Introduction
Ureteropelvic junction obstruction is the most
common obstructive urinary tract disease in pediatric urology, with
an incidence of 0.5–1/1,000 (1).
It is also one of the causes of obstructive nephropathy
characterized by renal fibrosis (2). The primary features of this
irreversible renal fibrosis are glomerular sclerosis and
tubulointerstitial fibrosis (3).
Previous studies have revealed that tubulointerstitial fibrosis is
associated with the process of epithelial-mesenchymal transition
(EMT) (4,5). EMT is characterized by a loss of
adhesion and polarity of epithelial cells and induction of α-smooth
muscle actin (α-SMA) (6,7). EMT is involved in numerous
pathological changes, including fibrosis and tumor metastasis
(8,9). The TGF-β signaling pathway serves a
key role in regulation of renal fibrosis (10). TGF-β1 expression levels are
significantly upregulated in the process of renal fibrosis caused
by unilateral ureteral obstruction (11).
A number of proteins with differential expression
levels between rat kidney tissue from sham operated group and those
with complete unilateral ureteral obstruction were identified in
our previous study (12). These
identified proteins have been reported to be involved in cell
apoptosis, energy metabolism and injury of mitochondria and
oxidative stress in a preliminary study (12). AMP-activated protein kinase (AMPK),
comprising α1/2, β1/2 and γ1/2/3 subunits, is a mitochondrial
energy sensor that detects changes in AMP levels (13). AMPK is involved in the maintenance
of cellular energy balance by affecting multiple factors during
metabolism (14). For example,
activation of AMPK increases the rate of catabolic (ATP-generating)
pathways and decreases the rate of anabolic (ATP-utilizing)
pathways (15). Increased
intracellular ratio of AMP to ATP activates AMPK; AMP binds to the
AMPK γ subunit, causing conformational changes in the protein and
allowing phosphorylation of the Thr-172 site in the α subunit
(16). Previous studies have
revealed that AMPK, particularly AMPKα2, regulates the EMT process
during liver and kidney fibrosis (17–19)
and serves an important role in tumor cell metastasis (20). However, the underlying mechanisms
for AMPK changes in renal tubular EMT remain unclear.
In the present study, the expression levels of
AMPKα2 in EMT-derived normal rat renal tubular epithelial (NRK-52E)
cells induced by TGF-β1 were investigated. Gene microarray was used
to analyze differential gene expression levels in EMT-derived
NRK-52E cells before and after AMPKα2 knockdown (KD). Ingenuity
pathway analysis (IPA) was performed to identify specific genes and
signaling pathways involved in the regulation of EMT by AMPKα2.
Finally, reverse transcription-quantitative PCR (RT-qPCR) and
western blotting were used to verify the prediction results.
Materials and methods
Cell culture, RNA interference and
TGF-β1 EMT induction
NRK-52E cells (Cell Bank of the Chinese Academy of
Sciences) were cultured in DMEM (high glucose; cat. no. SH30022.01;
Hyclone; Cytiva) supplemented with 10% FBS (cat. no. SH30071.03;
Hyclone; Cytiva) and 3% penicillin-streptomycin solution (cat. no.
SV30010; Hyclone; Cytiva). Cell culture was maintained at 37°C in a
humidified atmosphere at 5% CO2. Th cell medium was
changed every 2 days.
The sequence of small interfering RNA
(5′-GCTGACTTCGGACTCTCTA-3′) was designed by Shanghai GeneChem Co.,
Ltd. for targeting the AMPKα2 sequence (GenBank no. NM_023991). The
AMPKα2 hairpin oligonucleotide was inserted into the GV248-GFP
lentiviral vector (Shanghai GeneChem Co., Ltd.) to construct a
GV248-GFP-short hairpin (sh)AMPKα2 KD vector. The negative control
(shCtrl) sequence was 5′-TTCTCCGAACGTGTCACGT-3′, and when
incorporated into the lentiviral vector was referred to as
GV248-GFP-shCtrl. Then, GV248-GFP-shAMPKα2 KD and GV248-GFP-shCtrl
were respectively co-transfected with pHelper 1.0 (Shanghai
GeneChem Co., Ltd.) and pHelper 2.0 (Shanghai GeneChem Co., Ltd.)
into 293T cells (Shanghai GeneChem Co., Ltd.) to package and
produce the shRNA expressing and shCtrl lentivirus. The viral
titers of AMPKα2 shRNA lentivirus or control lentivirus reached
1×109 TU/ml for further studies.
NRK-52E cells were seeded in a 6-well tissue culture
plate with 1×105/well, 1 day prior to infection. The
complete culture solution was replaced by infection enhancing
solution with 5 µg/ml polybrene (Shanghai GeneChem Co., Ltd.) and
the packed AMPKα2 shRNA lentivirus or control LV was added to the
cells with multiplicity of infection 25. After 12 h, the lentivirus
solution was replaced with complete culture solution. At 72 h
post-transfection, the efficacy of AMPKα2 KD was validated via
RT-qPCR (Fig. S1).
For TGF-β1 treatment, all cells were stimulated with
10 ng/ml recombinant TGF-β1 (ProteinTech Group, Inc.) at 37°C for
24 h. Cells were divided into four groups: Negative control,
TGF-β1-treated, TGF-β1-treated + shCtrl and TGF-β1-treated +
shAMPKα2 KD. Protein expression levels and location were determined
using immunofluorescence staining. Total RNA was extracted and
analyzed using RT-qPCR. Proteins extracted from total cell lysates
were analyzed using western blotting. All measurements were
repeated ≥3 times.
Gene microarray
The present microarray dataset has been deposited in
National Center for Biotechnology Information Gene Expression
Omnibus (accession no. GSE141981). The genome-wide effect of AMPKα2
KD was assessed using the GeneChip™ Rat Genome 2302.0 Array
(Affymetrix; Thermo Fisher Scientific, Inc.) consisting of 28,000
genes. A total of three biological replicates of EMT-derived
NRK-52E cells transduced with shAMPKα2 or shCtrl LV (for 72 h) were
analyzed using a microarray. RNA was initially isolated using
TRIzol® reagent and quality was determined using the
NanoDrop 2000 spectrophotometer (NanoDrop; Thermo Fisher
Scientific, Inc.) and Agilent Bioanalyzer 2100 (Agilent
Technologies, Inc.). Individual microarrays were used for gene
expression level profiling of each sample. Briefly, 500 ng RNA
samples were reverse-transcribed and labeled with biotin using the
GeneChip3′ IVT labeling kit according to the manufacturer's
protocol. Labeled complementary (c)DNA was then hybridized onto the
GeneChip™Rat Genome 230 2.0 Array. Arrays were performed with the
GeneChip® Hybridization, Wash and Stain kit using the
GeneChip® Fluidics Station 450. All GeneChip®
products were obtained from Affymetrix (Thermo Fisher Scientific,
Inc.) and used according to the manufacturer's protocol. The chip
array was scanned directly post-hybridization using the
GeneChip® Scanner 3000. Microarray data were analyzed
using GeneSpring software (version 11; Agilent Technologies, Inc.).
P-values were determined using a linear model based on the
empirical Bayesian distribution (21). The false discovery rate (FDR) was
corrected using the Benjamini-Hochberg method (22). The screening criteria for
significantly differential genes were |fold-change|>3 and
FDR<0.05.
IPA
Datasets representing differentially expressed genes
derived from microarray analyses were imported into the IPA tool
(ingenuity.com; Ingenuity® Systems).
The ‘core analysis’ function in the IPA software was used to
interpret differentially expressed data, which included functional
signaling pathways. Differentially expressed genes were mapped onto
functional signaling pathways available in the Ingenuity database.
Z-score activation algorithms were computed using the IPA software.
Analyses performed within the IPA program included identification
of a particular dataset and its functional signaling pathways.
RT-qPCR
Total RNA was isolated from NRK-52E cells using
RNAiso Plus (Takara Bio, Inc.) according to the manufacturer's
instructions and subjected to reverse transcription into cDNA using
the PrimeScript™ RT Reagent kit (Takara Bio, Inc.) Reverse
transcription was performed at 37°C for 15 min and then 95°C for 5
sec. RT-qPCR analysis was performed using SYBR® Premix
Ex Taq™ (Takara Bio, Inc.) and the ABI ViiA7DX System (Applied
Biosystems; Thermo Fisher Scientific, Inc.). β-actin expression
levels were used as an internal reference for all PCR experiments.
RT-qPCR primers designed for specific target genes were synthesized
by Takara Bio, Inc. (Table SI).
PCR reactions were performed using the following cycling
conditions: 95°C for 30 sec, followed by 40 cycles at 95°C for 5
sec, 60°C for 30 sec and 72°C for 20 sec. The relative mRNA levels
for each sample were calculated by the 2−ΔΔcq method
(23).
Western blot analysis
Proteins were isolated using an isolation kit
(Beyotime Institute of Biotechnology) and quantified using the
2D-Quant kit (Beyotime Institute of Biotechnology) according to the
manufacturer's instructions. A total of 50 µg protein/lane
extracted from cells was separated by 8% SDS-PAGE and then
transferred in Tris-HCl methanol (20 mM Tris, 150 mM glycine and
20% methanol) onto PVDF membranes (EMD Millipore) using a
Trans-Blot electrophoresis transfer cell (Bio-Rad Laboratories,
Inc.). The membranes were subsequently blocked with 5% non-fat dry
milk in TBS containing 0.1% Tween-20 for 2 h at room temperature
and incubated with primary antibodies overnight at 4°C. Primary
antibodies included AMPKα2 (polyclonal rabbit; 1:2,000; product
code ab3760; Abcam), v-ets erythroblastosis virus E26 oncogene
homolog-1 (ETS-1) (monoclonal rabbit; 1:1,000; product no. 14069S;
Cell Signaling Technology, Inc.), homolog-1 and ribosomal protein
s6 kinase A1 (RPS6KA1) (monoclonal rabbit; 1:1,000; product code
ab32114; Abcam), E-cadherin (monoclonal mouse; 1:1,000; product no.
14472S; Cell Signaling Technology, Inc.), α-SMA (monoclonal mouse;
1:100; product code ab7817; Abcam), vimentin (monoclonal mouse;
1:1,000; product code ab8978; Abcam) and GAPDH (monoclonal rabbit;
1:10,000; product code ab181602; Abcam). Following three washes in
TBST (10 min/wash), the membranes were incubated with goat
anti-rabbit or anti-mouse IgG-HRP (both 1:2,000; product codes
ab6721 and ab6789, respectively; both Abcam) secondary antibodies
for 1 h at room temperature and were washed again. All immunoblots
were performed ≥3 times. The antigen-antibody complexes were
visualized using enhanced chemiluminescence reagents (Thermo Fisher
Scientific, Inc.). GAPDH was used as a loading control. Detected
bands were quantified using ImageJ 2× software (version 2.1.4.7;
National Institutes of Health). The relative density of each
protein was calculated by dividing the optical density value of
each protein by that of the loading control.
Immunofluorescence staining
NRK-52E cells (1×106) were washed with
PBS and fixed in 4% paraformaldehyde for 30 min at room
temperature. Fixed cells were washed again with PBS and
permeabilized in 0.5% Triton X-100 diluted in PBS for 10 min at
room temperature. The cells on the slides were subsequently blocked
with 5% bovine serum albumin (Beyotime Institute of Biotechnology)
for 1 h at room temperature. Subsequently, the negative control and
TGF-β1-treated groups were incubated with the AMPKα2 antibody
(polyclonal rabbit; 1:100; product code ab3760; Abcam) at 4°C
overnight. The TGF-β1-treated + shCtrl and TGF-β1-treated +
shAMPKα2 KD groups were incubated with the green fluorescent
protein (GFP) antibody (polyclonal rabbit antibody; 1:2,000;
product code ab6556; Abcam) at 4°C overnight, then incubated with
fluorescein isothiocyanate and rhodamine-conjugated goat
anti-rabbit (1:100; cat. no. sc-2012; Santa Cruz Biotechnology,
Inc.) secondary antibodies. 0.1% DAPI (BIOSS) was used to stain
cell nuclei on glass slides for 5 min at 37°C. The cells were then
examined using fluorescence microscopy (magnification, ×200; Nikon
CE1 Confocal Microscope; Nikon Corporation).
Statistical analysis
Data are presented as the mean ± SEM. The number of
repeats was ≥3 times. Statistical significance between two groups
was determined using unpaired Student's t-test and between four
groups by one-way ANOVA. P-values were calibrated using
Bonferroni's correction as a post hoc test. Analysis was performed
using SPSS software (version 23.0; IBM Corp.). P<0.05 was
considered to indicate a statistically significant difference.
Results
AMPKα2 expression levels are
upregulated following TGF-β1-induced EMT in NRK-52E cells and EMT
is impaired in NRK-52E cells following AMPKα2 knockdown
TGF-β1 was used to induce EMT in NRK-52E cells.
Resulting morphological changes in the NRK-52E cells included a
change from typical round and polygonal to fusiform shape,
indicating that EMT occurred (Fig.
1). In addition, western blot experiments demonstrated that EMT
protein markers α-SMA and vimentin were upregulated, whereas
epithelial cell protein marker E-cadherin was downregulated,
further confirming the occurrence of EMT (Fig. 2).
 | Figure 2.Western blot analysis of
EMT-associated proteins α-SMA, vimentin and E-cadherin. (A) Western
blot images of the EMT-associated proteins α-SMA, vimentin, and
E-cadherin, representative of ≥3 independent experiments. GAPDH was
used as a loading control. (B) Bar graph of western blot analysis.
*P<0.05 TGF-β1 treated vs. Ctrl; **P<0.05 TGF-β1-treated +
shAMPKα2 KD vs. TGF-β1-treated + shCtrl. EMT,
epithelial-mesenchymal transition; SMA, smooth muscle actin; Ctrl,
control; sh, short hairpin; AMPKα2, AMP-activated protein kinase
α2; KD, knockdown. |
Immunofluorescence experiments demonstrated that
AMPKα2 was primarily expressed in the cytoplasm and nucleus of
NRK-52E cells (Fig. 3A). Western
blotting and RT-qPCR experiments revealed that AMPKα2 protein and
mRNA expression levels were significantly upregulated during EMT in
NRK-52E cells (Figs. 3B and
4).
GFP expression levels were observed 72 h following
shAMPKα2 and shCtrl LV transfection in NRK-52E cells that underwent
EMT induced by TGF-β1, indicating that transfection was successful
(Fig. 5A). RT-qPCR demonstrated
that the expression levels of AMPKα2 mRNA were downregulated by 70%
in the TGF-β1-treated + shAMPKα2 group compared with the
TGF-β1-treated + shCtrl group, indicating that AMPKα2 was
specifically and effectively knocked down (Fig. 5B). In addition, western blot
experiments revealed that expression levels of AMPKα2 protein were
downregulated in the TGF-β1-treated + shAMPKα2 group compared with
the TGF-β1-treated + shCtrl group (Fig. 4). The expression levels of α-SMA
and vimentin were downregulated, whereas E-cadherin expression
levels were upregulated, suggesting that the EMT process was
inhibited following AMPKα2 knockdown (Fig. 2). These data indicated that AMPKα2
may play a key regulatory role in the process of EMT in NRK-52E
cells.
Differential gene expression levels
and IPA following AMPKα2 KD in NRK-52E cells with EMT
In order to detect which genes were altered
following AMPKα2 KD, the gene expression level profiles of
EMT-derived NRK-52E cells transduced with shAMPKα2 or
shCtrl-payload LVs were determined using the GeneChip Rat 230
2.0® PathArray™ Rat Gene Expression Array with three
biological replicates. A total of 1,588 differentially expressed
genes were identified, of which 1,510 were downregulated and 78
were upregulated (Fig. 6A).
Next, IPA was used to perform pathway analysis of
these 1,588 differentially expressed genes. The IPA demonstrated
that AMPKα2 may regulate EMT progression in NRK-52E cells via
multiple pathways, including ERK/MAPK pathway, Bone morphogenetic
protein signaling pathway and Ephrin Receptor Signaling. According
to the IPA internal algorithms and standards, a z-score >2
represents a significantly activated pathway, whereas z-score
<-2 represents a significantly inhibited pathway. In the present
study, the ERK/MAPK pathway was significantly inhibited
(z-score=−3.550; Fig. 6B).
Therefore, AMPKα2 regulation of renal tubular epithelial EMT was
achieved via the ERK/MAPK pathway.
IPA results indicated that 36 genes were inhibited
in the ERK/MAPK signaling pathway following AMPKα2 knockdown
(Table SII; Fig. 6C).
ETS1 and RPS6KA1 in ERK/MAPK signaling
pathway are upregulated in NRK-52E cells with EMT, whereas ETS1 and
RPS6KA1 are downregulated following AMPKα2 KD
A total of five significantly inhibited genes,
including HRas proto-oncogene, GTPase (HRAS), CRK proto-oncogene,
adaptor protein (CRK), ETS1, RPS6KA1 and cAMP responsive element
binding protein 1 (CREB1), were selected for validation. Using
RT-qPCR, ETS1 and RPS6KA1 mRNA levels were revealed to be
significantly upregulated in the TGF-β1-treated group compared with
the negative control. The relative mRNA expression levels of ETS1
and RPS6KA1 were significantly downregulated in the TGF-β1-treated
+ shAMPKα2 KD group compared with the TGF-β1-treated + shCtrl group
(Fig. 7A and B). Inconsistent with
microarray results, AMPKα2 downregulation had no effect on HRAS,
CRK and CREB1 (Fig. 7C-E). Western
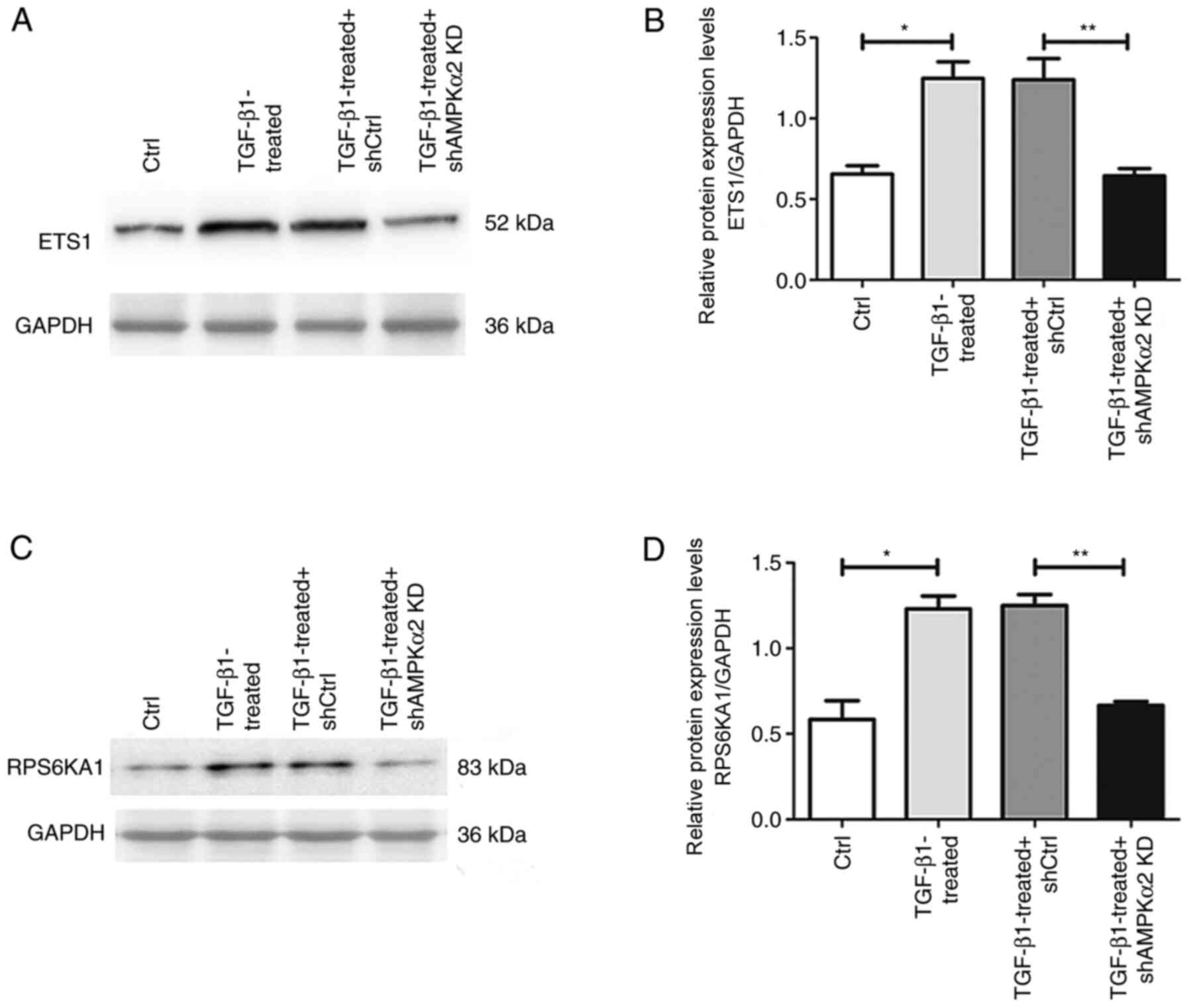
blot experiments revealed that expression levels of ETS1 and
RPS6KA1 protein in the TGF-β1-treated group were significantly
higher than in the negative control. The relative mRNA expression
levels of ETS1 and RPS6KA1 were significantly lower in the
TGF-β1-treated + shAMPKα2 KD group than in the TGF-β1-treated +
shCtrl group (Fig. 8). Therefore,
AMPKα2 may act by regulating ETS1 and RPS6KA1 in the ERK/MAPK
signaling pathway during EMT.
 | Figure 7.Differentially expressed mRNA levels
verified by reverse transcription-quantitative PCR. Relative (A)
ETS1, (B) RPS6KA1, (C) CREB1, (D) CRK and (E) HRAS mRNA expression
levels. Housekeeping gene β-actin was used as the endogenous
control. *P<0.05 TGF-β1-treated vs. Ctrl; **P<0.05
TGF-β1-treated + shAMPKα2 KD vs. TGF-β1-treated + shCtrl. ETS1,
v-ets erythroblastosis virus E26 oncogene homolog-1; RPS6KA1,
ribosomal protein S6 kinase A1; CREB1, cAMP responsive element
binding protein 1; CRK, CRK proto-oncogene, adaptor protein; HRAS,
HRas proto-oncogene, GTPase; Ctrl, control; sh, short hairpin;
AMPKα2, AMP-activated protein kinase α2; KD, knockdown. |
Discussion
Renal fibrosis is considered to be an irreversible
process that develops into end-stage renal failure (24,25).
EMT serves an important role in obstructive nephropathy and renal
fibrosis (26). Therefore, delay,
prevention and reversal of renal cell EMT are important for the
treatment of obstructive nephropathy (27). In the present study, AMPKα2 served
a key role in the EMT of NRK-52E cells and was upregulated in
TGF-β1-induced EMT-derived NRK-52E cells. Interference with AMPKα2
expression levels by shAMPKα2 LV significantly impaired EMT
progression. Moreover, using microarray and IPA, it was revealed
that AMPKα2 may play an important role in EMT process by regulating
ETS1 and RPS6KA1.
AMPK is an AMP-activated protein kinase that detects
changes in AMP levels and maintains cell energy balance via
multiple pathways, including Hippo-Yes-associated protein pathway
and Hedgehog signaling pathway, that affect cellular metabolism,
thus serving an important role in kidney disease (28,29).
However, current research on promotion or inhibition of EMT
following AMPK activation is controversial, which may be due to the
biological or cellular specificity of AMPK. The Wang et al
(17) study of rat liver cells
indicated that AMPK promoted hepatocyte EMT, which leads to liver
fibrosis, whereas AMPK-specific inhibitors can limit this process.
In addition, EMT also promotes tumor cell metastasis. Studies have
shown that in breast and lung cancer, as well as melanoma cells,
AMPK activation causes EMT, thereby promoting tumor cell metastasis
(20,30). The results of the present study are
consistent with these previous findings. However, other studies
contradict the hypothesis that AMPK activation increases EMT:
Previous research has revealed that activation of AMPK inhibited
trans-differentiation of myofibroblasts induced by TGF-β/SMAD
family member 3 and the occurrence of hepatic astrocytic fibrosis
(31,32). In addition, metformin was revealed
to activate AMPK to inhibit the TGF-β signaling pathway and
alleviate the EMT process in kidney in a rat model of renal
ischemia-reperfusion injury (18).
These inconsistent results may be due to tissue specificity or
distinct EMT models. In the present study, TGF-β1 was used to
induce EMT in NRK-52E cells and the expression levels of AMPKα2
were upregulated. EMT was inhibited following LV AMPKα2 KD,
indicating that AMPKα2 serves an important role in the EMT of
NRK-52E cells.
In order to investigate the expression levels of
differential genes in EMT-derived NRK-52E cells before and after
AMPKα2 KD, high-throughput analysis was used. According to the gene
chip and IPA results, genes in the ERK/MAPK pathway were strongly
inhibited following AMPKα2 KD, indicating that AMPKα2 modulates
renal tubular EMT by inhibiting the ERK/MAPK pathway. A total of
five genes that were strongly inhibited in the ERK/MAPK pathway,
including HRAS, CRK, ETS1, RPS6KA1 and CREB1, were selected for
validation. Using RT-qPCR, ETS1 and RPS6KA1 were found to be highly
expressed in EMT-derived NRK-52E cells. ETS1 and RPS6KA1 gene
expression levels decreased following AMPKα2 KD, which is
consistent with the microarray results, indicating that AMPKα2 KD
inhibits EMT by downregulating ETS1 and RPS6KA1 gene expression
levels. However, HRAS, CRK and CREB1 verification results may not
be consistent with the microarray results. CRK and CREB1 gene
expression levels decreased following AMPKα2 KD as shown in
microarray and RT-qPCR experiments, but there was no statistical
difference in RT-qPCR validation. HRAS gene expression was
decreased following AMPKα2 KD in microarray, but increased in
RT-qPCR validation.
RPS6KA1 is a member of the serine threonine kinase
family and has an N-terminal and a C-terminal kinase domain
(33). The C-terminal domain can
be activated by ERK1/2 phosphorylation and calcium-dependent kinase
(33). Activated RPS6KA1
phosphorylates CREB, NF-κB and other transcription factors
(34). RPS6KA1 cause apoptosis of
renal tubular epithelial cells during renal fibrosis (35), although to the best of our
knowledge, EMT of renal tubular epithelial cells has not yet been
reported. ETS1 is a downstream transcription factor of ERK
(36). ETS1 is widely expressed in
rat kidney and its normal expression levels ensure normal
differentiation and development of the kidney (37). A previous study demonstrated that
ETS1 may serve a role in the differentiation of liver cells via
regulation of the ERK pathway (38). ERK/MAPK cascade activation is
involved in a number of signaling pathways and comprises a class of
important molecules that receive membrane receptor signals and
transport them to the nucleus (39). This cascade serves a key role in
numerous differentiation-associated signaling pathways, including
oxidative stress (39). The
present study demonstrated that, in NRK52E cells with EMT, AMPKα2
KD resulted in downregulation of ETS1 and RPS6KA1 in the MAPK/ERK
pathway, and the EMT process was impaired. This indicated that
AMPKα2 may serve a key role in the EMT of renal tubular epithelial
cells by regulating ETS1 and RPS6KA1. AMPKα2 and RPS6KA1 are
located in the cytoplasm and nucleus, whereas ETS1 is located in
the nucleus. Hypothetically, when oxidative stress occurs,
phosphorylation of AMPKα2 is induced, which may directly or
indirectly interact with and activate ETS1 and RPS6KA1. Activated
ETS1 and RPS6KA1 may act as transcription factors, regulate the
production of EMT-associated proteins, and thereby participate in
regulating cell differentiation. The underlying mechanism and
association between AMPKα2 and its regulation require further
investigation.
The present study had certain limitations.
Subsequent experiments are required to verify whether AMPKα2
similarly regulates RPS6KA1 and ETS1 and whether AMPKα2
phosphorylation occurs and how AMPKα2 regulates RPS6KA1 and ETS1 in
the human renal tubular EMT. To the best of our knowledge, the
present study is the first to demonstrate that AMPKα2 KD may impair
renal tubular EMT by inhibiting the expression levels of RPS6KA1
and ETS1.
In summary, AMPKα2 serves an important regulatory
role in rat renal tubular EMT and this regulation may be achieved
by modulating ETS1 and RPS6KA1 in the ERK/MAPK pathway. However,
specific AMPKα2 regulation of ETS1 and RPS6KA1 requires further
study.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant. no. 81571514).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XY and YY conceived and designed the experiments.
XY, FM and XF performed the experiments. XY, YY, QZ and XL
performed data analysis and wrote the paper. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Weitz M, Portz S, Laube GF, Meerpohl JJ
and Bassler D: Surgery versus non-surgical management for
unilateral ureteric-pelvic junction obstruction in newborns and
infants less than two years of age. Cochrane Database Syst Rev.
7:CD0107162016.PubMed/NCBI
|
|
2
|
Klahr S: Obstructive nephropathy. Intern
Med. 39:355–361. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shihab FS: Do we have a pill for renal
fibrosis? Clin J Am Soc Nephrol. 2:876–878. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Iwano M: EMT and TGF-beta in renal
fibrosis. Front Biosci (Schol Ed). 2:229–238. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rastaldi MP: Epithelial-mesenchymal
transition and its implications for the development of renal
tubulointerstitial fibrosis. J Nephrol. 19:407–412. 2006.PubMed/NCBI
|
|
6
|
Puisieux A, Brabletz T and Caramel J:
Oncogenic roles of EMT-inducing transcription factors. Nat Cell
Biol. 16:488–494. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Martin-Belmonte F and Perez-Moreno M:
Epithelial cell polarity, stem cells and cancer. Nat Rev Cancer.
12:23–38. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu Y: Cellular and molecular mechanisms
of renal fibrosis. Nat Rev Nephrol. 7:684–696. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bronsert P, Enderle-Ammour K, Bader M,
Timme S, Kuehs M, Csanadi A, Kayser G, Kohler I, Bausch D, Hoeppner
J, et al: Cancer cell invasion and EMT marker expression: A
three-dimensional study of the human cancer-host interface. J
Pathol. 234:410–422. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kim SI and Choi ME: TGF-β-activated
kinase-1: New insights into the mechanism of TGF-β signaling and
kidney disease. Kidney Res Clin Pract. 31:94–105. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang D, Sun L, Xian W, Liu F, Ling G,
Xiao L, Liu Y, Peng Y, Haruna Y and Kanwar YS: Low-dose paclitaxel
ameliorates renal fibrosis in rat UUO model by inhibition of
TGF-beta/Smad activity. Lab Invest. 90:436–447. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhao Q, Yang Y, Wang CL, Hou Y and Chen H:
Screening and identification of the differential proteins in kidney
with complete unilateral ureteral obstruction. Int J Clin Exp
Pathol. 8:2615–2626. 2015.PubMed/NCBI
|
|
13
|
Hardie DG, Schaffer BE and Brunet A: AMPK:
An energy-sensing pathway with multiple inputs and outputs. Trends
Cell Biol. 26:190–201. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jeon SM: Regulation and function of AMPK
in physiology and diseases. Exp Mol Med. 48:e2452016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Carling D: AMPK signalling in health and
disease. Curr Opin Cell Biol. 45:31–37. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hawley SA, Boudeau J, Reid JL, Mustard KJ,
Udd L, Mäkelä TP, Alessi DR and Hardie DG: Complexes between the
LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are
upstream kinases in the AMP-activated protein kinase cascade. J
Biol. 2:282003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang X, Pan X and Song J: AMP-activated
protein kinase is required for induction of apoptosis and
epithelial-to-mesenchymal transition. Cell Signal. 22:1790–1797.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang M, Weng X, Guo J, Chen Z, Jiang G and
Liu X: Metformin alleviated EMT and fibrosis after renal
ischemia-reperfusion injury in rats. Ren Fail. 38:614–621. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Qiu S, Xiao Z, Piao C, Zhang J, Dong Y,
Cui W, Liu X, Zhang Y and Du J: AMPKα2 reduces renal epithelial
transdifferentiation and inflammation after injury through
interaction with CK2β. J Pathol. 237:330–342. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Saxena M, Balaji SA, Deshpande N,
Ranganathan S, Pillai DM, Hindupur SK and Rangarajan A:
AMP-activated protein kinase promotes epithelial-mesenchymal
transition in cancer cells through Twist1 upregulation. J Cell Sci.
131:cs2083142018. View Article : Google Scholar
|
|
21
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Benjamini Y and Hochberg Y: Controlling
the false discovery rate: A practical and powerful approach to
multiple testing. J R Statist Soc B. 57:289–300. 1995.
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zeisberg M, Maeshima Y, Mosterman B and
Kalluri R: Renal fibrosis: Extracellular matrix microenvironment
regulates migratory behavior of activated tubular epithelial cells.
Am J Pathol. 160:2001–2008. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sato M, Muragaki Y, Saika S, Roberts AB
and Ooshima A: Targeted disruption of TGF-beta1/Smad3 signaling
protects against renal tubulointerstitial fibrosis induced by
unilateral ureteral obstruction. J Clin Invest. 112:1486–1494.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Stahl PJ and Felsen D: Transforming growth
factor-beta, basement membrane, and epithelial-mesenchymal
transdifferentiation: Implications for fibrosis in kidney disease.
Am J Pathol. 159:1187–1192. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Grande MT and Lopez-Novoa JM: Fibroblast
activation and myofibroblast generation in obstructive nephropathy.
Nat Rev Nephrol. 5:319–328. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Garcia D and Shaw RJ: AMPK: Mechanisms of
cellular energy sensing and restoration of metabolic balance. Mol
Cell. 66:789–800. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tain YL and Hsu CN: AMP-activated protein
kinase as a reprogramming strategy for hypertension and kidney
disease of developmental origin. Int J Mol sci. 19:17442018.
View Article : Google Scholar
|
|
30
|
He K, Guo X, Liu Y, Li J, Hu Y, Wang D and
Song J: TUFM downregulation induces epithelial-mesenchymal
transition and invasion in lung cancer cells via a mechanism
involving AMPK-GSK3β signaling. Cell Mol Life Sci. 73:2105–2121.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mishra R, Cool BL, Laderoute KR, Foretz M,
Viollet B and Simonson MS: AMP-activated protein kinase inhibits
transforming growth factor-beta-induced Smad3-dependent
transcription and myofibroblast transdifferentiation. J Biol Chem.
283:10461–10469. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lim JY, Oh MA, Kim WH, Sohn HY and Park
SI: AMP-activated protein kinase inhibits TGF-β-induced fibrogenic
responses of hepatic stellate cells by targeting transcriptional
coactivator p300. J Cell Physiol. 227:1081–1089. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Anjum R and Blenis J: The RSK family of
kinases: Emerging roles in cellular signalling. Nat Rev Mol Cell
Biol. 9:747–758. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
34
|
Abe JI, Sandhu UG, Hoang NM, Thangam M,
Quintana-Quezada RA, Fujiwara K and Le NT: Coordination of cellular
localization-dependent effects of sumoylation in regulating
cardiovascular and neurological diseases. Adv Exp Med Biol.
963:337–358. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lin L, Shi C, Sun Z, Le NT, Abe JI and Hu
K: The Ser/Thr kinase p90RSK promotes kidney fibrosis by modulating
fibroblast-epithelial crosstalk. J Biol Chem. 294:9901–9910. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Plotnik JP, Budka JA, Ferris MW and
Hollenhorst PC: ETS1 is a genome-wide effector of RAS/ERK signaling
in epithelial cells. Nucleic Acids Res. 42:11928–11940. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lawrence MC, McGlynn K, Shao C, Duan L,
Naziruddin B, Levy MF and Cobb MH: Chromatin-bound
mitogen-activated protein kinases transmit dynamic signals in
transcription complexes in beta-cells. Proc Natl Acad Sci USA.
105:13315–13320. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Paumelle R, Tulasne D, Kherrouche Z, Plaza
S, Leroy C, Reveneau S, Vandenbunder B and Fafeur V: Hepatocyte
growth factor/scatter factor activates the ETS1 transcription
factor by a RAS-RAF-MEK-ERK signaling pathway. Oncogene.
21:2309–2319. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Darling NJ and Cook SJ: The role of MAPK
signalling pathways in the response to endoplasmic reticulum
stress. Biochim Biophys Acta. 1843:2150–2163. 2014. View Article : Google Scholar : PubMed/NCBI
|