-Hind ME and Hosford DA;
Rosiglitazone in Alzheimer's disease study group, : Efficacy of
rosiglitazone in a genetically defined population with
mild-to-moderate Alzheimer's disease. Pharmacogenomics J.
6:246–254. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Combs CK, Johnson DE, Karlo JC, Cannady SB
and Landreth GE: Inflammatory mechanisms in Alzheimer's disease:
Inhibition of β-amyloid-stimulated proinflammatory responses and
neurotoxicity by PPARγ agonists. J Neurosci. 20:558–567. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Quan Q, Qian Y, Li X and Li M:
Pioglitazone reduces β amyloid levels via inhibition of PPARγ
phosphorylation in a neuronal model of Alzheimer's disease. Front
Aging Neurosci. 11:1782019. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Aubert RE, Herrera V, Chen W, Haffner SM
and Pendergrass M: Rosiglitazone and pioglitazone increase fracture
risk in women and men with type 2 diabetes. Diabetes Obes Metab.
12:716–721. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Home PD, Pocock SJ, Beck-Nielsen H, Gomis
R, Hanefeld M, Jones NP, Komajda M and McMurray JJ; RECORD study
group, : Rosiglitazone evaluated for cardiovascular outcomes-an
interim analysis. New Engl J Med. 357:28–38. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Wang AT and Smith SA: ACP Journal Club. In
older patients, rosiglitazone was associated with increased risk
for stroke, heart failure, and mortality compared with
pioglitazone. Ann Intern Med. 153:JC6–JC11. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Lewis JD, Ferrara A, Peng T, Hedderson M,
Bilker WB, Quesenberry CP, Vaughn DJ, Nessel L, Selby J and Strom
BL: Risk of bladder cancer among diabetic patients treated with
pioglitazone interim report of a longitudinal cohort study.
Diabetes Care. 34:916–922. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Toyota T and Ueno Y: Clinical effect and
side effect of troglitazone. Nihon Rinsho. 58:376–382.
2000.PubMed/NCBI
|
|
59
|
Kim HJ, Kim P and Shin CY: A comprehensive
review of the therapeutic and pharmacological effects of ginseng
and ginsenosides in central nervous system. J Ginseng Res. 37:8–29.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Zhang S, Sun H, Wang C, Zheng X, Jia X,
Cai E and Zhao Y: Comparative analysis of active ingredients and
effects of the combination of Panax ginseng and Ophiopogon
japonicus at different proportions on chemotherapy-induced
myelosuppression mouse. Food Funct. 10:1563–1570. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Kim MH, Kim SH and Yang WM: Mechanisms of
action of phytochemicals from medicinal herbs in the treatment of
Alzheimer's disease. Planta Med. 80:1249–1258. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
LaFerla FM, Green KN and Oddo S:
Intracellular amyloid-beta in Alzheimer's disease. Nat Rev
Neurosci. 8:499–509. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Vassar R, Bennett BD, Babu-Khan S, Kahn S,
Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, et
al: Beta-secretase cleavage of Alzheimer's amyloid precursor
protein by the transmembrane aspartic protease BACE. Science.
286:735–741. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Yu G, Nishimura M, Arawaka S, Levitan D,
Zhang L, Tandon A, Song YQ, Rogaeva E, Chen F, Kawarai T, et al:
Nicastrin modulates presenilin-mediated notch/glp-1 signal
transduction and betaAPP processing. Nature. 407:48–54. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Xu H, Sweeney D, Wang R, Thinakaran G, Lo
AC, Sisodia SS, Greengard P and Gandy S: Generation of Alzheimer
β-amyloid protein in the trans-Golgi network in the apparent
absence of vesicle formation. Proc Natl Acad Sci USA. 94:3748–3752.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Gouras GK, Tsai J, Naslund J, Vincent B,
Edgar M, Checler F, Greenfield JP, Haroutunian V, Buxbaum JD, Xu H,
et al: Intraneuronal Abeta42 accumulation in human brain. Am J
Pathol. 156:15–20. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Pérez A, Morelli L, Cresto JC and Castano
EM: Degradation of soluble amyloid β-peptides 1–40, 1–42, and the
Dutch variant 1–40Q by insulin degrading enzyme from Alzheimer
disease and control brains. Neurochem Res. 25:247–255. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Helenius K, Yang Y, Alasaari J and Mäkelä
TP: Mat1 inhibits peroxisome proliferator-activated receptor
γ-mediated adipocyte differentiation. Mol Cell Biol. 29:315–323.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Iankova I, Petersen RK, Annicotte JS,
Chavey C, Hansen JB, Kratchmarova I, Sarruf D, Benkirane M,
Kristiansen K and Fajas L: Peroxisome proliferator-activated
receptor gamma recruits the positive transcription elongation
factor b complex to activate transcription and promote
adipogenesis. Mol Endocrinol. 20:1494–1505. 2006. View Article : Google Scholar : PubMed/NCBI
|