Introduction
Cholangiocarcinoma (CCA) is a fatal tumor that
arises from the biliary epithelium, which is characterized by late
diagnosis and poor outcome (1–4).
Over the past three decades the global incidence of CCA (1/100,000)
has steadily increased, whereas the 5-year survival rate of
diagnosed patients has remained ~10% (5–8).
Resection is the best option for the curative treatment of patients
with CCA. However, high rates of recurrence and short survival
times are associated with resection in patients with CCA (9–13).
Chemotherapy is used to improve the outcomes of
patients with CCA. Doxorubicin (Dox), which blocks DNA replication
(14,15), is a first-line drug widely used to
treat numerous types of tumors, including CCA. Nevertheless,
objective response rates of Dox treatment in clinical trials are
modest and the results vary (16,17).
Thus, it is crucial to investigate whether combination approaches
can enhance the antitumor activity of Dox.
GSK-3β is a serine/threonine kinase involved in
numerous disease processes, including tumorigenesis (18). Previous studies have indicated that
GSK-3β inhibition may be a potential therapeutic strategy for
cancer treatment (18,19). However, whether GSK-3β inhibition
affects the antitumor activity of Dox in patients with CCA remains
unknown. The present study investigated the effect of GSK-3β
inhibition on the antitumor activity of Dox in human CCA cells, and
suggested that GSK-3β inhibition promotes Dox-induced human CCA
cells apoptosis, in part by decreasing focal adhesion kinase
(FAK)/protein kinase B (AKT) activity. The present study
demonstrated that a combination of GSK-3β inhibitors and Dox may be
an effective therapeutic strategy for patients with CCA.
Materials and methods
Chemicals and antibodies
Dox and inhibitors of GSK-3β
[6-bromoindirubin-3′-oxime (BIO) and CHIR99021], mTOR (rapamycin
and AZD8055), PI3K (LY294002, PI828 and Wortmannin),
phosphoinositide-dependent protein kinase1 (PDK1; OSU-03012) and
focal adhesion kinase 1 (FAK; PF-573228) were purchased from
Selleck Chemicals. Human PDK1 small interfering (si)RNA,
rapamycin-insensitive companion of mTOR (RICTOR) siRNA, antibodies
against poly [ADP-ribose] polymerase, phosphorylated (p)-AKT (S473;
cat. no. 9271), p-AKT (T308; cat. no. 13038), AKT (cat. no. 9272),
p-ribosomal protein S6 (S6; cat. no. 4856), S6 (cat. no. 2217),
GSK-3β (cat. no. 12456), p-PDK1 (cat. no. 3438), PDK1 (cat. no.
5662), p-70 kDa ribosomal protein S6 kinase (P70S6K; cat. no.
9234), P70S6K (cat. no. 2708), RICTOR (cat. no. 2114), p-FAK (T397;
cat. no. 8556), p-FAK (T576/577; cat. no. 3281), FAK (cat. no.
3285), hemagglutinin (cat. no. 3724), cleaved caspase-3 (cat. no.
9661), caspase-3 (cat. no. 9662) and GAPDH (cat. no. 5174) were
purchased from Cell Signaling Technology, Inc., p-protein kinase C
(PKC; cat. no. ab180848), PKC (cat. no. ab32376) were purchased
from Abcam, and p-GSK-3a/β (cat. no. sc81496) was purchased from
Santa Cruz Biotechnology, Inc.
Cell culture and treatment
Human CCA cell lines QBC939 and RBE (The Cell Bank
of Type Culture Collection of the Chinese Academy of Sciences) were
grown in a humidified incubator at 37°C and 5% CO2 in
RPMI-1640 medium (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% fetal bovine serum (cat. no. 04-001-1ACS;
Biological Industries), 1% penicillin and 1% streptomycin. The
human CCA cells were treated with the following drugs: Dox (2 µM),
LY294002 (30 mM), PI828 (20 nM), BIO (5 µM), CHIR99021 (10 µM),
PF-573228 (10 µM), OSU-03012 (10 µM), rapamycin (100 nM), AZD8055
(1 µM) and Wortmannin (2 µM), for the indicated time periods at
37°C.
RNA interference
Cells were seeded into 6-well plates at a density of
1×106 cells per well and transiently transfected with
100 nM siRNA duplexes using Lipofectamine® 3000
(Invitrogen; Thermo Fisher Scientific, Inc.), according to the
manufacturer's instructions. Following addition of siRNA duplexes
at 37°C for 6 h, the cells were washed and recovered for 30 h.
GSK-3β siRNA sequences were synthesized by ViewSolid Biotech, Inc.,
as follows: siGSK-3β #1, 5′-CUCAAGAACUGUCAAGUAATT-3′; siGSK-3β #2,
5′-CGAGAGCUCCAGAUCAUGATT-3′; siGSK-3β #3,
5′-GCUAGAUCACUGUAACAUATT-3′. A scrambled siRNA sequence
(5′-UUCUCCGAACGUGUCACGTT-3′) was used as an internal negative
control.
Plasmid cell transfection
The constitutively activated AKT plasmid
(myr-HA-AKT) was provided by Professor JinQuan Cheng (Department of
Medical Oncology, Fox Chase Cancer Center, Philadelphia, USA), as
described in previous studies (20,21).
Cells were seeded into 6-well plates at a density of
1×106 cells per well and transfected with 2.5 mg plasmid
per well using Lipofectamine 3000 (Invitrogen; Thermo Fisher
Scientific, Inc.), according to the manufacturer's instructions.
Following the addition of the plasmid at 37°C for 6 h, the cells
were washed and recovered for 30 h.
Western blotting
Cells were lysed in Triton lysis buffer (cat. no.
P0013; Beyotime Institute of Biotechnology), and protein
concentration was determined via the BCA method. Protein samples
were denatured with 4X SDS-loading buffer at 100°C for 8 min and an
equal amount of protein (40 µg) was loaded and separated via
SDS-PAGE on a 10% gel. After being transferred to a PVDF membrane,
5% non-fat milk was used to block non-specific sites at room
temperature for 1 h. proteins were probed with specific antibodies
(1:1,000) at room temperature for 2 h or 4°C overnight. Then, the
blots were incubated at room temperature for 1 h with the following
secondary antibodies: IRDye 800CW Goat anti-Mouse (1:10,000; cat.
no. 926-32210; LI-COR Biosciences) and IRDye 800CW Goat anti-Rabbit
(1:10,000; cat. no. 926-32211; LI-COR Biosciences). Blots were then
visualized in the Odyssey® CLx-1279 system (LI-COR
Biosciences). ImageJ software (version 1.51; National Institutes of
Health) was used to semi-quantify the western blotting results.
Lactate dehydrogenase (LDH) release
assay
QBC939 and RBE cells were seeded into 96-well plates
at a density of 4×104 cells per well. LDH Release Assay
kit (cat. no. C0017) was purchased from Beyotime Institute of
Biotechnology. The ratio of cell death was detected according to
the manufacturer's instructions.
Reverse transcription quantitative
(RT-q)PCR analysis
Total RNA was isolated from transfected human CCA
cells with TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions.
cDNA was generated using M-MLV reverse transcriptase (Promega
Corporation) according to the manufacturer's protocol. RT-qPCR
analysis was performed using SYBR Premix Ex Taq (Takara Bio, Inc.).
PCR was performed as follows: 94°C for 5 min, 35 cycles of 94°C for
20 sec, 60°C for 30 sec and 72°C for 30 sec. RPL13A was used as an
internal control for normalization. The primers sequences are
presented in Table SI. The
results were calculated by the 2−∆∆Cq method (22).
PI3K activity
QBC939 and RBE cells were seeded into 96-well plates
at a density of 4×104 cells per well. The
phosphatidylinositol (3,4,5)-trisphosphate (PIP3)
concentration was detected by PI3-Kinase Activity ELISA:Pico kit
(cat. no. K-1000S; Echelon Biosciences, Inc.), according to the
manufacturer's instructions.
Statistical analysis
Data are presented as the mean ± SD of ≥3
independent repeats. SPSS software (version 17.0; SPSS, Inc.) was
used for statistical analysis using ANOVA followed by Tukey's post
hoc test. P<0.05 was considered to indicate a statistically
significant difference.
Results
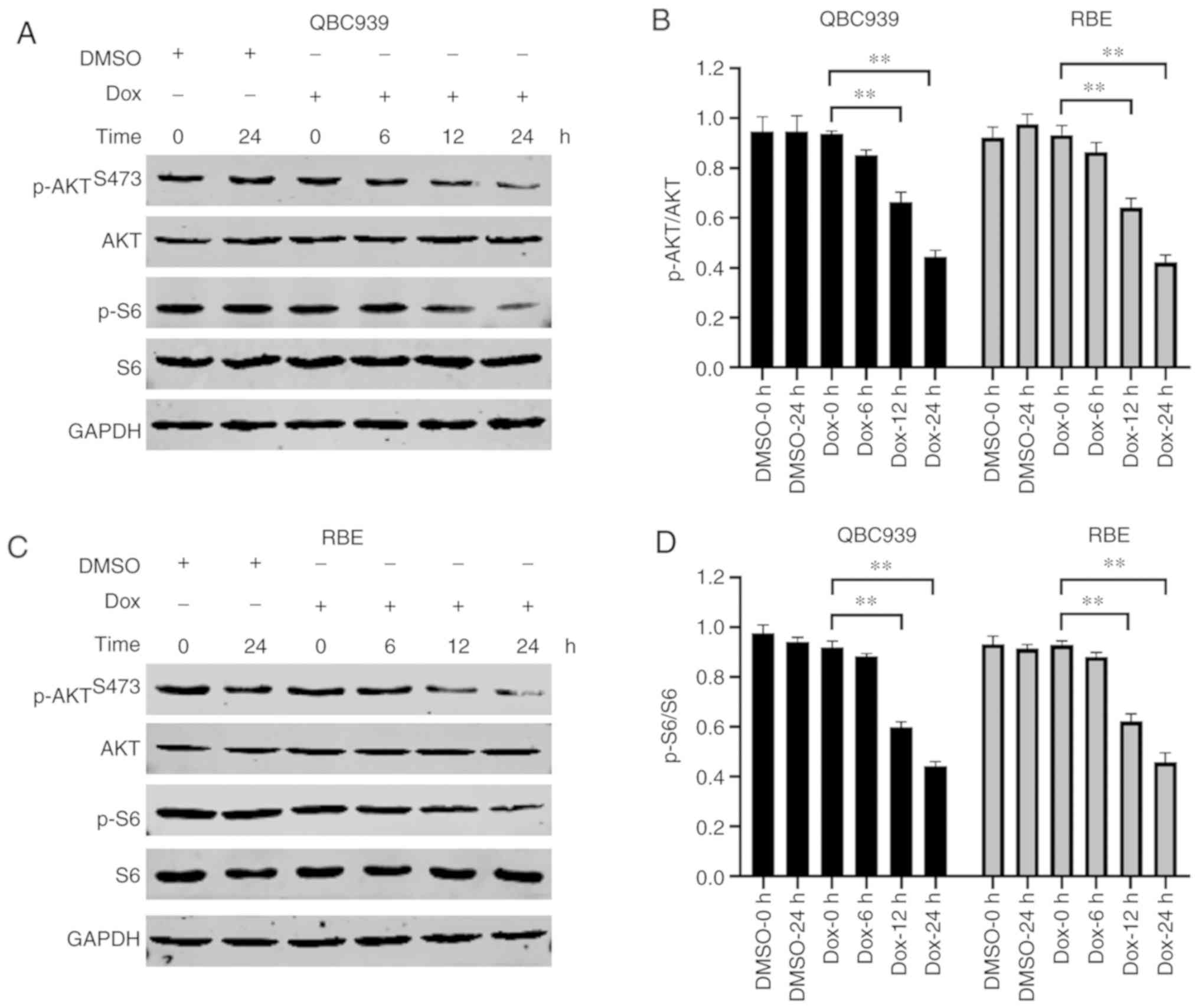
AKT involvement in Dox-treated human
CCA cells
AKT signaling is reported to serve a key role in
drug resistance in numerous types of cancer (23,24).
In order to investigate the role of AKT, phosphorylation levels of
AKT and S6 were detected in Dox-treated human CCA cells.
Phosphorylation levels of AKT and S6, a classical downstream
protein of AKT signaling, were decreased in Dox-treated human CCA
cells (Fig. 1). These results
indicated that AKT protected human CCA cells from Dox
treatment.
AKT inhibition promotes Dox-induced
apoptosis in human CCA cells
In order to investigate the role of AKT in
Dox-treated human CCA cells, QBC939 and RBE cells were treated with
Dox in the presence or absence of PI3K inhibitors LY294002 and
PI828. The western blotting results demonstrated that LY294002 and
PI828 pretreatment increased Dox-induced QBC939 and RBE cell
apoptosis, as indicated by the increased expression of
apoptosis-associated proteins (Fig.
2). The level of p-S6 was used as a marker of the effect of
PI3K inhibitors. In addition, LDH analysis also indicated that AKT
inhibition, via the inhibition of AKT upstream kinase PI3K,
promoted cell death following Dox treatment (Fig. S1). These results suggested that
AKT protected human CCA cells against Dox-induced apoptosis.
GSK-3β inhibition promotes Dox-induced
apoptosis in human CCA cells
Numerous studies have demonstrated that GSK-3β is a
critical molecule involved in cell growth, proliferation and drug
resistance (25,26). In order to determine the role of
GSK-3β in Dox-induced CCA cell apoptosis, western blotting was used
to detect the expression of p-GSK-3β. The level of p-GSK-3β was
unchanged following Dox treatment (Fig. 3A). QBC939 and RBE cells were
treated with Dox in the presence or absence of GSK-3β inhibitors
BIO or CHIR99021. Immunoblotting results demonstrated that BIO
pretreatment increased Dox-induced QBC939 and RBE cell apoptosis
(Fig. 3B and D). Moreover,
CHIR99021 treatment also increased Dox-induced QBC939 and RBE cell
apoptosis (Fig. 3C and E). In
order to further confirm the role of GSK-3β in Dox-mediated QBC939
and RBE cell apoptosis, three siRNAs targeting against GSK-3β (#1,
#2, #3) were transfected into QBC939 cells, and knockdown
efficiency was validated via RT-qPCR and western blotting. Although
all of these siRNAs significantly decreased the expression of
GSK-3β, the target sequence of #3 demonstrated the highest
interference effect (Fig. 3F).
Based on these results, #3 was selected for further investigation.
The data showed that knockdown of GSK-3β expression significantly
enhanced QBC939 and RBE cell apoptosis following Dox treatment
(Fig. 3G and H). Moreover, LDH
analysis also demonstrated that GSK-3β inhibition promoted cell
death following Dox treatment (Fig.
S2). These results indicated that GSK-3β protected human CCA
cells against Dox-induced apoptosis.
 | Figure 3.GSK-3β inhibition promotes
Dox-induced apoptosis in human cholangiocarcinoma cells. (A) QBC939
and RBE cells were treated with Dox (2 µM) for the indicated time
periods, then the cell lysates were subjected to western blotting
and protein expression was semi-quantified. QBC939 and RBE cells
were pre-treated with vehicle (DMSO) or GSK-3β inhibitor (B) BIO (5
µM) or (C) CHIR990219 (10 µM) for 1 h before Dox (2 µM) treatment
for 24 h, then cell lysates were subjected to western blotting and
the change in protein expression was semi-quantified. GAPDH was
used as a loading control. Western blotting results of cells
treated with (D) BIO were semi-quantified. GSK-3β inhibition
promotes Dox-induced apoptosis in human cholangiocarcinoma cells.
Western blotting results of cells treated with (E) CHIR990219 were
semi-quantified. NC and three GSK-3β-targeted siRNAs were
transfected into QBC939 and RBE cells for 6 h, then allowed to
recover for 30 h in normal media, and the cell lysates were
subjected to (F) reverse transcription-quantitative PCR or western
blotting analysis. (G) Following transfection with siRNAs targeting
against NC and GSK-3β #3, QBC939 and RBE cells were treated with
Dox (2 µM) for 24 h, and the cell lysates were subjected to western
blotting and (H) protein expression was semi-quantified.
**P<0.01. Dox, doxorubicin; BIO, 6-bromoindirubin-3′-oxime; NC,
negative control; siRNA, small interfering RNA; C-caspase-3,
cleaved caspase-3; p-, phosphorylated. |
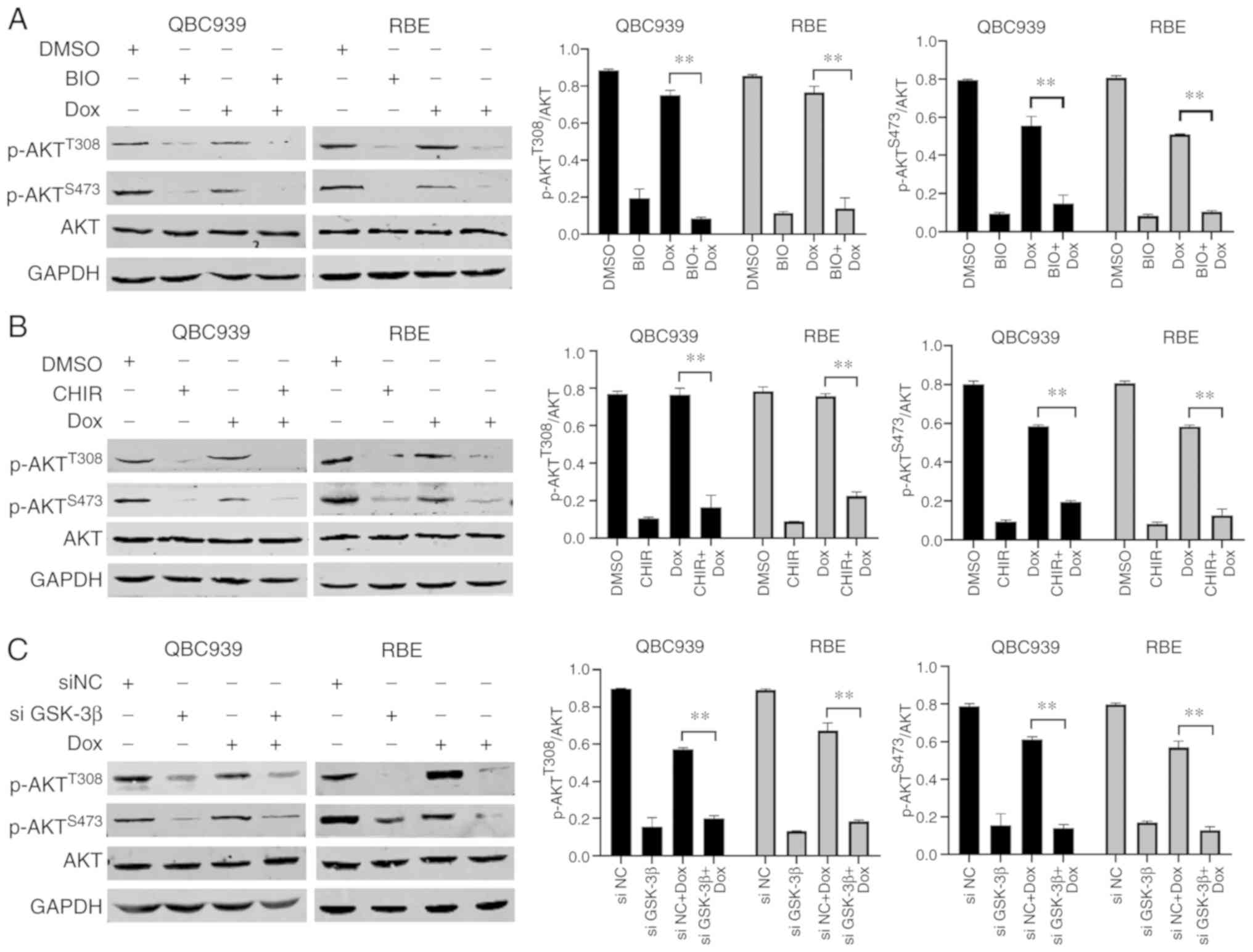
GSK-3β inhibition suppresses AKT
signaling in human CCA cells
In order to determine whether the AKT signaling
pathway was associated with GSK-3β signaling in human CCA cells,
the phosphorylation levels of AKT were detected in Dox-treated
QBC939 and RBE cells following GSK-3β inhibition. BIO and CHIR99021
pretreatment significantly decreased expression levels of p-AKT
(S473 and T308) in Dox-treated RBE and QBC939 cells (Fig. 4A and B). This suggested that GSK-3β
inhibition suppressed the activation of the AKT signaling pathway.
In addition, knockdown of GSK-3β expression levels by siRNA also
significantly decreased the expression levels of p-AKT (S473 and
T308) in Dox-treated QBC939 and RBE cells (Fig. 4C). Altogether, these data indicated
that GSK-3β serves an important role in the activation of the AKT
pathway in Dox-treated human CCA cells.
FAK protects human CCA cells against
Dox-induced apoptosis via AKT signaling
In the present study, although it was demonstrated
that the phosphorylation of T308 and S473 of AKT was regulated by
Serine/Threonine kinase 3-PDK1 and mTOR complex 2 (mTORC2),
respectively, in human CCA cells (Figs. S3 and S4), GSK-3β inhibition did not exhibit a
notable effect on PDK1 and mTORC2 activation (Fig. S5). Since FAK has been reported to
serve a key role in the regulation of AKT activity (27–29),
the present study investigated the role of FAK in AKT activity in
human CCA cells. The levels of p-AKT were detected in human CCA
cells in the presence or absence of an FAK inhibitor. FAK inhibitor
PF-573228 significantly decreased AKT phosphorylation (Fig. 5A). Moreover, inhibiting FAK
promoted Dox-induced human CCA cell apoptosis (Figs. 5B and S6). In order to further confirm the role
of FAK in AKT regulation in human CCA cells, myr-HA-AKT, the
phosphorylation of which is independent of PIP3, was
transiently transfected into QBC939 and RBE cells. The results
demonstrated that FAK inhibitor PF-573228 significantly decreased
the expression of p-AKT (Fig. 5C).
In addition, the concentration of PIP3 was detected
following inhibition of GSK-3β, PI3K or FAK in human CCA cells. The
PI3K inhibitor, Wortmannin, decreased the concentration of
PIP3, whereas GSK-3β or FAK inhibition exhibited no
notable effect on PIP3 concentration in human CCA cells
(Fig. S7). These data indicated
that FAK inhibition promoted Dox-induced apoptosis by suppressing
the activity of AKT independently of PI3K.
 | Figure 5.FAK protects human cholangiocarcinoma
cells against Dox-induced apoptosis via AKT signaling. (A) QBC939
and RBE cells were treated with vehicle (DMSO) or FAK inhibitor
PF-573228 (10 µM) for 12 h, then cell lysates were subjected to
western blotting and protein expression was semi-quantified. (B)
QBC939 and RBE cells were pre-treated with vehicle (DMSO) or FAK
inhibitor, PF (10 µM), for 1 h before being treated with Dox (2 µM)
for 24 h, then cell lysates were subjected to western blotting and
protein expression was semi-quantified. (C) myr-HA-AKT expression
plasmid was transfected into QBC939 and RBE cells for 6 h, then
allowed to recover for 30 h in normal media before PF (10 µM)
treatment for 12 h. Cell lysates were subjected to western blotting
and protein expression was semi-quantified. **P<0.01. FAK, focal
adhesion kinase; Dox, doxorubicin; PF, PF-573228; C-, cleaved; p-,
phosphorylated; PARP, poly [ADP-ribose] polymerase. |
GSK-3β inhibition suppresses FAK
signaling in human CCA cells
In order to investigate whether GSK-3β regulates AKT
activation via FAK in human CCA cells, the phosphorylation levels
of FAK were detected in QBC939 and RBE cells following GSK-3β
inhibitor treatment. The results demonstrated that GSK-3β
inhibitors BIO and CHIR99021 significantly decreased the expression
of p-FAK in QBC939 and RBE cells (Fig.
6A). Knockdown of GSK-3β expression levels by siRNA also
decreased the expression of p-FAK in QBC939 and RBE cells (Fig. 6B), and these protein levels were
also downregulated in the presence of Dox with si-GSK-3β (Fig. 6C). Together, these data indicated
that GSK-3β sustained AKT activation via FAK in human CCA
cells.
Discussion
Although Dox is an important chemotherapeutic drug
used to treat numerous types of tumors, including CCA, resistance
to Dox is a significant impediment to successful chemotherapy
(30–32). Therefore, it is crucial to develop
a combination therapy to improve Dox efficacy. The present study
demonstrated that the combination of GSK-3β inhibitor and Dox may
be a potential therapeutic strategy for treating CCA.
It has been confirmed that abnormal activity of
GSK-3β is involved in a number of disease processes, such as
diabetes and cancer (33–36), but the role of GSK-3β in cancer
chemotherapy is unclear. Although the expression of p-GSK-3β was
not notably altered following Dox treatment, GSK-3β inhibition
effectively promoted human CCA cell death following Dox treatment.
Thus, it was hypothesized that GSK-3β protected human CCA cells
against Dox-induced apoptosis.
GSK-3β inhibition notably decreased the activity of
AKT. As the AKT pathway serves a key role in promoting cancer cell
survival following chemotherapeutic drug treatment (37,38),
GSK-3β inhibition-mediated AKT suppression may be involved in the
synergistic effects of GSK-3β inhibition on Dox-induced human CCA
cell apoptosis. This hypothesis was supported by the present
results, which demonstrated that PI3K inhibitors promoted
Dox-induced apoptosis in human CCA cells. These data indicated that
GSK-3β inhibition promoted Dox-induced cell death, at least in part
via suppression of AKT signaling.
To the best of our knowledge, the mechanism
underlying GSK-3β regulation of the activation of AKT in human CCA
cells has not previously been elucidated. PDK1 and mTORC2 are
responsible for the phosphorylation of AKT at T308 and S473,
respectively (39–43), the present study therefore
investigated the role of GSK-3β in PDK1 and mTORC2 regulation.
According to the current results, GSK-3β had no effect on PDK1 and
mTORC2 in human CCA cells. Thus, the present study demonstrated
that GSK-3β mediated AKT activation in human CCA cells
independently of PDK1 and mTORC2.
FAK, which regulates cell migration, invasion,
adhesion, proliferation and survival, has been reported to serve a
key role in cancer (28,29). Moreover, it has been demonstrated
that FAK is a pivotal upstream regulator of AKT signaling in
various types of tumors (44,45).
In order to investigate whether GSK-3β regulated the activation of
AKT via FAK in human CCA cells, the association between GSK-3β and
FAK in human CCA cells was investigated. GSK-3β inhibition notably
decreased the activation of FAK, indicating that GSK-3β may promote
AKT activation via FAK in human CCA cells. The present results
demonstrated that FAK inhibition decreased AKT activity in human
CCA cells. Thus, it was hypothesized that GSK-3β induces AKT
activation, at least in part, via FAK in human CCA cells. It has
been reported that FAK promotes AKT activation via PI3K activity
regulation (46). Moreover, FAK
inhibition notably decreased the levels of AKT phosphorylation on
S473 and had no effect on PI3K activity, confirming that FAK
sustained AKT activation independently of PI3K. Taken together,
these results indicated that GSK-3β inhibition promoted apoptosis
following Dox treatment by suppressing the FAK/AKT pathway
independently of PI3K activity in human CCA cells. Further
investigation is required to investigate the association between
GSK-3β and FAK in human CCA cells.
In summary, GSK-3β inhibition increased human CCA
cell death following Dox treatment. The synergistic role of GSK-3β
inhibition under Dox treatment was mediated, at least in part, by
FAK/AKT pathway inactivation. These results provided a basis for
the development of a novel combination treatment strategy against
human CCA.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
National Natural Science Foundation of China (grant no. 81472312),
Innovation Team of Education Department of Sichuan Province (grant
no. 16TD0021), Luzhou City-Southwest Medical University Foundation
(grant nos. 2016LZXNYD-T02 and 2015LZCYD-S01-8/15), Sichuan
Province-Luzhou City-Southwest Medical University Foundation (grant
no. 14ZC0070), Sichuan Science and Technology Program (grant nos.
2017JY0134 and 2019YJ0482) and Team of Education Department of
Sichuan Province (grant no. 17ZA0437).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LL, YCX, RYD and YPL conceived and designed the
experiments, analyzed the data and wrote the manuscript. YZ, CYD,
XMX, TZ and YQZ carried out the experiments, and prepared and
analyzed the figures and tables. BX and WJY obtained the study
materials and reagents in preparation for the experiments, and
participated in the design of the experiment and checked the
statistics. All authors reviewed drafts of the paper. All authors
read and approved the manuscript and agree to be accountable for
all aspects of the research in ensuring that the accuracy or
integrity of any part of the work are appropriately investigated
and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Razumilava N and Gores GJ:
Cholangiocarcinoma. Lancet. 383:2168–2179. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Razumilava N and Gores GJ: Combination of
gemcitabine and cisplatin for biliary tract cancer: A platform to
build on. J Hepatol. 54:577–578. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Esnaola NF, Meyer JE, Karachristos A,
Maranki JL, Camp ER and Denlinger CS: Evaluation and management of
intrahepatic and extrahepatic cholangiocarcinoma. Cancer.
122:1349–1369. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Charbel H and Al-Kawas FH:
Cholangiocarcinoma: Epidemiology, risk factors, pathogenesis, and
diagnosis. Curr Gastroenterol Rep. 13:182–187. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Khan SA, Davidson BR, Goldin RD, Heaton N,
Karani J, Pereira SP, Rosenberg WM, Tait P, Taylor-Robinson SD,
Thillainayagam AV, et al: Guidelines for the diagnosis and
treatment of cholangiocarcinoma: An update. Gut. 61:1657–1669.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Everhart JE and Ruhl CE: Burden of
digestive diseases in the United States Part III: Liver, biliary
tract, and pancreas. Gastroenterology. 136:1134–1144. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tyson GL and El-Serag HB: Risk factors for
cholangiocarcinoma. Hepatology. 54:173–184. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen C, Nelson LJ, Avila MA and Cubero FJ:
Mitogen-activated protein kinases (MAPKs) and cholangiocarcinoma:
The Missing Link. Cells. 8:11722019. View Article : Google Scholar
|
|
9
|
Blechacz B, Komuta M, Roskams T and Gores
GJ: Clinical diagnosis and staging of cholangiocarcinoma. Nat Rev
Gastroenterol Hepatol. 8:512–522. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bridgewater J, Galle PR, Khan SA, Llovet
JM, Park JW, Patel T, Pawlik TM and Gores GJ: Guidelines for the
diagnosis and management of intrahepatic cholangiocarcinoma. J
Hepatol. 60:1268–1289. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rizvi S and Gores GJ: Pathogenesis,
diagnosis, and management of cholangiocarcinoma. Gastroenterology.
145:1215–1229. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang H, Yang T, Wu M and Shen F:
Intrahepatic cholangiocarcinoma: Epidemiology, risk factors,
diagnosis and surgical management. Cancer Lett. 379:198–205. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Moeini A, Sia D, Bardeesy N, Mazzaferro V
and Llovet JM: Molecular pathogenesis and targeted therapies for
intrahepatic Cholangiocarcinoma. Clin Cancer Res. 22:291–300. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fornari FA, Randolph JK, Yalowich JC,
Ritke MK and Gewirtz DA: Interference by doxorubicin with DNA
unwinding in MCF-7 breast tumor cells. Mol Pharmacol. 45:649–656.
1994.PubMed/NCBI
|
|
15
|
Momparler RL, Karon M, Siegel SE and Avila
F: Effect of adriamycin on DNA, RNA, and protein synthesis in
cell-free systems and intact cells. Cancer Res. 36:2891–2895.
1976.PubMed/NCBI
|
|
16
|
QuanJun Y, GenJin Y, LiLi W, YongLong H,
Yan H, Jie L, JinLu H, Jin L, Run G and Cheng G: Protective effects
of dexrazoxane against doxorubicin-induced cardiotoxicity: A
metabolomic study. PLoS One. 12:e01695672017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Theodoulou M and Hudis C: Cardiac profiles
of liposomal anthracyclines: Greater cardiac safety versus
conventional doxorubicin? Cancer. 100:2052–2063. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yoshino Y and Ishioka C: Inhibition of
glycogen synthase kinase-3 beta induces apoptosis and mitotic
catastrophe by disrupting centrosome regulation in cancer cells.
Sci Rep. 5:132492015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fang G, Zhang P, Liu J, Zhang X, Zhu X, Li
R and Wang H: Inhibition of GSK-3β activity suppresses HCC
malignant phenotype by inhibiting glycolysis via activating
AMPK/mTOR signaling. Cancer Lett. 463:11–26. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dai R, Chen R and Li H: Cross-talk between
PI3K/Akt and MEK/ERK pathways mediates endoplasmic reticulum
stress-induced cell cycle progression and cell death in human
hepatocellular carcinoma cells. Int J Oncol. 34:1749–1757.
2009.PubMed/NCBI
|
|
21
|
Sun M, Wang G, Paciga JE, Feldman RI, Yuan
ZQ, Ma XL, Shelley SA, Jove R, Tsichlis PN, Nicosia SV and Cheng
JQ: AKT1/PKBalpha kinase is frequently elevated in human cancers
and its constitutive activation is required for oncogenic
transformation in NIH3T3 cells. Am J Pathol. 159:431–437. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Polivka J Jr and Janku F: Molecular
targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol
Ther. 142:164–175. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Guerrero-Zotano A, Mayer IA and Arteaga
CL: PI3K/AKT/mTOR: Role in breast cancer progression, drug
resistance, and treatment. Cancer Metastasis Rev. 35:515–524. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Edderkaoui M, Chheda C, Soufi B, Zayou F,
Hu RW, Ramanujan VK, Pan X, Boros LG, Tajbakhsh J, Madhav A, et al:
An Inhibitor of GSK3B and HDACs kills pancreatic cancer cells and
slows pancreatic tumor growth and metastasis in mice.
Gastroenterology. 155:1985–1998.e5. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shen W, Taylor B, Jin Q, Nguyen-Tran V,
Meeusen S, Zhang YQ, Kamireddy A, Swafford A, Powers AF, Walker J,
et al: Inhibition of DYRK1A and GSK3B induces human β-cell
proliferation. Nat Commun. 6:83722015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shang N, Arteaga M, Zaidi A, Stauffer J,
Cotler SJ, Zeleznik-Le NJ, Zhang J and Qiu W: FAK is required for
c-Met/β-catenin-driven hepatocarcinogenesis. Hepatology.
61:214–226. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Levy A, Alhazzani K, Dondapati P, Alaseem
A, Cheema K, Thallapureddy K, Kaur P, Alobid S and Rathinavelu A:
Focal adhesion kinase in ovarian cancer: A potential therapeutic
target for platinum and taxane-resistant tumors. Curr Cancer Drug
Targets. 19:179–188. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sulzmaier FJ, Jean C and Schlaepfer DD:
FAK in cancer: Mechanistic findings and clinical applications. Nat
Rev Cancer. 14:598–610. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Speth PA, van Hoesel QG and Haanen C:
Clinical pharmacokinetics of doxorubicin. Clin Pharmacokinet.
15:15–31. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rivankar S: An overview of doxorubicin
formulations in cancer therapy. J Cancer Res Ther. 10:853–858.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Genovese I, Fiorillo A, Ilari A,
Masciarelli S, Fazi F and Colotti G: Binding of doxorubicin to
Sorcin impairs cell death and increases drug resistance in cancer
cells. Cell Death Dis. 8:e29502017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gao C, Holscher C, Liu Y and Li L: GSK3: A
key target for the development of novel treatments for type 2
diabetes mellitus and Alzheimer disease. Rev Neurosci. 23:1–11.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Juhaszova M, Zorov DB, Yaniv Y, Nuss HB,
Wang S and Sollott SJ: Role of glycogen synthase kinase-3beta in
cardioprotection. Circ Res. 104:1240–1252. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fu Y, Hu D, Qiu J, Xie X, Ye F and Lu WG:
Overexpression of glycogen synthase kinase-3 in ovarian carcinoma
cells with acquired paclitaxel resistance. Int J Gynecol Cancer.
21:439–444. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kawazoe H, Bilim VN, Ugolkov AV, Yuuki K,
Naito S, Nagaoka A, Kato T and Tomita Y: GSK-3 inhibition in vitro
and in vivo enhances antitumor effect of sorafenib in renal cell
carcinoma (RCC). Biochem Biophys Res Commun. 423:490–495. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Szymonowicz K, Oeck S, Malewicz NM and
Jendrossek V: New insights into protein kinase B/Akt signaling:
Role of localized akt activation and compartment-specific target
proteins for the cellular radiation response. Cancers (Basel).
10:782018. View Article : Google Scholar
|
|
38
|
Brognard J, Clark AS, Ni Y and Dennis PA:
Akt/protein kinase B is constitutively active in non-small cell
lung cancer cells and promotes cellular survival and resistance to
chemotherapy and radiation. Cancer Res. 61:3986–3997.
2001.PubMed/NCBI
|
|
39
|
Kilic U, Caglayan AB, Beker MC, Gunal MY,
Caglayan B, Yalcin E, Kelestemur T, Gundogdu RZ, Yulug B, Yılmaz B,
et al: Particular phosphorylation of PI3K/Akt on Thr308 via PDK-1
and PTEN mediates melatonin's neuroprotective activity after focal
cerebral ischemia in mice. Redox Biol. 12:657–665. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bellacosa A, Chan TO, Ahmed NN, Datta K,
Malstrom S, Stokoe D, McCormick F, Feng J and Tsichlis P: Akt
activation by growth factors is a multiple-step process: The role
of the PH domain. Oncogene. 17:313–325. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yang G, Murashige DS, Humphrey SJ and
James DE: A Positive Feedback Loop between Akt and mTORC2 via SIN1
Phosphorylation. Cell Rep. 12:937–943. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sarbassov DD: mTOR-rictor Phosphorylation
and Regulation of Akt/PKB by the Rictor-mTOR Complex. Science.
307:1098–1101. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chen CH, Shaikenov T, Peterson TR,
Aimbetov R, Bissenbaev AK, Lee SW, Wu J, Lin HK and Sarbassov dos
D: ER stress inhibits mTORC2 and Akt signaling through
GSK-3β-mediated phosphorylation of rictor. Sci Signal. 4:ra102011.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wang S and Basson MD: Protein kinase B/AKT
and focal adhesion kinase: Two close signaling partners in cancer.
Anticancer Agents Med Chem. 11:993–1002. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Paul R, Luo M, Mo X, Lu J, Yeo SK and Guan
JL: FAK activates AKT-mTOR signaling to promote the growth and
progression of MMTV-Wnt1-driven basal-like mammary tumors. Breast
Cancer Res. 22:592020. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Casar B, Rimann I, Kato H, Shattil SJ,
Quigley JP and Deryugina EI: In vivo cleaved CDCP1 promotes early
tumor dissemination via complexing with activated beta1 integrin
and induction of FAK/PI3K/Akt motility signaling. Oncogene.
33:255–268. 2014. View Article : Google Scholar : PubMed/NCBI
|