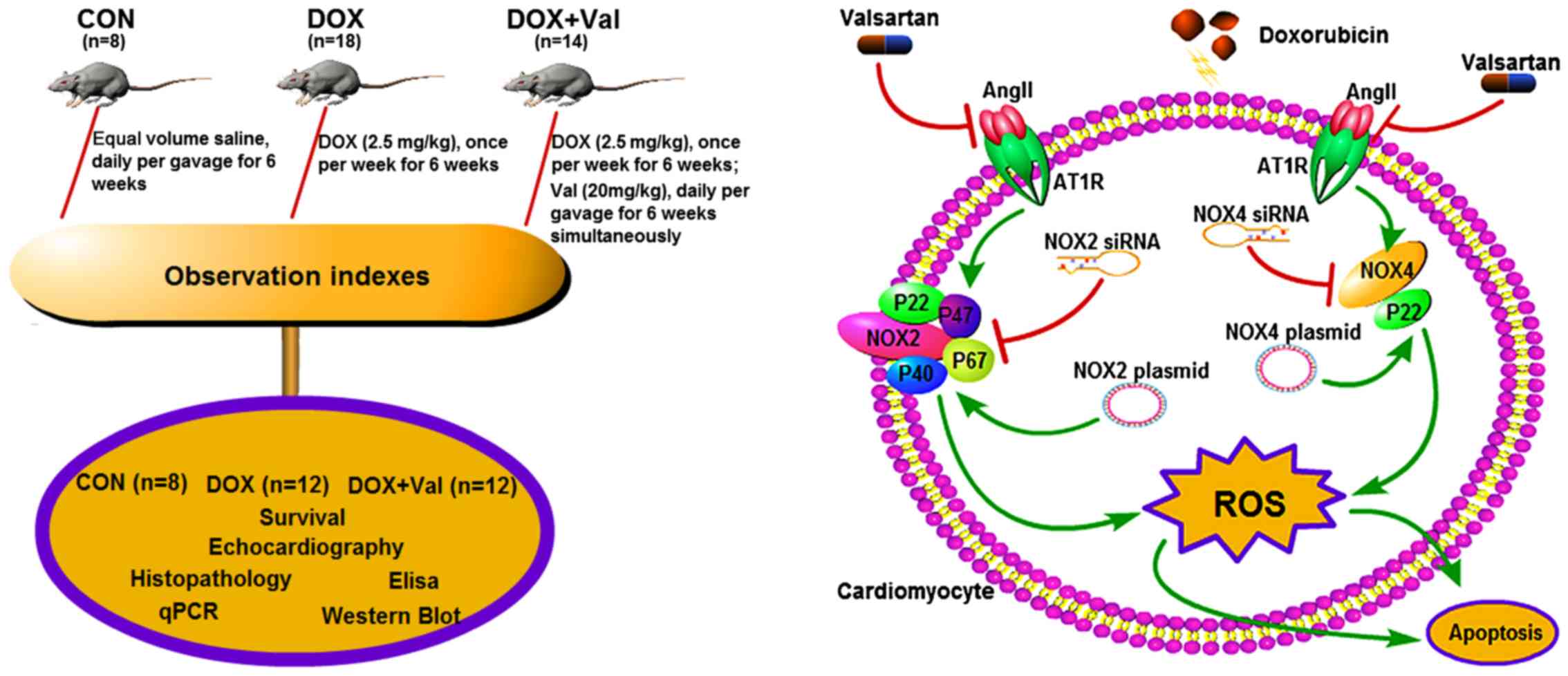
Introduction
Doxorubicin (DOX) is highly effective treatment
against acute lymphoblastic and myeloblastic leukemia, and numerous
types of solid tumors, including breast cancer, sarcomas and
childhood solid tumors (1).
However, clinical use of DOX is hampered by its potential
cardiotoxicity (1–5). Angiotensin receptor blockers (ARBs)
may attenuate DOX-induced cardiomyopathy (6–9);
however, the underlying mechanisms are not fully understood and
there are very few studies examining the effects of simultaneous
treatment with DOX and ARBs for the prevention of DOX-induced
myocardial injury. Valsartan (Val), which is a type of ARB, is
widely used to treat patients with hypertension and heart failure
(10). A clinical observational
study demonstrated that Val could prevent the acute cardiotoxicity
induced by cyclophosphamide, doxorubicin, vincristine and
prednisolone, which are standard chemotherapeutic options for
treatment of non-Hodgkin lymphoma (11). More recently, Sakr et al
(12) investigated the effect of
Val on DOX-induced cardiotoxicity in rats, and found that
concurrent or post-but not pre-treatment with Val attenuated
DOX-induced cardiotoxicity by inhibiting oxidative stress,
apoptosis and senescence. Another study reported that Val
alleviated DOX-induced cardiac dysfunction via regulation of the
TGF-β signaling pathway (13).
Excessive reactive oxygen species (ROS) production
is a known risk factor responsible for the initiation and
development of heart failure (14,15)
and DOX-induced cardiotoxicity. NAD(P)H oxidase (NOX) is one of
several contributing sources responsible for increased ROS
generation (16). NOX may be
activated by growth factors or inflammatory cytokines (17–19).
Previous studies confirmed that angiotensin II may stimulate ROS
production by activating NOX (20–23).
Therefore, it may be hypothesized that reduced NOX and ROS
signaling is involved in the beneficial effects of ARB on
attenuating DOX-induced cardiotoxicity.
The aim of the present study was to test the
hypothesis that simultaneous treatment with Val could prevent
DOX-induced myocardial injury by downregulating myocardial NOX
expression and reducing ROS production in rats (Fig. 1).
Materials and methods
Reagents
DOX was purchased from Zhejiang Hisun Pharmaceutical
Co., Ltd. Val was purchased from Beijing Novartis Pharmaceutical
Co., Ltd. (https://www.novartis.com.cn/).
Animal model and study protocol
A total of 40 specific pathogen free-grade
8-week-old male Sprague-Dawley rats (body weight, 200–250 g) were
purchased from the Experimental Animal Center of Dalian Medical
University. Rats were housed in 530 cm2 cages with
wood-shaving bedding (2 rats per cage) in a temperature-controlled
room (25±2°C), the humidity was maintained at 50–70%, the noise was
<85 decibels, with 14 h of light and 10 h of darkness every day.
Rats were fed under standard conditions with free access to food
and drinking water. Rats were randomly divided into three groups:
i) Control group (CON, n=8), rats treated with equal volume saline
daily via gavage for 6 weeks; ii) DOX group (n=18) rats received
intraperitoneal DOX (2.5 mg/kg) injection once per week for 6
weeks; iii) DOX+Val group (n=14), rats received intraperitoneal DOX
(2.5 mg/kg) injection once per week plus Val (20 mg/kg) daily via
gavage for 6 weeks. After another 4 weeks, surviving rats underwent
echocardiography examination. Subsequently, rats were sacrificed
under deep anesthesia (intramuscular ketamine hydrochloride
injection, 100 mg/kg), and the heart was isolated and weighed. The
atria and right ventricle were separated from the left ventricle
(LV), and the LV was cut into three sections along the LV long-axis
at a thickness of 3 mm. The middle section of the LV was processed
for histological examination. The basal and the apical sections
were stored at −80°C for immunohistological and biochemical
analysis. The experimental protocol is presented in Fig. 1. All experiments were performed in
compliance with the ARRIVE guidelines as well as the Guide for the
Care and Use of Laboratory Animals of the National Academy of
Sciences. The Institutional Animal Research and Ethics Committee of
Dalian University approved the protocols of the animal
experiments.
Echocardiography
Under light anesthesia (intramuscular ketamine
hydrochloride injection, 22 mg/kg), rats underwent echocardiography
using a Vivid E9 dimension system (General Electric Company),
equipped with a 12.0 MHz transducer. Two-dimensional and M-mode
echocardiography images were obtained in the parasternal long-axis
and short-axis views of the heart. All measurements were performed
online with optimal images from >10 cardiac cycles taken by an
experienced sonographer who was blinded to the study protocol and
grouping (24). Left ventricular
end-diastolic (LVEDD) and end-systolic diameters (LVESD) were
measured with M-mode in the parasternal short-axis images at the
papillary muscle level. Left ventricular fractional shortening
(LVFS) was calculated as follows: LVFS (%) = (LVEDD-LVESD)/LVEDD
×100. Left ventricular ejection fraction (LVEF) was calculated
according to the Teichholz formula (25).
Histopathological evaluation
The LV tissue assigned for histological examination
(middle section) was fixed in 4% paraformaldehyde for 24 h at 4°C,
embedded in paraffin, sectioned into 5 µm slices and stained with
Masson (Shanghai Bogoo Biotechnology Co., Ltd.; http://www.bgswkj.com/) stain A for 15 min and stain B
for 20 min. Then, slices were stained with Picrosirius Red (Beijing
Solarbio Science & Technology Co., Ltd.) for 1 h at room
temperature and observed with a light microscope (magnification,
×100). Interstitial collagen volume fraction (CVF) was determined
using Image-Pro Plus 6.0 (Media Cybernetics, Inc.). CVF was
calculated as follows: CVF (%) = area of stained collagen/total
area of field of vision ×100.
ELISA
Specimens from left ventricular tissues of rats in
each group were weighed and cut into pieces, mixed with pre-cooled
PBS at a ratio of 1:9 (weight:volume) to prepare the tissue
homogenate. Myocardial levels of renin (Sigma-Aldrich; Merck KGaA;
cat. no. RAB1162), TNF-α (Sigma-Aldrich; Merck KGaA; cat. no.
RAB0479), IL-6 (Sigma-Aldrich; Merck KGaA; cat. no. RAB0311), brain
natriuretic peptide (BNP; Sigma-Aldrich; Merck KGaA; cat. no.
RAB0386), aldosterone (ALD; Abcam; cat. no. Ab136933),
malondialdehyde (MDA; Abcam; cat. no. Ab238537), ROS (Jianglai Bio;
http://www.laibio.com/; cat. no. JL21051),
superoxide dismutase (SOD; Jianglai Bio; cat. no. JL11065), NOX1
(Jianglai Bio; cat. no. JL36449), NOX2 (Jianglai Bio; cat. no.
JL50110) and NOX4 (Jianglai Bio; cat. no. JL23194) were measured.
ELISA was performed according to the manufacturer's protocol and a
DNA Eraser was used (Takara Bio, Inc.).
Reverse transcription-quantitative
(RT-q)PCR
All primer sequences were designed and synthesized
by Sangon Biotech Co., Ltd., which are presented in Table I. PrimeScript™ RT Reagent kit with
a gDNA Eraser (Takara Bio, Inc.) was used for reverse-transcription
at 37°C for 15 min and 85°C for 5 sec. qPCR was performed using
SYBR®Premix Ex Taq™ II (Takara Bio, Inc.). qPCR was
performed as described previously (26). Briefly, qPCR was performed in 20 µl
reaction containing 10 µl SYBR Premix Ex Taq II (Tli RNaseH Plus),
Bulk, 0.8 µl of each primer (10 µM), 0.4 µl ROX Reference Dye (50X)
and 4 µl dH2O and 2 µl cDNA. The thermocycling
conditions were: Pre-denaturation for 30 sec at 95°C; followed by
30 cycles of 95°C for 5 sec and 60°C for 30 sec. Relative
quantities of all targets in test samples were normalized to the
respective GAPDH levels. 2−∆∆Cq was calculated as
follows (27): ∆Cq=DOX group
(target gene Cq value-made in Cq)-control group (target gene Cq
value-made in Cq value). For different groups of the genes, the
internal change ratio=2−∆∆Cq. The expression levels were
estimated using the integrated optical density (OD) of the positive
cells. Integrated OD was the average cumulative OD of the positive
staining area of each group determined by Image Pro Plus version
6.0 (Media Cybernetics, Inc.). RT-qPCR experiments were performed
in triplicate.
 | Table I.Primer sequences for reverse
transcription-quantitative PCR. |
Table I.
Primer sequences for reverse
transcription-quantitative PCR.
| Gene | Primer sequence
(5′→3′) |
|---|
| Bax | F:
GTGGTTGCCCTCTTCTACTTTG |
|
| R:
CCACAAAGATGGTCACTGTCTG |
| Collagen I | F:
CGGTGGTTATGACTTCAG CTTC |
|
| R:
AGAGGGCTGAGTGGGGAAC |
| BNP | F:
GCAGGCCAGCAGGGTCCACTACAC |
|
| R: GACCTC
GCCATTCCGCTGATTCT |
| Beclin-1 | F:
GACAAATCTAAGGAGTTGCCG |
|
| R:
AGAACTGTGAGGACACCCA |
| BCL2 | F:
AGTTCGGTGGGGTCATGTGTG |
|
| R:
CCAGGTATGCACCCAGAGTG |
| NOX4 | F:
GAACCCAAGTTCCAAGCTCA |
|
| R: GCA
CAAAGGTCCAGAAATCC |
| NOX1 | F:
GGCATCCCTTTACTCTGACCT |
|
| R:
TGCTGCTCGAATATGAATG |
| NOX2 | F:
TGTGCACCACGATGAGGAGAAAGATG |
|
| R:
ATTCTGGTGTTGGGGTGTGACTFGC |
| Caspase-3 | F: CTTTGCGCCATGCTG
AAACT |
|
| R:
TCAAATTCCGTGGCCACTT |
| MMP9 | F:
TCCCCAGAGCGTTACTCG CT |
|
| R: ACCTGG
TTCACCCGGTTGTG |
| MMP2 | F:
GAGTAAGAACAAGAAGACATACATC |
|
| R:
GTAATAAGCACCCTTGAA GAAATAG |
| ANP | F: GGGTGTGAACCA
CGAGAAAT |
|
| R:
ACTGTGGTCATGAGCCCTTC |
| β-MHC | F: GGGCAA
AGGCAAAGCAAAGA |
|
| R:
AAAGTGAGGGTGCGTGGAGC |
| BGN | F:
ACAACAAGCTGTCCCGGGTG |
|
| R: AGC
CCATGGGGCAGAAATCG |
| TPM1 | F:
GCTGGTTGAGGAGGAGTT |
|
| R:
ACTTGTTCGTCACCGTTT |
| Collagen III | F:
GGAGTCGGAGGAATGGGTG |
|
| R:
ATTGCGTCCATCAAAGCCTC |
| CORIN | F: GTACAG
TGCGGTGTCCAACA |
|
| R:
ATCCTGTCAATCCTACCCCC |
| TGF-β1 | F:
CCAACTATTGCTTCAGCTCCA |
|
| R:
GTGTCCAGGCTCCAAATGT |
| GDF15 | F:
CTGCTCTTGCTGCTTCTGCT |
|
| R:
TCGTCCGGGTTGAGTTGG |
| POSTN | F:
AGGAGCCGTGTTTGAGACCAT |
|
| R: CGGTGAAAGTGGTTT
GCTGTTT |
| GAPDH | F:
GGTGCTGAGTATGTCGTGGAGT |
|
| R: CACAGT
CTTCTGAGTGGCAGTG |
Western blot analysis
Myocardial protein expression levels of NOX1, NOX2
and NOX4 were determined by western blotting as described
previously (28). Extracted
membrane proteins from LV tissue was quantified using a BCA protein
assay kit and protein (25 µg/lane) was separated via SDS-PAGE on a
20% gel, and subsequently transferred to a polyvinylidene
difluoride membrane. Then, the membrane was blocked with 5% BSA
blocking buffer (Nanjing KeyGen Biotech Co., Ltd.) at room
temperature for 1 h, and incubated overnight at 4°C with rabbit
anti-human NOX1 (1:5,000; cat. no. GTX103888), rabbit anti-human
NOX2 (1:5,000; cat. no. GTX133715) and rabbit anti-human NOX4
antibodies (1:1,000; cat. no. GTX121929; all purchased from
GeneTex, Inc.), and the loading control rabbit anti-human
Na+/K±ATPase α-1 antibodies (1:10,000; cat.
no. ab76020; Abcam), followed by secondary goat anti-rabbit IgG
(1:5,000; cat. no. ab6721; Abcam) or at room temperature for 1.5 h.
The signal was developed by applying goat anti-rabbit IgG
conjugated to horseradish peroxidase, and thereafter exposed to
X-ray films that were scanned and determined by ImageJ software
v1.8.0 (National Institutes of Health) to quantify protein
expression.
In vitro experiments
Cell culture
H9C2 cardiomyocytes (China Center for Type Culture
Collection) were maintained at 37°C in a humidified incubator with
5% CO2. Cells were cultured in DMEM (Gibco; Thermo
Fisher Scientific, Inc.) supplemented with 10% FBS (AusGeneX,
Ltd.), 100 U/ml penicillin and 100 µg/ml streptomycin, 0.25%
trypsin and 0.02% EDTA digestive solution were used to trypsinize
the cells. After the third passage, H9C2 cells were harvested for
further experiments.
Downregulation of NOX2 and NOX4 via
small interfering RNA (siRNA) transfection
siRNA targeting NOX2 (20 µM; 10 µl/well), siRNA
targeting NOX4 (20 µM; 10 µl/well) and the negative random siRNA
(20 µM; 10 µl/well; Shanghai GenePharma Co., Ltd.) were transiently
transfected into H9C2 cells (1×105/well) using
Lipofectamine® 2000 (Thermo Fisher Scientific, Inc.),
according to the manufacturer's protocols and a previous study
(29). The sequences of the siRNAs
were as follows: NOX2-rat-663 siRNA sense,
5′-CCAUGGAGCUGAACGAAUUTT-3′ and anti-sense,
5′-AAUUCGUUCAGCUCCAUGGTT-3′; and NOX4-rat-576 siRNA sense,
5′-GCUUCUACCUAUGCAAUAATT-3′ and anti-sense,
5′-UUAUUGCAUAGGUAGAAGCTT-3′.
Upregulation of NOX2 and NOX4 via
construction and transfection of eukaryotic overexpression
plasmids
Total RNA was extracted from cultured H9C2 cells
using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.).
cDNA was synthesized and the PCR product was inserted into a
pMD18-T plasmid (Takara Bio, Inc.). Insertion was confirmed using
the restriction enzymes XhoI and KpnI (Takara Bio,
Inc.) and DNA sequencing. The NOX2 and NOX4 genes were cloned into
a pEX-4 (pGCMV/MCS/T2A/EGFP/Neo) vector (Shanghai GenePharma Co.,
Ltd.). Recombinant plasmids were obtained and identified by
digestion with XhoI and KpnI. The following primers
were designed with specific restriction enzyme sites to clone the
complete coding region of NOX2 and NOX4: NOX2 forward,
5′-GCGCTACCGGACTCAGATCTCGAGGCCACCATGGGGAACTGGGCTGTGAATGAGGGACTC-3′
(XhoI) and reverse,
5′-ACTTCCTCTGCCCTCGGTACCGAAGTTTTCCTTGTTGAAGATGAAGTGGACTCCACGTGG-3′
(KpnI); and NOX4 forward,
5′-GCTACCGGACTCAGATCTCGAGGCCACCATGGCGCTGTCCTGGAGGAGCTGGCTGGCCAA-3′
(XhoI) and reverse,
5′-ACTTCCTCTGCCCTCGGTACCGCTGAAAGATTCTTTATTGTATTCAAATTTTGTCCCATA-3′
(KpnI). The optimized PCR amplification conditions were:
Annealing at 58°C and extension at 72°C for 35 cycles. H9C2 cells
(1×105/well) were transfected with pEX-4-NOX2/NOX4 (1
µg/µl; 2.5 µl/well) or empty vectors (1 µg/µl; 2.5 µl/well), which
was added to balance the total amount of transfected DNA using
Lipofectamine 2000 according to the manufacturer's protocols and a
previous study (30).
Untransfected cells were used as controls. Cells were cultured for
24 h before use in subsequent experiments.
Cell viability
H9C2 cells (3×104/well) were treated with
DOX (0.1, 0.3, 0.5, 1, 3, 5, 10, 30 or 50 µM) and Val (0.1, 0.5, 1,
3, 5, 7.5, 10, 15 or 30 µM) for 12, 24, 48 or 72 h) with 5%
CO2, at 37°C in an incubator, and subsequently harvested
for further molecular and biochemical analyses. Untreated H9C2
cells and DMSO-pre-treated H9C2 cells were used as the control
groups. All in vitro experiments were performed in
triplicate. Cell viability was determined using a modified MTT
assay as described previously (31). Briefly, MTT solution in PBS (5
mg/ml) was added to each well at a final concentration of 0.05%.
After 3 h, the formazan precipitate was dissolved in DMSO. The
absorbance was measured at 570 and 620 nm (background) using
Epoch™microplate spectrophotometer (BioTek Instruments, Inc.).
In vitro measurement of intracellular
ROS production
To measure intracellular ROS production, H9C2 cells
(6×104/well) were seeded in a 24-well culture plate and
incubated with dichlorofluorescein diacetate (DCFH-DA; 10 mM;
Sigma-Aldrich; Merck KGaA) for 1 h at 37°C in the dark. After the
various aforementioned treatments, cells were washed immediately
and resuspended in PBS solution. Viable cells incorporate
2′,7′-DCFH-DA, which is not fluorescent, but in the presence of
ROS, DCFH-DA reacts with oxygen species to produce the fluorescent
dye 2′,7′-DCF. Fluorescence emission was measured by flow cytometry
(BD FACSCanto™ II Flow Cytometer; BD Biosciences) using a 525 nm
band pass filter, which provides an index of the intracellular
oxidative metabolism (32). ROS
levels were detected by performing a DCFH assay and
semi-quantitative analysis (BD FACSDiva software; version 7.0; BD
Biosciences). The cells were divided into ten groups and treated as
follows: i) Control group included H9C2 cells without any
treatment; ii) H9C2 cells exposed to 1 µM DOX for 24 h; iii) cells
exposed to 5 µM Val for 1 h; iv) cells pre-treated with 5 µM Val
for 1 h followed by treatment with 1 µM DOX for 24 h; v) cells
transiently transfected with NOX2-siRNA for 24 h followed by
treatment with 1 µM DOX for 24 h; vi) cells transiently transfected
with NOX4-siRNA for 24 h followed by treatment with 1 µM DOX for 24
h; vii) cells transiently transfected with negative siRNA for 24 h
followed by treatment with 1 µM DOX for 24 h; viii) cells
transiently transfected with an empty vector for 24 h; ix)
NOX2-overexpressing cells treated with 1 µM DOX for 24 h; and x)
NOX4-overexpressing cells were treated with 1 µM DOX for 24 h.
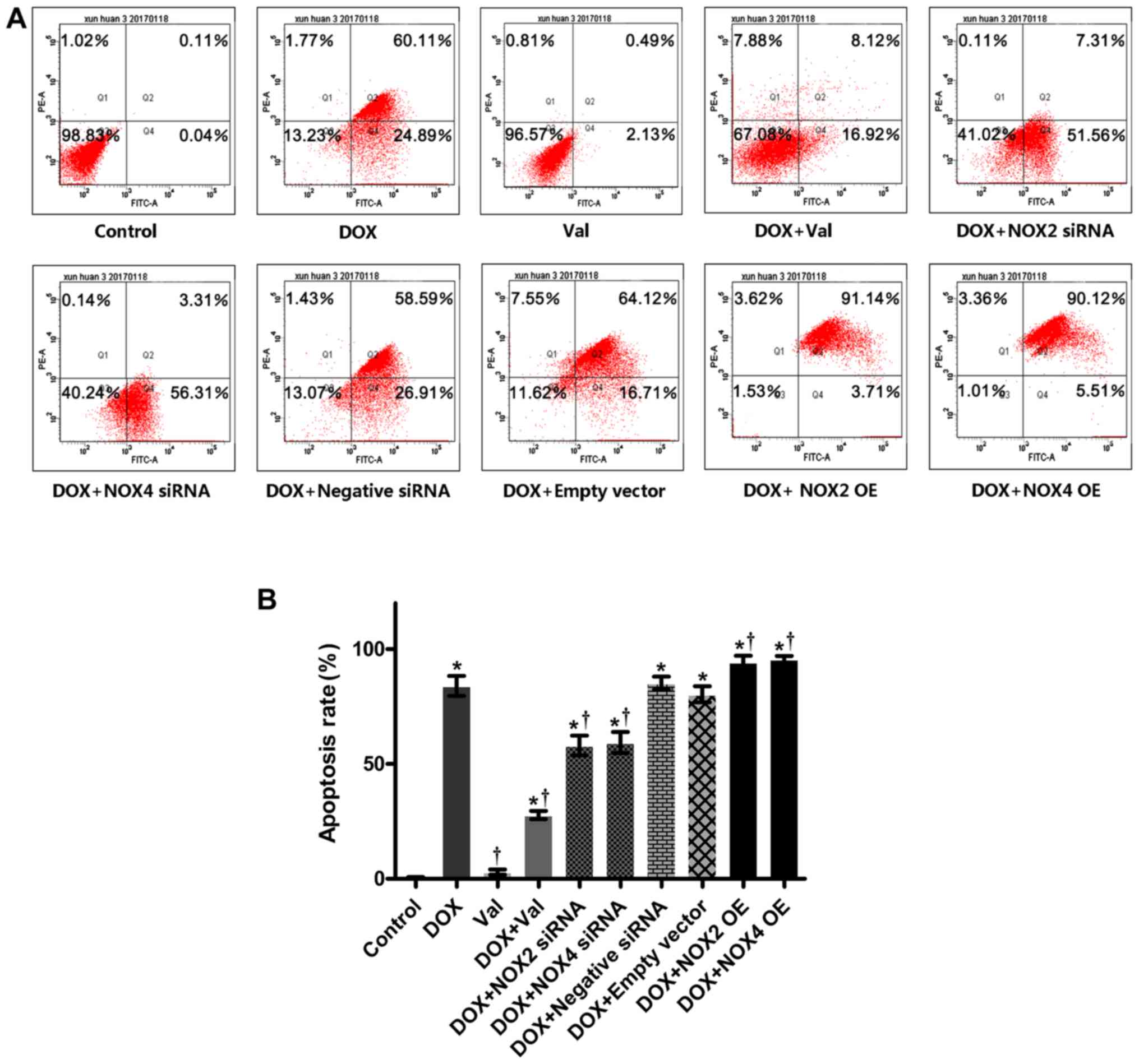
Apoptosis and flow cytometry
Cardiomyocyte apoptosis was evaluated using flow
cytometry and DNA electrophoresis. A total of 1×105 H9C2
cells/well were seeded in 12-well culture plates and cardiomyocytes
under various treatments were trypsinized, washed with PBS
solution, centrifuged at 800 × g for 6 min at 4°C, resuspended in
ice-cold 70% ethanol/PBS, centrifuged at 800 × g for another 6 min
at 4°C, and resuspended in PBS. Cells were then incubated with
propidium iodide (PI) and FITC-labeled Annexin V for 30 min at
37°C. Cells were washed to remove excess PI and Annexin V, and then
analyzed by flow cytometry using a FACSCalibur™ flow cytometer (BD
Biosciences) with a 488 nm argon laser light source, a 525 nm band
pass filter for FITC fluorescence, and a 625 nm band pass filter
for PI-fluorescence. Flow cytometry data were analyzed using
CellQuest software version 3.0 (BD Biosciences). Dot plots of PI
fluorescence (y-axis) vs. FITC fluorescence (x-axis) are presented.
The assessment of apoptosis rate was the sum of early and late
apoptosis. Apoptosis analysis was performed in the aforementioned
ten groups that were used to measure intracellular ROS
production.
Statistical analysis
Data are presented as the mean ± standard deviation
and were analyzed using one-way or two-way ANOVA followed by
Bonferroni's pos hoc comparisons using SPSS software (version 20.0;
IBM Corp.). Kaplan-Meier curves were used to plot and estimate
survival. P<0.05 was considered to indicate a statistically
significant difference.
Results
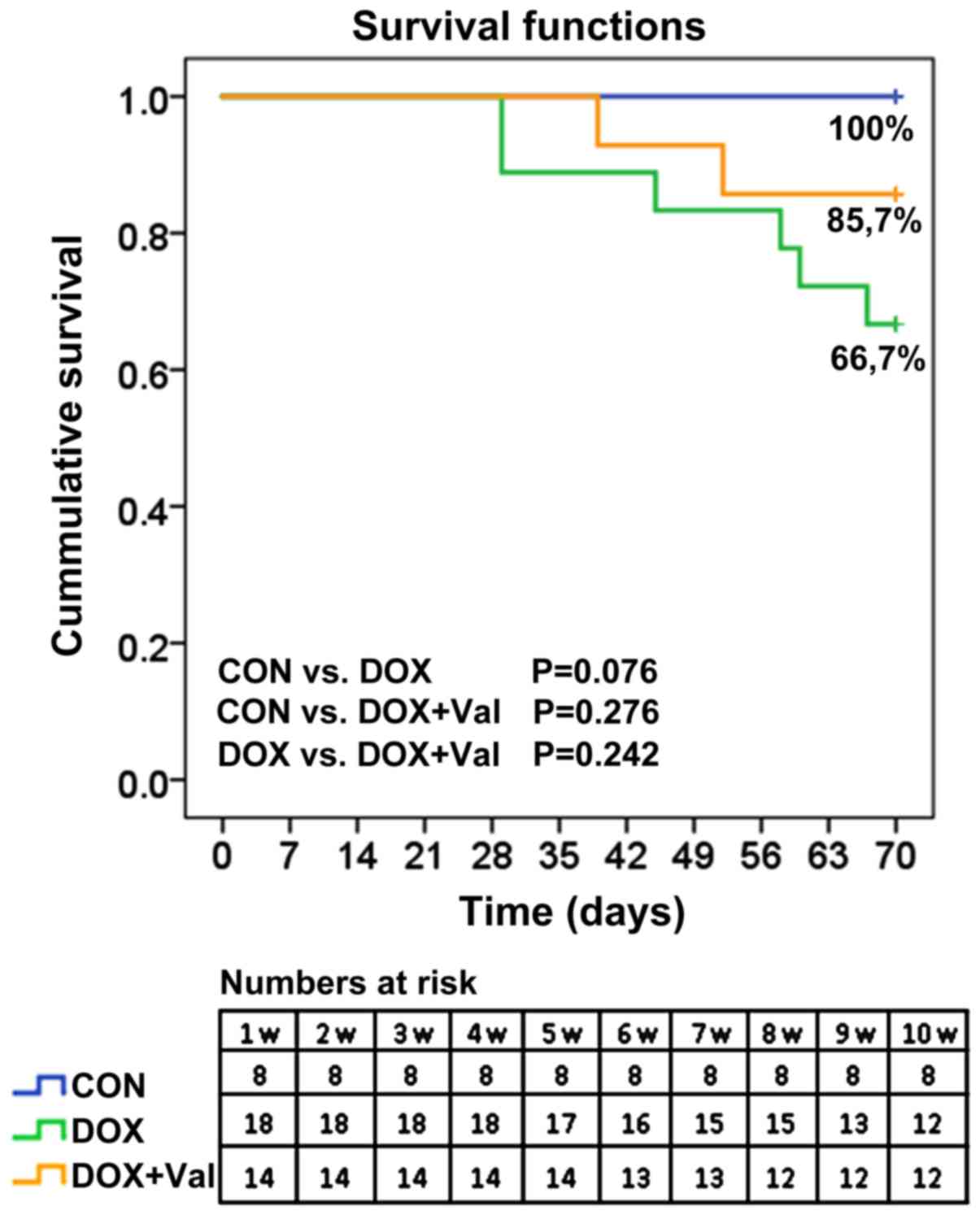
Cumulative survival rates of rats
All rats (n=8) in the CON group survived to the end
of the study. In the DOX group, 16 of the 18 (88.9%) rats were
alive at week 7 and 12 (66.7%) rats were alive at week 10. In the
DOX+Val group, 13 of the 14 (92.9%) rats were alive at week 6 and
12 (85.7%) rats were alive at week 10 (Fig. 2).
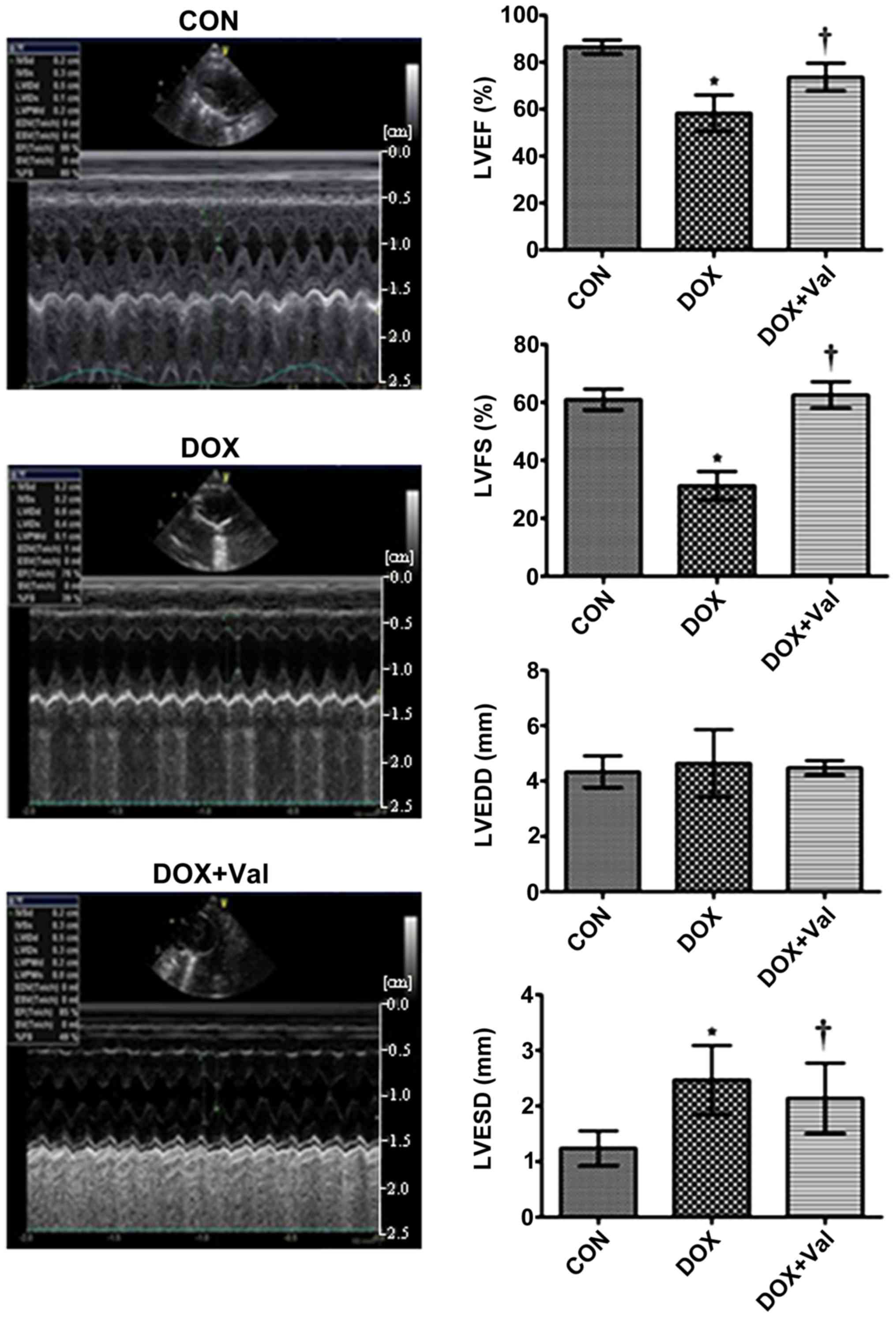
Effect of DOX+Val on LV function and
CVF
At week 10 of the study, LVEF and LVFS were
significantly lower, whereas LVESD was significantly higher in the
DOX group compared with the CON group. LVEF and LVFS were
significantly higher and LVESD was significantly lower in the
DOX+Val group compared with the DOX group (Fig. 3).
Interstitial CVF values measured using Masson
staining and Sirius Red staining were significantly higher in the
DOX group compared with the CON group. In the DOX+Val group, the
values were significantly lower compared with the DOX group
(Fig. 4A and B). Overall, these
results suggest that simultaneous application of Val with DOX
attenuated DOX-induced myocardial injury in rats, as shown by
improved cardiac function and attenuated cardiac remodeling.
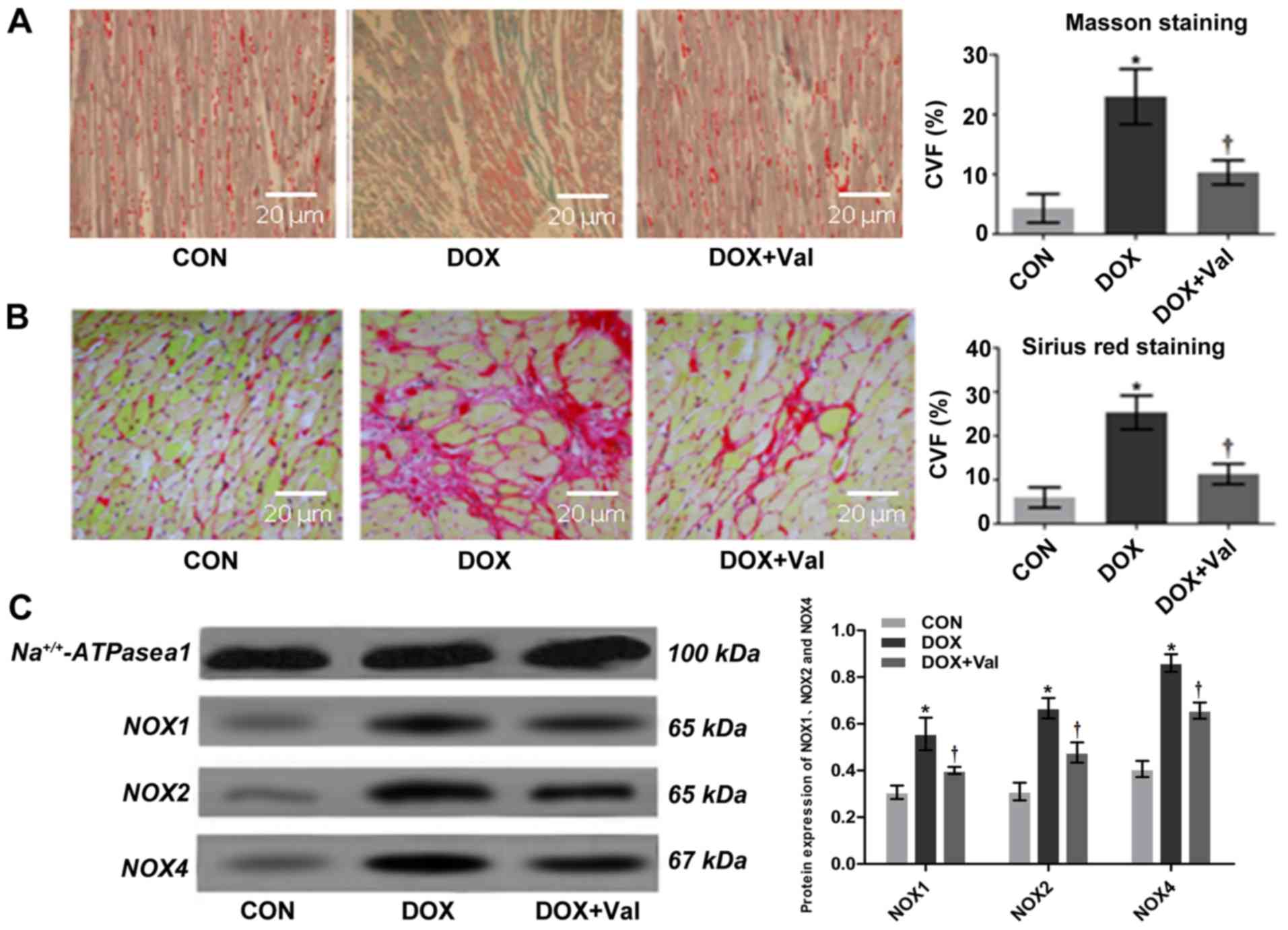 | Figure 4.Myocardial histopathology and protein
expression of NOX1, NOX2 and NOX4. (A) Masson staining and CVF
(magnification, ×200). (B) Sirius red staining and CVF
(magnification, ×200). Collagen stained with Masson staining (blue)
and Sirius red staining (red) are significantly increased in the
DOX group compared with those in the CON group, which is attenuated
in the DOX+Val group. (C) Myocardial protein expression of NOX1,
NOX2 and NOX4 detected by western blotting. *P<0.05 vs. CON;
†P<0.05 vs. DOX.CON, control; DOX, doxorubicin; Val,
valsartan; NOX, NAD(P)H oxidase; CVF, collagen volume fraction. |
Effect of DOX+Val on myocardial mRNA
expression levels of various signaling molecules
The myocardial mRNA expression levels of NOX2, NOX4,
Bax, Caspase-3, matrix metallopeptidase (MMP)2, MMP9,collagen I,
Beclin-1, brain natriuretic peptide (BNP), atrial natriuretic
peptide (ANP), β myosin heavy chain (β-MHC), growth differentiation
factor 15 (GDF15), tropomyosin 1 (TPM1), biglycan (BGN) and
periostin (POSTN) were significantly lower in the DOX+Val group,
whereas the mRNA expression levels of BCL2 and collagen III were
significantly higher in the DOX+Val group compared with the DOX
group (all P<0.05; Table II).
Myocardial mRNA expression levels of NOX1, atrial natriuretic
peptide-converting enzyme (CORIN) and transforming growth factor
(TGF)-β1 were similar between DOX and DOX+Val groups. The results
suggested that the simultaneous application of Val with DOX could
reduce the DOX-induced myocardial mRNA expressions of various
NAD(P)H oxidase, apoptosis and collagen signaling molecules, and
increase the mRNA expression levels of anti-apoptotic signaling
molecule in rats.
| Table II.mRNA expression of myocardial genes
in CON (n=8), DOX (n=12) and DOX+Val (n=12) groups. |
Table II.
mRNA expression of myocardial genes
in CON (n=8), DOX (n=12) and DOX+Val (n=12) groups.
| Gene | CON (mean ±
SD) | DOX (mean ±
SD) | DOX+Val (mean ±
SD) |
|---|
| Bax |
0.005472±0.002241 |
0.579347±0.015441a |
0.016487±0.002301a,b |
| Collagen I |
0.000159±0.000117 |
0.003708±0.001421a |
0.000743±0.000246a,b |
| BNP |
0.008863±0.000374 |
0.033877±0.011124a |
0.015468±0.003612b |
| Beclin-1 |
0.070218±0.038941 |
0.496883±0.027112a |
0.408002±0.014891a,b |
| BCL2 |
0.069447±0.024328 |
0.005516±0.002308a |
0.011095±0.006306b |
| NOX4 |
2.660319±0.722017 |
23.744164±9.831247a |
4.283176±0.274911a,b |
| NOX1 |
0.329351±0.027661 |
0.715372±0.196175a |
0.616009±0.201272 |
| NOX2 |
0.005916±0.002794 |
0.085991±0.008259a |
0.008974±0.000806a,b |
| Caspase-3 |
0.000813±0.000582 |
0.004873±0.003057a |
0.002485±0.001034a,b |
| MMP9 |
0.000110±0.000222 |
0.001814±0.000453a |
0.000134±0.000177a,b |
| MMP2 |
0.106209±0.082461 |
0.409883±0.172409a |
0.160207±0.036443a,b |
| ANP |
1.263391±0.404303 |
7.823333±2.880115a |
2.786022±0.582717a,b |
| β-MHC |
1.473521±0.610019 |
24.508593±9.604317a |
3.712543±1.140049a,b |
| BGN |
3.914408±0.593123 |
0.594062±0.097032a |
0.038563±0.004427a,b |
| TPM1 |
0.007855±0.003824 |
0.002717±0.001296a |
0.000702±0.000241a,b |
| Collagen III |
0.017781±0.003225 |
0.000461±0.000158a |
0.001982±0.000791a,b |
| CORIN |
0.245495±0.074003 |
0.030678±0.028541a |
0.016321±0.008023a |
| TGF-β1 |
0.013657±0.004844 |
0.039625±0.011513a |
0.022389±0.012937 |
| GDF15 |
0.003563±0.001879 |
0.044461±0.013455a |
0.004693±0.000264a,b |
| POSTN |
0.003533±0.002247 |
0.530036±0.110447a |
0.020532±0.006552a,b |
Effect of DOX+Val on myocardial
protein expression levels of NOX1, NOX2 and NOX4
Myocardial protein expression levels of NOX1, NOX2
and NOX4 were significantly higher in the DOX treated group
compared with the CON group, whereas the expression levels were
significantly lower in the DOX+Val group compared with the DOX
group (all P<0.05; Fig. 4C).
The results indicated that simultaneous application of Val with DOX
resulted in a reduction of DOX-induced myocardial protein
expression levels of NOX1, NOX2 and NOX4. Myocardial levels of
renin, ALD, TNF-α, IL-6, BNP, ROS, MDA, SOD, NOX1, NOX2 and NOX4
were measured using ELISA (Table
III). Myocardial levels of renin, ALD, TNF-α, IL-6, BNP, ROS,
MDA, NOX1, NOX2 and NOX4 were significantly higher in the DOX group
compared with the CON group (all P<0.05), and the levels of
these factors were significantly lower in the DOX+Val group
compared with the DOX group (all P<0.05). Myocardial SOD levels
were significantly lower in the DOX group compared with the CON
group, and significantly higher in the DOX+Val group compared with
the DOX group (both P<0.05). These results suggest that
simultaneous application of Val with DOX decreased DOX-induced
changes in the myocardial levels of renin, ALD, TNF-α, IL-6, BNP,
ROS, MDA, NOX1, NOX2 and NOX4 and increased SOD levels.
| Table III.Myocardial content of genes in CON
(n=8), DOX (n=12) and DOX+Val (n=12) groups. |
Table III.
Myocardial content of genes in CON
(n=8), DOX (n=12) and DOX+Val (n=12) groups.
| Protein, ng/ml | CON (mean ±
SD) | DOX (mean ±
SD) | DOX+Val (mean ±
SD) |
|---|
| Renin | 27.27±2.32 |
41.82±3.76a |
34.02±3.14a,b |
| ALD | 246.55±68.03 |
376.87±85.34a |
264.25±71.05b |
| MDA | 2.55±0.72 |
3.99±0.93a |
2.72±0.77b |
| SOD | 144.61±32.15 |
105.48±22.65a |
138.84±18.16b |
| ROS | 275.42±71.86 |
355.64±100.17a |
327.41±69.88b |
| TNF-α | 173.52±20.88 |
260.24±23.47a |
211.77±20.59b |
| IL-6 | 77.09±17.26 |
88.28±21.73a |
73.27±15.15b |
| BNP | 208.31±33.62 |
331.77±22.45a |
275.73±26.21a,b |
| NOX1 | 59.77±8.94 |
68.87±10.16a |
59.13±7.65b |
| NOX2 | 44.22±9.55 |
51.41±7.31a |
49.21±3.80b |
| NOX4 | 21.76±5.76 |
27.37±6.41a |
21.08±5.49b |
Effect of DOX+Val on cell
viability
H9C2 cells were treated with various concentrations
of DOX (0.1–50 µM) for 24 h. DOX treatment resulted in decreased
cell viability in a dose-dependent manner, and cell viability was
significantly decreased by 1 µM DOX treatment. Thus, 1 µM DOX was
chosen as the target dose for subsequent experiments (Fig. 5A). H9C2 cells were also treated
with various concentrations of Val (0.1–30 µM) for 24 h, and the
results showed that Val did not affect cell survival (Fig. 5B). As shown in Fig. 5C, DOX-induced H9C2 cytotoxicity was
significantly attenuated after 1 h of pre-treatment with 5 and 10
µM/l Val.
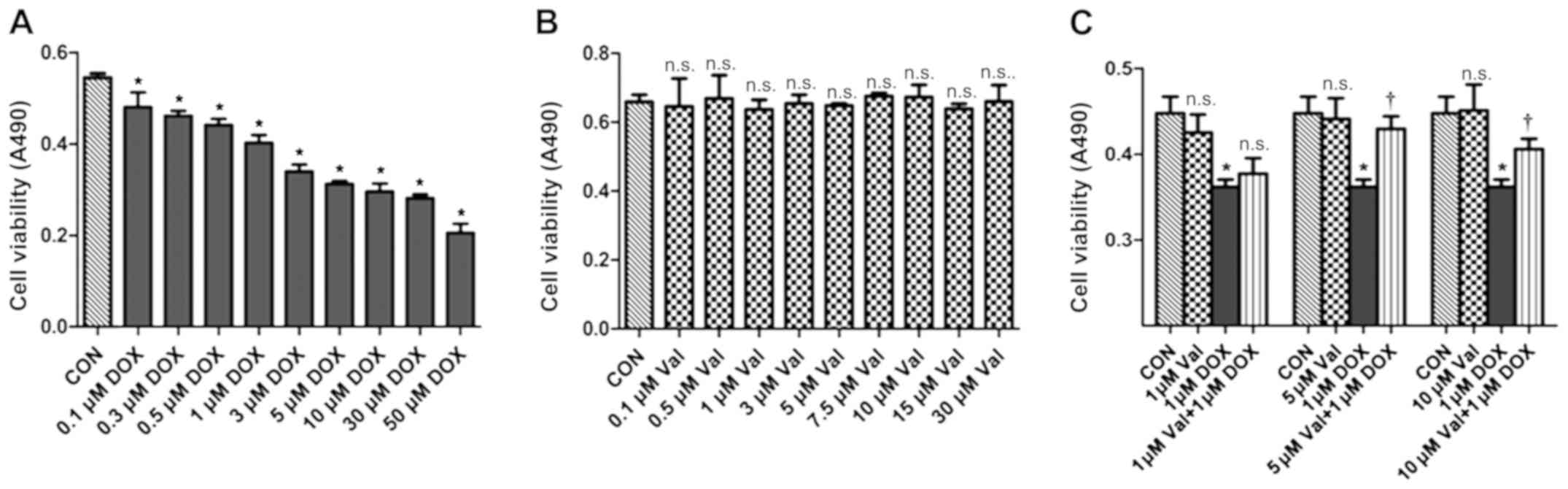 | Figure 5.Cell viability measured by MTT assay.
(A) H9C2 cells were treated with various concentrations of DOX for
24 h, DOX resulted in cell death in a dose-dependent manner. Cell
viability significantly decreased with 1 µM DOX, so this
concentration was chosen as the target dose. (B) H9C2 cells were
treated with numerous concentrations of Val, the results revealed
that the nine different concentrations of Val did not affect the
survival of H9C2 cells within 24 h. (C) Val concentration required
to protect against DOX-induced cytotoxicity was calculated by
performing a dose-response study in the presence of 1, 5 and 10 µM
Val. The cytotoxic effects of DOX were significantly attenuated by
pre-treatment with 5 and 10 µM Val. Based on these results, 5 µM
Val was selected as the target dose for further study. *P<0.05
vs. CON; †P<0.05 vs. DOX; n.s., not significant. CON,
control; DOX, doxorubicin; Val, valsartan; NOX, NAD(P)H
oxidase. |
Verification of transfection efficacy of siRNA and
OE vector. As presented in Fig.
S1, NOX2 and NOX4 mRNA expressions in the negative siRNA group
and Empty vector group were similar with the Control group. It was
apparent that the designed siRNA, vector and transfection process
do not affect the expression of the targeted gene in H9c2 cells.
The expressions of NOX2 and NOX4 in NOX2 siRNA group and NOX4 siRNA
groups were significantly lower compared with the negative siRNA
group, and their expressions in the NOX2 OE group and NOX4 OE group
were significantly higher compared with the Empty vector group.
These results indicated that siRNA and OE vectors transfection is
high effective.
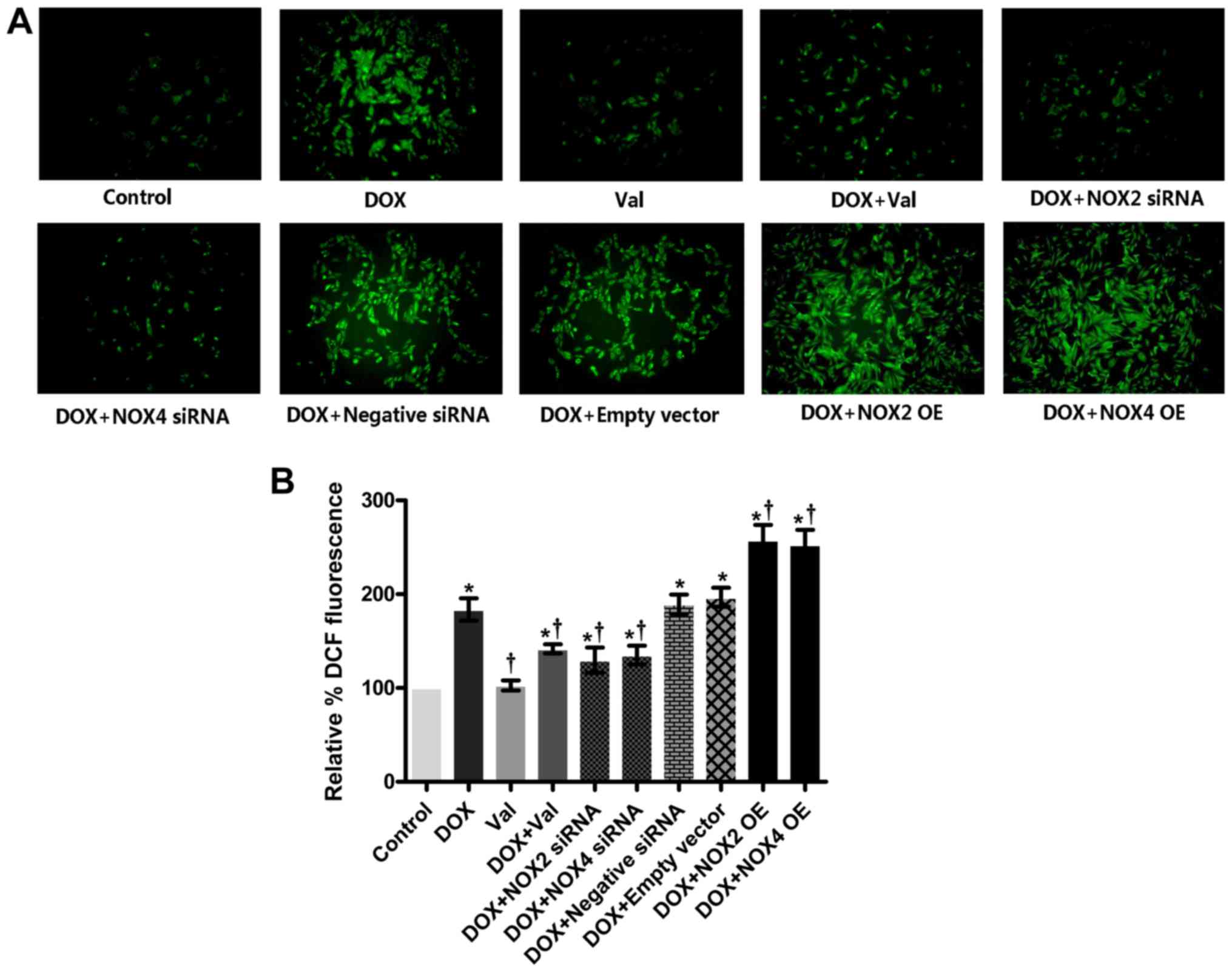
Effect of DOX+Val on intracellular ROS
production in vitro
As shown in Fig. 6A and
B, intracellular ROS production was significantly increased in
the DOX-treated H9C2 cells compared with the CON group. ROS
production was significantly reduced in the DOX+Val-treated H9C2
cells compared with the DOX group. Similarly, intracellular ROS
production in the DOX-treated H9C2 cells was significantly reduced
by knockdown of NOX2 and NOX4, and intracellular ROS production in
the DOX-treated H9C2 cells was further increased following NOX2 and
NOX4 overexpression. Intracellular ROS production in DOX-treated
H9C2 cells was not significantly affected by the control siRNA or
empty vector transfection.
Effect of DOX+Val on cardiomyocyte
apoptosis
As shown in Fig. 7A and
B, cardiomyocyte apoptosis was significantly increased in the
DOX-treated H9C2 cells compared with the CON group. Apoptosis was
significantly reduced by DOX+Val treatment, as well as NOX2 and
NOX4 knockdown. Apoptosis rates in the DOX-treated H9C2 cells were
further increased following NOX2 and NOX4 overexpression.
Cardiomyocyte apoptosis in the DOX-treated H9C2 cells was not
affected by the control siRNA or empty vector transfection.
Discussion
The results of the present study showed that a
combined treatment of Val and DOX significantly attenuated
DOX-induced myocardial injury and reduced DOX-induced ROS
production and apoptosis, potentially via downregulation of the
NOX2/NOX4 signaling pathway.
In agreement with previous studies (7,33–35),
it was shown that simultaneous application of Val with DOX
attenuated DOX-induced myocardial injury in rats, as shown by the
improvement in cardiac function and reduction in cardiac
remodeling. Compared with the CON group, the CVF was significantly
increased, along with upregulated expression of MMP2, MMP9 and
collagen I in the DOX group, suggesting enhanced fibrosis was
responsible for reduced cardiac function in this model. Treatment
with Val and DOX significantly reversed the aforementioned changes
induced by DOX, suggesting that Val protected the rats from
DOX-induced myocardial injury by reducing collagen remolding and
myocardial fibrosis, possibly by downregulating myocardial
expression of MMP2, MMP9 and collagen I. This result is in
agreement with a previous study that demonstrated that
co-administration of telmisartan with DOX decreased the levels of
cardiotoxicity-associated biochemical markers (lactate
dehydrogenase and creatine kinase myocardial band) and attenuated
the effects of DOX on oxidative stress parameters and nitric oxide
production, as well as myocardial fibrosis (36). Taken together, these results
suggest that application of ARBs with DOX at the time of drug
initiation may be a clinically feasible strategy to
prevent/attenuate the potential cardiac damage induced by DOX in
patients with tumors.
Increased ROS production and reduced SOD levels are
frequently observed pathological features of various cardiovascular
diseases, such as atherosclerosis and hypertension (37,38).
In the present study, MDA levels were increased and SOD levels were
decreased in myocardial tissues from the DOX-treated rats in
vivo, and increased ROS production and apoptosis were observed
in the DOX-treated H9C2 cells in vitro. Similarly,
simultaneous treatment with Val significantly reversed these
changes both in vivo and in vitro. Furthermore, it
was shown that mRNA expression levels of the pro-apoptotic genes
Caspase-3 and Bax were upregulated, whereas the mRNA expression
levels of BCL2 were downregulated in the DOX group, and Val
treatment reversed these changes. Taken together, these results
suggest that treatment with Val may effectively reduce the enhanced
myocardial apoptosis induced by DOX, possibly through modulating
the expression of apoptosis-related genes. Additionally, these
results suggest that the beneficial effects of Val in this model
are at least partly associated with the capacity of Val to reduce
DOX-induced myocardial injury via the reduction of myocardial
apoptosis.
The myocardial mRNA and protein expression levels of
NOX1, NOX2 and NOX4 following DOX treatment were measured. The
results showed that DOX treatment significantly upregulated the
myocardial protein expression of NOX1, NOX2 and NOX4, and mRNA
expression of NOX2 and NOX4. Simultaneous treatment with Val
significantly reduced the mRNA expression levels of NOX2 and NOX4,
as well as the myocardial protein expression levels of NOX1, NOX2
and NOX4. These results suggest that upregulated NOX expression
levels, particularly NOX2 and NOX4 signaling, may contribute to
DOX-induced cardiac injury, and the observed beneficial effects of
simultaneous application of Val may partially be associated with
its capacity to downregulate NOX2 and NOX4 signaling in this
model.
To explore the mechanism underlying the protective
effects of Val in DOX-induced myocardial injury, ROS production and
apoptosis of DOX-treated H9C2 cells were observed following up- or
downregulation of NOX2 and NOX4 expression, as Val significantly
downregulated the myocardial mRNA expression of these two genes.
The results suggested that ROS production and apoptosis were
significantly reduced by downregulating NOX2 and NOX4, whereas
overexpression of NOX2 and NOX4 increased ROS production and
apoptosis in DOX-treated H9C2 cells, suggesting that downregulation
of NOX2 and NOX4 expression may be mechanism by which Val
alleviates DOX-induced myocardial injury. Although the present
study observed changes in intracellular ROS and cardiomyocyte
apoptosis under the conditions of NOX-overexpressing and
NOX-silencing, we did not detect them at the protein level, which
is a limitation of this study. In our previous study examining the
mechanisms of DOX toxicity, the expression of angiotensin (Ang)II
receptor protein (AT1R) was significantly upregulated following DOX
stimulation in H9C2 cells, and AngII increased the protein
expression levels of NOX2 and NOX4 and the production of ROS (data
not yet published). The protein expression levels of ERK, JNK and
P38, which lie downstream of the mitogen-activated protein kinase
(MAPK) signaling pathway, were increased significantly (data not
yet published). Pre-treatment with Val reduced the expression of
AT1R, NOX2, NOX4 and ROS, and activity of the MAPK signaling
pathway was decreased (data not yet published). Thus, it was
hypothesized that the AngII-NOX-ROS-MAPK signaling pathway may
underlie DOX-induced myocardial toxicity.
In summary, co-treatment with Val and DOX
significantly reduced DOX-induced myocardial injury, potentially
through downregulation of NOX2 and NOX4 signaling. The present
study provides experimental evidence supporting the simultaneous
use of ARBs to prevent DOX-induced cardiotoxicity in the clinical
setting.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the Natural
Science Funds of Liaoning Province (grant no. 201602033) and the
National Natural Science Foundation of China (grant no.
81770405).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
DC, LC and WT performed the experiments and data
analysis, and prepared the manuscript. HW and QW contributed to the
conception and design of the study and critically revised the
manuscript for important intellectual content and supervised the
experimental process. LM and ZL prepared the figures, and analyzed
and interpreted the data. QY designed this study and proofread the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
All experiments were performed in compliance with
the ARRIVE guidelines as well as the Guide for the Care and Use of
Laboratory Animals of the National Academy of Sciences. The
Institutional Animal Research and Ethics Committee of Dalian
University approved the protocols of the animal experiments.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Razavi-Azarkhiavi K, Iranshahy M, Sahebkar
A, Shirani K and Karimi G: The protective role of phenolic
compounds against doxorubicin-induced cardiotoxicity: A
comprehensive review. Nutr Cancer. 68:892–917. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Swain SM, Whaley FS and Ewer MS:
Congestive heart failure in patients treated with doxorubicin: A
retrospective analysis of three trials. Cancer. 97:2869–2879. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ganame J, Claus P, Uyttebroeck A, Renard
M, D'hooge J, Bijnens B, Sutherland GR, Eyskens B and Mertens L:
Myocardial dysfunction late after low-dose anthracycline treatment
in asymptomatic pediatric patients. J Am Soc Echocardiogr.
20:1351–1358. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tebbi CK, London WB, Friedman D, Villaluna
D, De Alarcon PA, Constine LS, Mendenhall NP, Sposto R, Chauvenet A
and Schwartz CL: Dexrazoxane-associated risk for acute myeloid
leukemia/myelodysplastic syndrome and other secondary malignancies
in pediatric Hodgkin's disease. J Clin Oncol. 25:493–500. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shaikh AY and Shih JA:
Chemotherapy-induced cardiotoxicity. Curr Heart Fail Rep.
9:117–127. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Iqbal M, Dubey K, Anwer T, Ashish A and
Pillai KK: Protective effects of telmisartan against acute
doxorubicin-induced cardiotoxicity in rats. Pharmacol Rep.
60:382–390. 2008.PubMed/NCBI
|
|
7
|
Zong WN, Yang XH, Chen XM, Huang HJ, Zheng
HJ, Qin XY, Yong YH, Cao K, Huang J and Lu XZ: Regulation of
angiotensin-(1–7) and angiotensin II type 1 receptor by telmisartan
and losartan in adriamycin-induced rat heart failure. Acta
Pharmacol Sin. 32:1345–1350. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Angsutararux P, Luanpitpong S and
Issaragrisil S: Chemotherapy-induced cardiotoxicity: Overview of
the roles of oxidative stress. Oxid Med Cell Longev.
2015:7956022015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chang SA, Lim BK, Lee YJ, Hong MK, Choi JO
and Jeon ES: A Novel Angiotensin Type I receptor antagonist,
fimasartan, prevents doxorubicin-induced cardiotoxicity in rats. J
Korean Med Sci. 30:559–568. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Manohar P and Pina IL: Therapeutic role of
angiotensin II receptor blockers in the treatment of heart failure.
Mayo Clin Proc. 78:334–338. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nakamae H, Tsumura K, Terada Y, Nakane T,
Nakamae M, Ohta K, Yamane T and Hino M: Notable effects of
angiotensin II receptor blocker, valsartan, on acute cardiotoxic
changes after standard chemotherapy with cyclophosphamide,
doxorubicin, vincristine, and prednisolone. Cancer. 104:2492–2498.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sakr HF, Abbas AM and Elsamanoudy AZ:
Effect of valsartan on cardiac senescence and apoptosis in a rat
model of cardiotoxicity. Can J Physiol Pharmacol. 94:588–598. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen L, Yan KP, Liu XC, Wang W, Li C, Li M
and Qiu CG: Valsartan regulates TGF-β/Smads and TGF-β/p38 pathways
through IncRNA CHRF to improve doxorubicin-induced heart failure.
Arch Pharm Res. 41:101–109. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hafstad AD, Nabeebaccus AA and Shah AM:
Novel aspects of ROS signalling in heart failure. Basic Res
Cardiol. 108:3592013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dietl A and Maack C: Targeting
mitochondrial calcium handling and reactive oxygen species in heart
failure. Curr Heart Fail Rep. 14:338–349. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Meitzler JL, Antony S, Wu Y, Juhasz A, Liu
H, Jiang G, Lu J, Roy K and Doroshow JH: NADPH oxidases: A
perspective on reactive oxygen species production in tumor biology.
Antioxid Redox Signal. 20:2873–2889. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bedard K and Krause KH: The NOX family of
ROS-generating NADPH oxidases: Physiology and pathophysiology.
Physiol Rev. 87:245–313. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tsutsui H, Kinugawa S and Matsushima S:
Oxidative stress and heart failure. Am J Physiol Heart Circ
Physiol. 301:H2181–H2190. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhao QD, Viswanadhapalli S, Williams P,
Shi Q, Tan C, Yi X, Bhandari B and Abboud HE: NADPH oxidase 4
induces cardiac fibrosis and hypertrophy through activating
Akt/mTOR and NFκB signaling pathways. Circulation. 131:643–655.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ruzicka M, Yuan B, Harmsen E and Leenen
FH: The renin-angiotensin system and volume overload-induced
cardiac hypertrophy in rats. Effects of angiotensin converting
enzyme inhibitor versus angiotensin II receptor blocker.
Circulation. 87:921–930. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bendall JK, Cave AC, Heymes C, Gall N and
Shah AM: Pivotal role of a gp91(phox)-containing NADPH oxidase in
angiotensin II-induced cardiac hypertrophy in mice. Circulation.
105:293–296. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Murdoch CE, Zhang M, Cave AC and Shah AM:
NADPH oxidase-dependent redox signalling in cardiac hypertrophy,
remodelling and failure. Cardiovasc Res. 71:208–215. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Seddon M, Looi YH and Shah AM: Oxidative
stress and redox signalling in cardiac hypertrophy and heart
failure. Heart. 93:903–907. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Watson LE, Sheth M, Denyer RF and Dostal
DE: Baseline echocardiographic values for adult male rats. J Am Soc
Echocardiogr. 17:161–167. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Barauna VG, Rosa KT, Irigoyen MC and de
Oliveira EM: Effects of resistance training on ventricular function
and hypertrophy in a rat model. Clin Med Res. 5:114–120. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu HN, Wang HR, Cheng D, Pei ZW, Zhu N
and Yu Q: Potential role of a disintegrin and metalloproteinase-17
(ADAM17) in age-associated ventricular remodeling of rats. RSC
Advances. 9:14321–14330. 2019. View Article : Google Scholar
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ye L, Haider HKH, Jiang S, Ge R, Law PK
and Sim EK: In vitro functional assessment of human skeletal
myoblasts after transduction with adenoviral bicistronic vector
carrying human VEGF165 and angiopoietin-1. J Heart Lung Transplant.
24:1393–1402. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Xie F, Wu D, Huang SF, Cao JG, Li HN, He
L, Liu MQ, Li LF and Chen LX: The endoplasmic reticulum
stress-autophagy pathway is involved in apelin-13-induced
cardiomyocyte hypertrophy in vitro. Acta Pharmacol Sin.
38:1589–1600. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Huang YL, Zhao F, Luo CC, Zhang X, Si Y,
Sun Z, Zhang L, Li QZ and Gao XJ: SOCS3-mediated blockade reveals
major contribution of JAK2/STAT5 signaling pathway to lactation and
proliferation of dairy cow mammary epithelial cells in vitro.
Molecules. 18:12987–13002. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Priya LB, Baskaran R, Huang CY and Padma
VV: Neferine ameliorates cardiomyoblast apoptosis induced by
doxorubicin: Possible role in modulating NADPH oxidase/ROS-mediated
NFkB redox signaling cascade. Sci Rep. 7:122832017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gao J, Chen T, Zhao D, Zheng J and Liu Z:
Ginkgolide B Exerts Cardioprotective properties against
doxorubicin-induced cardiotoxicity by regulating reactive oxygen
species, Akt and calcium signaling pathways in vitro and in vivo.
PLoS One. 11:e01682192016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yu Q, Li Q, Na R, Li X, Liu B, Meng L,
Liutong H, Fang W, Zhu N and Zheng X: Impact of repeated
intravenous bone marrow mesenchymal stem cells infusion on
myocardial collagen network remodeling in a rat model of
doxorubicin-induced dilated cardiomyopathy. Mol Cell Biochem.
387:279–285. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bakhtiari E, Hosseini A and Mousavi SH:
Protective effect of Hibiscus sabdariffa against serum/glucose
deprivation-induced PC12 cells injury. Avicenna J Phytomed.
5:231–237. 2015.PubMed/NCBI
|
|
35
|
Huang CY, Chen JY, Kuo CH, Pai PY, Ho TJ,
Chen TS, Tsai FJ, Padma VV, Kuo WW, Huang CY, et al: Mitochondrial
ROS-induced ERK1/2 activation and HSF2-mediated AT1 R
upregulation are required for doxorubicin-induced cardiotoxicity. J
Cell Physiol. 233:463–475. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ibrahim MA, Ashour OM, Ibrahim YF,
El-Bitar HI, Gomaa W and Abdel-Rahim SR: Angiotensin-converting
enzyme inhibition and angiotensin AT(1)-receptor antagonism equally
improve doxorubicin-induced cardiotoxicity and nephrotoxicity.
Pharmacol Res. 60:373–381. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Khosravi M, Poursaleh A, Ghasempour G,
Farhad S and Najafi M: The effects of oxidative stress on the
development of atherosclerosis. Biol Chem. 400:711–732. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wu J, Saleh MA, Kirabo A, Itani HA,
Montaniel KR, Xiao L, Chen W, Mernaugh RL, Cai H, Bernstein KE, et
al: Immune activation caused by vascular oxidation promotes
fibrosis and hypertension. J Clin Invest. 126:50–67. 2016.
View Article : Google Scholar : PubMed/NCBI
|