Introduction
Multiple endocrine neoplasia type 1 (MEN1) is a
genetic disorder that has an autosomal dominant transmission. It
has an estimated prevalence of ~1 in 30,000. The disease is
characterized by the occurrence of multiple endocrine system tumors
affecting the parathyroid glands, pituitary gland and pancreatic
islets. Some patients may also develop thymic, adrenal cortical,
bronchopulmonary or carcinoid tumors, as well as angiofibromas,
lipomas, collagenomas or meningiomas (1,2).
Chandrasekharappa et al (3) identified the tumor suppressor gene
MEN1 in 1997. The MEN1 gene (OMIM *613733) consists
of 10 exons and it encodes the nuclear scaffold protein menin
(4). Menin is a 610 amino acid
protein that is involved in regulating gene transcription via the
coordination of chromatin remodeling (5). Germline mutations in MEN1 have
been associated with the occurrence of MEN1. Sanger sequencing of
the MEN1 gene is an effective and established method for
diagnosing MEN1 (1,6,7). The
present paper describes the case of a patient with MEN1 that was
associated with a novel frameshift mutation based on the deletion
of a single nucleotide in the MEN1 gene.
Case report
A 52-year-old woman (II-2; Fig. 1) and her daughter (III-1; Fig. 1) viewed the website of our division
and subsequently visited Nihon University Itabashi Hospital to
receive genetic testing and counseling in July 2016. The mother
(II-2; Fig. 1) had already been
diagnosed with MEN1 in another hospital. Her family and medical
history were obtained by interviewing her and drawing the pedigree,
shown in Fig. 1, according to the
Standardized Human Pedigree Nomenclature by the National Society of
Genetic Counselors (8). She
developed hypercalcemia five years previously and visited a general
hospital. She underwent examinations to detect the cause of
hypercalcemia and a single parathyroid tumor was identified. She
received a minimally invasive parathyroidectomy and the resected
tumor was pathologically revealed to be an adenoma. She was
subsequently diagnosed with primary hyperparathyroidism. Brain
magnetic resonance imaging (MRI) indicated there was a 2 mm
pituitary microadenoma. Abdominal computed tomography and MRI also
detected a 10 mm tumor in the pancreas. These two tumors were not
associated with any endocrine disorder; therefore, they were
surveilled using radiological and laboratory tests.
Hyperparathyroidism and the parathyroid adenoma did not recur for
five years. The patient's father (I-1; Fig. 1) died due to laryngeal cancer at
the age of 59 years. The patient's daughter (III-1; Fig. 1) underwent medical examinations
that included blood tests every year and was never diagnosed with
hypercalcemia. In addition, no other members of the patient's
family, including her father and daughter, showed any symptoms of
MEN1.
Blood samples (7 ml) were obtained from both the
patient and her daughter, and they received genetic counseling from
a clinical geneticist. Unfortunately, the patient's son (III-2;
Fig. 1) lived far away and he did
not agree to undergo genetic testing. An aliquot of the blood (1
ml) was processed by QIAamp® DNA Blood Mini Kit (Qiagen
GmbH) and genomic DNA was isolated. Oligonucleotide primers which
were applicable to both amplification by PCR and direct sequencing
by the dideoxy method were designed. The sequence of primers are
shown in Table I. The
concentration of genomic DNA was determined using a NanoDrop OneC
Spectrophotometer (Thermo Fisher Scientific, Inc.). The DNA was
diluted to a final concentration of 100 ng/ml using nuclease-free
water. PCR was performed using a Veriti™ 200 thermal cycler (Thermo
Fisher Scientific, Inc.) with AmpliTaq Gold® 360 Master
Mix (Thermo Fisher Scientific, Inc.). Genomic DNA was used as the
template with primers flanking the target gene. The PCR reaction
conditions were 95°C for 3 min followed by 35 cycles of 95°C for 30
sec, 62°C for 30 sec, and 72°C for 30 sec. Following PCR
amplification, the amplification products were checked by agarose
gel electrophoresis and purified using an ExoSAP-IT purification
kit for PCR products (Affymetrix; Thermo Fisher Scientific, Inc.).
The purification reaction conditions were 37°C for 15 min and 80°C
for 15 min. Bidirectional sequencing was performed using forward
and reverse primers. The reaction was carried out in a Veriti™ 200
thermal cycler using a BigDye® Terminator v1.1 Cycle
Sequencing kit (Thermo Fisher Scientific, Inc.). The reaction
conditions were 96°C for 1 min followed by 25 cycles of 96°C for 10
sec and 60°C for 2 min. The sequencing reaction products were
purified using a BigDye Xterminator™ Purification kit (Thermo
Fisher Scientific, Inc.). Amplicons were examined with a direct
sequencing apparatus and protocol according to Sanger's method
(3130 DNA Analyzer; Thermo Fisher Scientific). Sequence data were
processed using GeneMapper™ ID-X software v1.3 (Thermo Fisher
Scientific, Inc.) and the sequence results were mapped to the human
genome sequence. Reference sequences of the MEN1 gene used
in this analysis were NM_130799.2 for mRNA and NP_570711.1 for
protein from National Center for Biotechnology Information database
(https://www.ncbi.nlm.nih.gov/nuccore/NM_130799.2/;
accessed November 8, 2019).
 | Table I.Sequence of primers. |
Table I.
Sequence of primers.
| Exon | Forward primer | Reverse primer |
|---|
| Exon 2_1 |
5′-GAACCTTAGCGGACCCTGGGAG-3′ |
5′-GAAAGTAGGTGAGGCCGCCAGG-3′ |
| Exon 2_2 |
5′-AGCATTTTCTGGCTGTCAACC-3′ |
5′-AAGGGTTCTGTAAACCATGGAGG-3′ |
| Exon 3 |
5′-CCTTTCCCCATGTTAAAGCAC-3′ |
5′-GTGGCTTGGGCTACTACAGTATG-3′ |
| Exon 4 |
5′-CTTTTCCTGGCTGTCATTCCCTG-3′ |
5′-GTCCCACAGCAAGTCAAGTCTGG-3′ |
| Exon 5 and 6 |
5′-CGATAGGCTAAGGACCCGTTCTC-3′ |
5′-CCCTGCCTCAGCCACTGTTAGG-3′ |
| Exon 7 | 5′-
CATTTGTGCCAGCAGGGCAGCT-3′ |
5′-GAGGGTGGTTGGAAACTGATGGAG-3′ |
| Exon 8 |
5′-CCGATGGTGAGACCCCTTCAGACC-3′ |
5′-CAGCCCCCATGGCCTGTGGAAG-3′ |
| Exon 9 |
5′-CTCTGCTAAGGGGTGAGTAAGAGAC-3′ |
5′-CTGGGCCAGAAAAGTCTGACAAGC-3′ |
| Exon 10_1 |
5′-GGTCCTGGAGTTCCAGCCACTG-3′ |
5′-CTGGAAAGTGAGCACTGGACCCT-3′ |
| Exon 10_2 |
5′-AAGCCTCCTGGGACTGTCGCTG-3 |
′5′-GCTCAGAGTTGGGGGACTAAGG-3′ |
Sequence analysis of the patient's MEN1 gene
revealed a heterozygous c.930delG mutation in exon 5 (Fig. 2A). In the sense strand of the
MEN1 gene, nucleotides 929 and 930 were both guanine
residues. The single nucleotide deletions were deemed to be
c.930delG rather than c. 929delG according to Sequence Variant
Nomenclature, version 19.01, by the Human Genome Variation Society
(https://varnomen.hgvs.org/recommendations/DNA/variant/deletion/;
accessed November 8, 2019). Sequencing of the antisense strand
showed a complementary deletion of cytosine 930, confirming the
analysis of the sense strand (Fig.
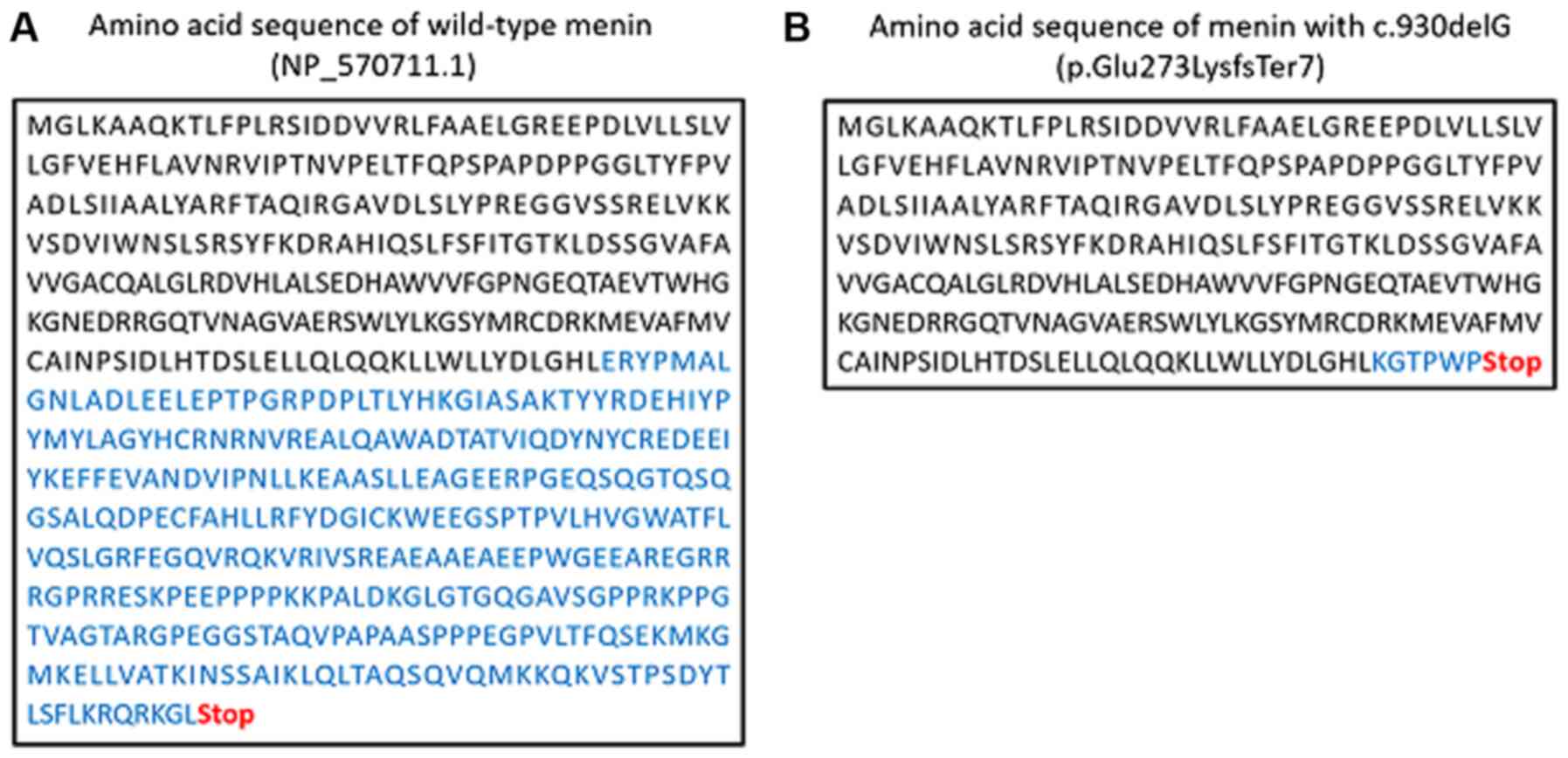
2B). Deletion of this single nucleotide caused a frameshift
that generated a stop codon in the downstream sequence
(NP_570711.1: p.Glu273LysfsTer7; Fig.
3). The tertiary structure of the mutant menin protein
predicted using Swiss-Model revealed that the amino acid sequence
of the mutant protein was markedly shorter than that of the
wild-type protein (Fig. 4)
(9). However, analysis of the
MEN1 gene of the patient's daughter showed no evidence of
any mutation, including c.930delG. Considering no particular
disease symptoms were present, the patient's daughter was not
diagnosed with MEN1.
Discussion
A novel heterozygous MEN1 gene
deletion/frameshift mutation (c.930delG) was identified in a
Japanese patient with MEN1. To the best of our knowledge, this is
the first report on this mutation.
Diagnostic criteria for MEN1 include the presence of
multiple primary MEN1-related tumors (including parathyroid
adenoma, entero-pancreatic endocrine tumor and pituitary adenoma)
(1,10). As described above, the patient in
the present case (II-1; Fig. 1)
developed a parathyroid adenoma and a pituitary adenoma. Depending
on categorization, the patient had two MEN1-related tumors and was
therefore definitively diagnosed with MEN1. The deletion/frameshift
mutation identified in the present case should be considered
pathogenic.
The definition of familial MEN1 is the presence of
at least one MEN1 case plus at least one first-degree relative with
one of three MEN1-related tumors (10). As shown in Fig. 1, familial MEN1 was not observed in
the present family and the patient seemed to be a sporadic case.
Hai et al (11) previously
revealed that, even in sporadic cases, the MEN1 gene
sometimes has pathogenic mutations, which is similar to the present
case. The patient's son (III-2; Fig.
1) did not undergo genetic testing; however, he may be at risk
of developing MEN1 in the future.
Some patients with MEN1 who undergo MEN1 gene
testing are shown to be negative for a MEN1 mutation
(11). De Laat et al
(12) analyzed 322 Dutch patients
with MEN1 and reported that 90.7% of cases were MEN1
mutation positive, whereas the remaining 9.3% were MEN1
mutation negative. Their study also demonstrated that
mutation-positive cases were diagnosed earlier (age 33 years vs.
age 46 years) and exhibited a poorer prognosis (survival to age 73
years vs. survival to age 87 years) than mutation-negative cases.
In addition, mutation-negative patients developed no third primary
MEN1-related tumor during the course of follow-up, despite ~half of
the mutation-positive patients showing all three primary
manifestations (12). Ellard et
al (13) described mutation
detection rates of 79, 37 and 15% in patients with three, two and
one primary MEN1-related tumor, respectively. Therefore, the
mutation detection rate was shown to be associated with the number
of MEN1-related tumors (13). The
patient in the present case was mutation positive and presented
with two primary MEN1 manifestations. In the future it may be
revealed that the patient's pancreatic tumor is a pancreatic
endocrine tumor.
Several articles have reviewed previously described
MEN1 gene mutations. Marini et al (14) examined over 400 different germline
or somatic mutations and reported that 50% of MEN1 mutations
were frameshift mutations, 20% were nonsense mutations, 20% were
missense mutations and 7% were splice-site defects. Lemos and
Thakker (15) also reviewed 1,133
germline mutations in the MEN1 gene and classified 41% as
frameshift mutations, 23% as nonsense mutations, 20% as missense
mutations, 9% as splice-site mutations, 6% as in-frame deletions or
insertions and 1% as gross deletions. In addition, Concolino et
al (16) reviewed 576
MEN1 mutations and reported frameshift mutations in 42% of
the cases, missense mutations in 25.5% of the cases, nonsense
mutations in 14% of the cases, splice-site mutations in 10.5% of
the cases, in-frame deletions or insertions in 5.5% of the cases,
and gross deletions in the remaining 2.5% of the cases. These
reviews described very similar results, with frameshift mutations
representing the most frequent type of mutation at 40–50%. Thus,
the novel mutation identified in the present case was in the most
frequent class of MEN1 mutations.
The nuclear scaffold protein menin is the product of
the MEN1 gene. Menin acts as a tumor suppressor and
interacts with numerous proteins, including transcription factor
JunD, NF-κB, SMAD3, protein-energy malnutrition, non-metastatic
protein 23 homolog 1, replication protein A2, non-muscle myosin
heavy chain IIA, Fanconi anemia group D2, paired amphipathic helix
protein Sin3, histone deacetylase 1, activator of S-phase kinase
and checkpoint suppressor 1 (15).
Menin with the p.Glu273LysfsTer7 mutation lacks the amino acid
sequence spanning from the middle of exon 5 to the C-terminus,
which is hypothesized to be essential for binding with the 11
above-mentioned proteins (15).
It must be noted that the lack of functional
experiments to validate the results of the present study is a
limitation of this research. Future studies aimed at confirming the
pathogenicity of the c.930delG frameshift mutation are thus
warranted.
In conclusion, a novel frameshift heterozygous
c.930delG mutation in the MEN1 gene that is associated with
MEN1 was identified.
Acknowledgements
Not applicable.
Funding
The present study was supported by a Grant-in-Aid
from the Japanese Ministry of Education, Culture, Sports, Science
and Technology (grant no. 16K08978) and by a grant from the Health
Sciences Research Institute, Inc., Yokohama, Japan, for the
Division of Companion Diagnostics, Department of Pathology of
Microbiology, Nihon University School of Medicine, Tokyo, Japan
grant no. 20131001).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HN, HU and TN designed the study. TN supervised the
project. HN performed the experiments. HN and HU analyzed the data.
HN and HU drafted and wrote the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Genetic testing and the overall study protocol for
advanced medical care were approved by the Director of the Nihon
University Itabashi Hospital on February 1, 2016, according to the
Ministry of Health, Labor and Welfare Japan. Written informed
consent was obtained from both subjects prior to collection of the
samples, which were used for genomic analysis.
Patient consent for publication
Written informed consent was obtained from both
subjects for publication.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Thakker RV, Newey PJ, Walls GV, Bilezikian
J, Dralle H, Ebeling PR, Melmed S, Sakurai A, Tonelli F and Brandi
ML; Endocrine Society, : Clinical practice guidelines for multiple
endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab.
97:2990–3011. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mark SJ: Recent topics around multiple
endocrine neoplasia type 1. J Clin Endocrinol Metab. 103:1296–1301.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chandrasekharappa SC, Guru SC, Manickam P,
Olufemi SE, Collins FS, Emmert-Buck MR, Debelenko LV, Zhuang Z,
Lubensky IA, Liotta LA, et al: Positional cloning of the gene for
multiple endocrine neoplasia-type 1. Science. 276:404–407. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Guru SC, Goldsmith PK, Burns AL, Marx SJ,
Spiegel AM, Collins FS and Chandrasekharappa SC: Menin, the product
of the MEN1 gene, is a nuclear protein. Proc Natl Acad Sci USA.
95:1630–1634. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Canaff L, Vanbellinghen JF, Kaji H,
Goltzman D and Hendy GN: Impaired transforming growth factor-β
(TGF-β) transcriptional activity and cell proliferation control of
a menin in-frame deletion mutant associated with multiple endocrine
neoplasia type 1 (MEN1). J Biol Chem. 287:8584–8597. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hyde SM, Cote GJ and Grubbs EG: Genetics
of multiple endocrine neoplasia type 1/multiple endocrine neoplasia
type 2 syndromes. Endocrinol Metab Clin North Am. 46:491–502. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Luo Y, Sun Y, Zhu X and Li X: Analysis of
MEN1 c.482G>A (p.Gly161Asp) mutation in a pedigree with familial
multiple endocrine neoplasia type 1. Mol Med Rep. 16:8973–8976.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bennett RL, French KS, Resta RG and Doyle
DL: Standardized human pedigree nomenclature: Update and assessment
of the recommendations of the National Society of Genetic
Counselors. J Genet Couns. 17:424–433. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Guex N, Peitsch MC and Schwede T:
Automated comparative protein structure modeling with SWISS-MODEL
and Swiss-PdbViewer: A historical perspective. Electrophoresis. 30
(Suppl 1):S162–S173. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Brandi ML, Gagel RF, Angeli A, Bilezikian
JP, Beck-Peccoz P, Bordi C, Conte-Devolx B, Falchetti A, Gheri RG,
Libroia A, et al: Guidelines for diagnosis and therapy of MEN type
1 and type 2. J Clin Endocrinol Metab. 86:5658–5671. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hai N, Aoki N, Shimatsu A, Mori T and
Kosugi S: Clinical features of multiple endocrine neoplasia type 1
(MEN1) phenocopy without germline MEN1 gene mutations: Analysis of
20 Japanese sporadic cases with MEN1. Clin Endocrinol (Oxf).
52:509–518. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
De Laat JM, van der Luijt RB, Pieterman
CR, Oostveen MP, Hermus AR, Dekkers OM, de Herder WW, van der
Horst-Schrivers AN, Drent ML, Bisschop PH, et al: MEN1 redefined, a
clinical comparison of mutation-positive and mutation-negative
patients. BMC Med. 14:1822016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ellard S, Hattersley AT, Brewer CM and
Vaidya B: Detection of an MEN1 gene mutation depends on clinical
features and supports current referral criteria for diagnostic
molecular genetic testing. Clin Endocrinol (Oxf). 62:169–175. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Marini F, Falchetti A, Del Monte F,
Carbonell Sala S, Gozzini A, Luzi E and Brandi ML: Multiple
endocrine neoplasia type 1. Orphanet J Rare Dis. 1:382006.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lemos MC and Thakker RV: Multiple
endocrine neoplasia type 1 (MEN1): Analysis of 1336 mutations
reported in the first decade following identification of the gene.
Hum Mutat. 29:22–32. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Concolino P, Costella A and Capoluongo E:
Multiple endocrine neoplasia type 1 (MEN1): An update of 208 new
germline variants reported in the last nine years. Cancer Genet.
209:36–41. 2016. View Article : Google Scholar : PubMed/NCBI
|