Introduction
Ischemia-reperfusion (I-R) injury plays an important
role in the pathogenesis of several diseases, such as stroke,
coronary heart disease and multiorgan failure (1–3). Due
to the high metabolic demands of renal tubular cells, the kidney is
exceptionally vulnerable to I-R, and I-R injury is the most common
cause of acute kidney injury (4).
I-R injury consists of two consecutive, but distinct, stages.
During ischemia, cell energy collapse induces cell death, whereas
during reperfusion, cell death results from increased reactive
oxygen species (ROS) production (1–3).
Regarding renal tubular epithelial cells, a study demonstrated that
under anoxia, cells die through apoptosis, whereas reoxygenation
induces lipid peroxidation-induced cell death, also known as
ferroptosis (5,6). The latter has also been detected in
isolated mouse renal tubules, confirming the different
pathophysiology of ischemia- and reperfusion-induced cell injury
(7).
Ferroptosis is a type of regulated iron-dependent
cell necrosis mainly caused by increased redox imbalance and
characterized by extensive lipid peroxidation and, eventually,
severe cell membrane dysfunction (8). Unlike uncontrolled cell necrosis,
which occurs acutely after a severe physical, chemical, or
mechanical insult, ferroptosis and other types of regulated cell
necrosis involve a genetically encoded machinery, occur in a
delayed manner, and could be considered as a part of an adaptive
response that unsuccessfully attempts to restore cellular
homeostasis (8). Thus, contrary to
uncontrolled necrosis, for ferroptosis, pharmacological
intervention is possible (8).
Apoptosis is another type of programmed cell death, which differs
in many ways from the various types of regulated cell necrosis. A
notable difference is that during apoptosis, cell membrane rupture
does not take place, and the release of cytoplasmic content and the
ensuing inflammation are avoided (8).
When metabolizing their substrates, the enzymes of
the cytochrome P450 superfamily (CYP) produce ROS (9). By oxidizing their substrates, often
drugs or xenobiotics, CYPs increase their polarity, aiding to their
excretion. Substrate oxidation by CYPs is achieved by a six-step
reaction during which heme-thiolate iron fluctuates between ferric
(Fe+3) and ferrous (Fe+2) form, interacts
with oxygen, and eventually oxidize the substrate. The general
reaction is RH + 2H+ + 2e− → ROH +
H2O. However, two shunts exist in the six-step CYP
catalytic pathway. The first takes place in step 3 of the reaction
and gives rise to superoxide. The second occurs in step 4 of the
reaction and releases hydrogen peroxide (9,10). In
addition to the above mechanism of CYP-induced ROS production, a
supplementary mechanism has also been identified. CYPs metabolize
arachidonic acid to produce 20-hydroxyeicosatetraenoic acid
(20-HETE). The latter increases NADPH oxidase activity resulting in
further ROS production (11,12).
The role of CYPs in producing ROS and eventually cell death has
been confirmed in experimental models of heart and liver I-R injury
(13–17).
Certain CYPs are under the transcriptional control
of the aryl hydrocarbon receptor (AhR). Non-ligand bound AhR is
retained in the cytoplasm as an inactive complex with various
factors, which also protect AhR from proteasomal degradation. When
activated by exogenous or endogenous ligands, AhR is released,
translocates into the nucleus, forms a complex with HIF-1β, and
transcribes the CYPs CYP1A1, CYP1A2, and CYP1B1 (18,19).
Interestingly, activated AhR becomes vulnerable to proteasomal
degradation, leading to decreased AhR cellular levels (20–24).
Several studies have suggested that AhR-induced CYP
overexpression results in ROS production (25–29).
For instance, in human aortic endothelial cells,
2,3,7,8-tetrachlorodibenzo-p-dioxin activates AhR and induces
CYP1A1 expression and ROS production. In this model, small
interfering RNA targeting of AhR or CYP1A1 decreased ROS production
significantly, confirming that CYPs could mediate AhR-induced ROS
production (27). However, the role
of this pathway during reperfusion remains unclear.
Although AhR induces oxidative stress by increasing
CYPs, it also promotes the expression of nuclear factor erythroid
2-related factor 2 (Nrf2) (30–32).
Nrf2 is kept in the cytoplasm by kelch-like ECH-associated protein
1 (KEAP1) and cullin-3. Cullin-3 ubiquitinates Nrf2 leading it to
proteasomal degradation. Under increased ROS levels, the
conformation of KEAP1 changes releasing Nrf2, which translocates
into the nucleus and transcribes genes encoding antioxidant and
anti-ferroptotic proteins, as well as proteins required for
detoxification (33,34). At first glance, AhR-induced Nrf2
might attenuate the oxidative stress produced by AhR-induced CYPs
(30–32). A study has demonstrated that the
kynurenine-AhR pathway mediates brain damage after experimental
stroke, a condition that involves both phases of I-R injury
(35). Moreover, inhibition of AhR
through downregulation of CYPs and ROS protects newborn rat lungs
from hyperoxia (25). Thus,
evidence indicates that in the context of oxidative stress, the
balance between AhR-induced CYPs and AhR-induced Nrf2 tilts towards
the former. Besides the detection of a possible detrimental effect
of AhR activation in I-R injury or hyperoxia, the aforementioned
studies did not assess Nrf2, nor the effect the reperfusion per se.
Thus, the precise interaction between AhR and Nrf2 during the
reperfusion stage of I-R-injury has not been evaluated.
Another transcription factor that is inevitably
affected by I-R is hypoxia-inducible factor-1α (HIF-1α). Under
normoxia, HIF-prolyl-hydroxylases are active, leading to HIF-1α
proteasomal degradation. Under anoxia, hydroxylation does not take
place, and HIF-1α is released from the Von Hippel-Lindau E3
ubiquitin ligase, escaping proteasomal degradation. The accumulated
HIF-1α forms a heterodimer with HIF-1β and transcribes many genes
necessary for cellular adaptation to anoxia (36,37).
However, activated AhR also forms a heterodimer with HIF-1β (33,
34). In certain experimental models of AhR activation, the
competition between AhR and HIF-1α for HIF-1β results in reduced
HIF-1α transcriptional activity, and vice versa (38–40).
However, this interaction between AhR and HIF-1α has not been
evaluated in models of I-R injury.
Most studies have evaluated I-R-injury as a one-step
process (1–3), yet I-R consists of two consecutive but
different stages, the first of ischemia, and the second of
reperfusion (1–3). The present study aimed to evaluate the
role of AhR during the reoxygenation phase, and more precisely, its
effect on ROS producing CYPs, Nrf2, HIF-1α and cell survival. A
cell culture model was established, in which primary murine renal
proximal tubular epithelial cells (RPTECs) were subjected to
anoxia, a condition that imitates ischemia, and then to
reoxygenation and replenishment with fresh culture medium, a
condition that mimics reperfusion. RPTECs are particularly
vulnerable to I-R injury (4).
To evaluate the role of AhR in reoxygenation-induced
cell death the specific AhR inhibitor CH223191 was applied at the
onset of reoxygenation (41). The
HIF-prolyl-hydroxylase inhibitor roxadustat was used to assess the
possible role of interaction between AhR and HIF-1α (42). Finally, the ferroptosis inhibitor
α-tocopherol (43) was applied at
the onset of reoxygenation to examine the effect of AhR on
ferroptotic cell death specifically.
Materials and methods
Cell culture and treatment
Primary C57BL/6 murine RPTECs (cat. no. C57-6015)
and Complete Epithelial Cell Medium kit (cat. no. M6621) were
purchased from Cell Biologics. Primary cells were used instead of
genetically altered cell lines in order to obtain reliable results.
Second-passage RPTECs were cultured in 96-well plates
(104 cells/well) or 6-well plates (3×105
cells/well) at 37°C. In order to reproduce ischemic conditions,
cells were placed in a GasPak™ EZ Anaerobe Container System with
Indicator (cat. no. 26001; BD Biosciences), which reduces the
concentration of oxygen to <1%. To replicate reperfusion, cells
were then removed from the Anaerobe Container System and washed
with PBS. Fresh culture medium was added, and cells were cultured
in a humidified atmosphere containing 5% CO2.
The selection of appropriate culture time points was
based on a previous study demonstrating that primary mouse RPTECs
died after 48 h of anoxia or 4 h of reoxygenation (5). The latter was also confirmed in the
present study. Thus, in this study, cells were subjected to anoxia
for 24 h and subsequently to reoxygenation for 2 h, as after these
time points, cell death precludes any further reliable
analysis.
At the onset of the reoxygenation period, 3 µM of
the AhR inhibitor CH223191 (cat. no. C8124; Sigma-Aldrich; Merck
KGaA) or 100 µM of the ferroptosis inhibitor α-tocopherol (cat. no.
T3251; Sigma-Aldrich; Merck KGaA) or the HIF-1α activator
roxadustat at a concentration of 10 µg/ml (cat. no. FG-4592;
Selleck Chemicals) were added.
RPTECs cultured under normoxic conditions for a
total of 26 h were used as control. Two hours before the end of the
26-h period, control cells were washed with PBS and fresh culture
medium was added.
Cell imaging
Cell images were captured at the onset of
reoxygenation, then at 2-h intervals for 24 h. An Axiovert 40C
inverted microscope (Carl Zeiss AG) equipped with a digital camera
(3MP USB2.0 Microscope Digital Camera; Amscope) and related
software (Amscope; v. ×64, 3.7.3036) were used.
Assessment of ROS production, lipid
peroxidation and cell necrosis
To assess ROS production, RPTECs were cultured in
96-well plates. At the end of the reoxygenation period, 5 µM of the
fluorogenic probe CellROX® Deep Red Reagent (Invitrogen;
Thermo Fisher Scientific, Inc.) was added, and cells were incubated
at 37°C for 30 min. Then, cells were washed with PBS, and
fluorescence signal intensity was measured with an
EnSpire® Multimode plate reader (PerkinElmer, Inc.).
To assess lipid peroxidation, RPTECs were cultured
in 6-well plates. At the end of the reoxygenation period, the end
product of lipid peroxidation malondialdehyde (MDA) was measured
fluorometrically in cell lysates using the Lipid Peroxidation (MDA)
Assay Kit (cat. no. ab118970; Abcam). The required cell lysis
buffer was provided along with the aforementioned kit. The lower
detection limit of the kit is 0.1 nmol. Prior to MDA measurement, a
Bradford assay was performed to adjust lysate volumes to an equal
protein mass of 100 µg.
To assess cell necrosis and the effect of AhR,
RPTECs were cultured in 96-well plates in the presence or absence
of the ferroptosis inhibitor α-tocopherol or the AhR inhibitor
CH223191. The above inhibitors were added into the cell cultures at
the beginning of the reoxygenation period. At the end of the
reoxygenation period, cell necrosis was evaluated using a lactate
dehydrogenase (LDH) release assay with the Cytotox Non-Radioactive
Cytotoxic Assay kit (Promega Corporation). Cell necrosis was
calculated as (LDH in the supernatant/Total LDH) × 100.
Western blot analysis
RPTECs were cultured in 6-well plates. Once the
reoxygenation period was over, cells were lysed with the T-PER
tissue protein extraction reagent (Thermo Fisher Scientific, Inc.)
supplemented with phosphatase inhibitor (Roche Diagnostics) and
protease inhibitor (Sigma-Aldrich; Merck KGaA). Protein was
quantified using a Bradford assay (Sigma-Aldrich; Merck KGaA). For
western blotting, 10 µg of protein from each sample were
electrophoresed in SDS-PAGE (4–12% Bis-Tris) gels (Thermo Fisher
Scientific, Inc.) and transferred to PVDF membranes (Thermo Fisher
Scientific, Inc.). Tris-buffered saline with 0.1% Tween-20 and 5%
non-fat dry milk was used as blocking buffer. Blots were incubated
at 4°C for 16 h with the primary antibody, then at room temperature
for 30 min with the secondary antibody. For the enhanced
chemiluminescent detection of the western blot bands, the
LumiSensor Plus Chemiluminescent HRP Substrate kit (GenScript
Corporation) was used. Whenever reprobing of the PVDF blots was
necessary, the Restore Western Blot Stripping Buffer (Thermo Fisher
Scientific, Inc.) was used. The Image J software v. 1.51t (National
Institutes of Health) was used for densitometric analysis.
Primary antibodies were specific for AhR (1:200;
cat. no. sc-133088; Santa Cruz Biotechnology, Inc.), cytochrome
P450 family 1 subfamily A member 1 (CYP1A1; 1:500; cat. no.
sc-25304; Santa Cruz Biotechnology, Inc.), Nrf2 (1:1,000; cat. no.
TA343586; OriGene Technologies, Inc.), superoxide dismutase 3
(SOD-3; 1:100; cat. no. sc-271170; Santa Cruz Biotechnology, Inc.),
cystine-glutamate antiporter (xCT, also known as SLC7A11; 1:1,000;
cat. no. ANT-111; Alomone Labs), HIF-1α (1:500; cat. no. sc-10790;
Santa Cruz Biotechnology, Inc.), LDH-A (1:1,000; cat. no. 2012;
Cell Signaling Technology, Inc.), activated cleaved caspase-3 (CC3;
1:1,000; cat. no. ab13847; Abcam) and β-actin (1:2,500; cat. no.
4967; Cell Signaling Technology, Inc.). Anti-mouse IgG, HRP-linked
antibody (1:1,000; cat. no. 7076; Cell Signaling Technology, Inc.)
or anti-rabbit IgG, HRP-linked antibody (1:1,000; cat. no. 7074;
Cell Signaling Technology, Inc.) were used as secondary
antibodies.
Statistical analysis
One-sample Kolmogorov-Smirnov test verified that
all, except one, variables were normally distributed. One-way
analysis of variance and Bonferroni's correction test were used for
comparison of means. Results are presented as the mean ± SEM of six
experiments. The analysis of the cell imaging results was carried
out using the Kruskal-Wallis H test and Dunn's post hoc test, since
this variable did not follow the normal distribution. Statistical
analysis was performed with the SPPS version 20 (IBM Corp.).
P<0.05 was considered to indicate a statistically significant
difference.
Results
Reoxygenation activates AhR and
increases CYP1A1 expression, while CH223191 prevents both
Reoxygenation decreased AhR levels to 0.68±0.05 of
the control (P=0.001). The AhR inhibitor CH223191 inhibited the
reoxygenation-induced changes in AhR levels, increasing the AhR
levels 0.99±0.08 of the control (P not significant compared with
the control; P=0.003 compared with cells subjected to reoxygenation
only). Of note, under normoxic conditions, CH223191 increased AhR
level to 1.29±0.04 of the control (P=0.03). Since AhR is degraded
after its activation, these results indicated that reoxygenation
activated AhR, and that this effect was inhibited by the AhR
specific inhibitor CH223191 (Fig. 1A
and B).
The expression of the AhR transcriptional target
CYP1A1 confirmed the aforementioned conclusion. Reoxygenation
increased CYP1A1 expression to 1.44±0.01 of the control (P=0.033).
CH223191 prevented the reoxygenation-induced change of CYP1A1
expression since, in this case, CYP1A1 level equaled 1.04±0.07 of
the control (P not significant compared with the control; P=0.014
compared with cells subjected to reoxygenation only). Notably,
under normoxic conditions, CH223191 decreased CYP1A1 expression
levels to 0.66±0.05 of the control (P=0.033; Fig. 1A and C).
Inhibition of AhR prevents
reoxygenation-induced ROS production and ameliorates lipid
peroxidation
Reoxygenation increased ROS production. Considering
the ROS production in control cells as 100%, in cells subjected to
reoxygenation, ROS levels doubled, compared with the control
(199.9±9.6%; P<0.001). The AhR inhibitor CH223191 prevented
reoxygenation-induced ROS production, with ROS level only reaching
106.0±0.6% of the control (P not significant compared with the
control; P<0.001 compared with cells subjected to reoxygenation
only). Under normoxic conditions, CH223191 did not affect ROS
production (102.5±2.9% of the control; P not significant; Fig. 2A).
 | Figure 2.Inhibition of AhR prevents
Reox-induced ROS production and attenuates lipid peroxidation and
ferroptosis. RPTECs were cultured under ctrl conditions or
subjected to Reox, with or without the AhR inhibitor CH223191 or
the ferroptosis inhibitor tocophe. (A and B) Reox induced ROS
production and lipid peroxidation, assessed by MDA levels. CH223191
prevented Reox-induced ROS production and attenuated lipid
peroxidation. Reox caused cell necrosis. Necrosis was abrogated by
α-tocopherol, demonstrating ferroptosis. *P<0.05 vs. ctrl;
#P<0.05 vs. ctrl CH223191; ^P<0.05 vs.
Reox; +P<0.05 vs. Reox CH223191. (C) CH223191
ameliorated Reox-induced cell necrosis. Data are presented as the
mean ± SEM of six independent experiments. *P<0.05 vs. ctrl;
#P<0.05 vs. ctrl tocophe; ^P<0.05 vs.
ctrl CH223191; +P<0.05 vs. Reox,
&P<0.05 vs. Reox tocophe; $P<0.05
vs. Reox CH223191. AhR, arylhydrocarbon receptor; RPTEC, renal
proximal tubular epithelial cell; ROS, reactive oxygen species;
MDA, malondialdehyde; ctrl, control; Reox, reoxygenation; tocophe;
α-tocopherol. |
Lipid peroxidation, assessed by MDA levels, followed
the same trend as ROS. In control cells, cellular MDA concentration
was 3.0±0.2 µM, while under reoxygenation, MDA concentration
increased to 42.6±0.5 µM (P<0.001). CH223191 decreased MDA in
RPTECs subjected to reoxygenation to 21.3±0.7 µM (P<0.001,
compared with cells subjected to reoxygenation only). Notably,
inhibition of AhR under control conditions did not alter MDA
significantly (3.6±0.3 µM; Fig.
2B).
Inhibition of AhR attenuates
reoxygenation-induced ferroptosis, while apoptosis was not
observed
Cell imaging indicated that RPTECs were extremely
vulnerable to reoxygenation since in all performed experiments they
died after 4 h. The specific AhR inhibitor CH223191 rescued RPTECs
from reoxygenation induced cell death since, in this case, in all
experiments, cells remained alive at the 24-h timepoint
(P<0.001; Fig. 3A and B).
In the control cells, LDH release assay indicated a
percentage of necrotic cells equal to 10.7±0.6%. In cells subjected
to reoxygenation, the percentage of necrotic cells increased
significantly to 23.3±0.6%, compared with control cells
(P<0.001). The inhibitor of ferroptosis α-tocopherol prevented
reoxygenation-induced cell necrosis completely since, in this case,
the percentage of cell necrosis was equal to that detected in
control cells (10.7±0.2%; P not significant compared to the control
cells; P<0.001 compared to cells subjected to reoxygenation
only). Thus, reoxygenation-induced cell necrosis may be mediated
through the ferroptotic pathway. Moreover, the AhR inhibitor
CH223191 ameliorated reoxygenation-induced cell necrosis
considerably. Compared with the cells subjected to reoxygenation,
in cells subjected to reoxygenation and treated with the AhR
inhibitor, cell necrosis percentage decreased from 23.3±0.6 to
15.3±0.4% (P<0.001). Of note, under control conditions, CH223191
was not toxic for RPTECs, since the percentage of cell necrosis was
10.7±0.8% (P not significant; Fig.
2C).
Apoptosis was assessed using the levels of CC3.
Neither reoxygenation nor CH223191 had any effect on CC3 levels.
Considering the level of CC3 in the control cells as 1, CC3 level
equaled 1.0±0.05 in cells treated with the inhibitor, 1.1±0.08 in
RPTECs subjected to reoxygenation, and 1.0±0.03 in cells subjected
to reoxygenation and treated with CH223191 (P not significant;
Fig. 4).
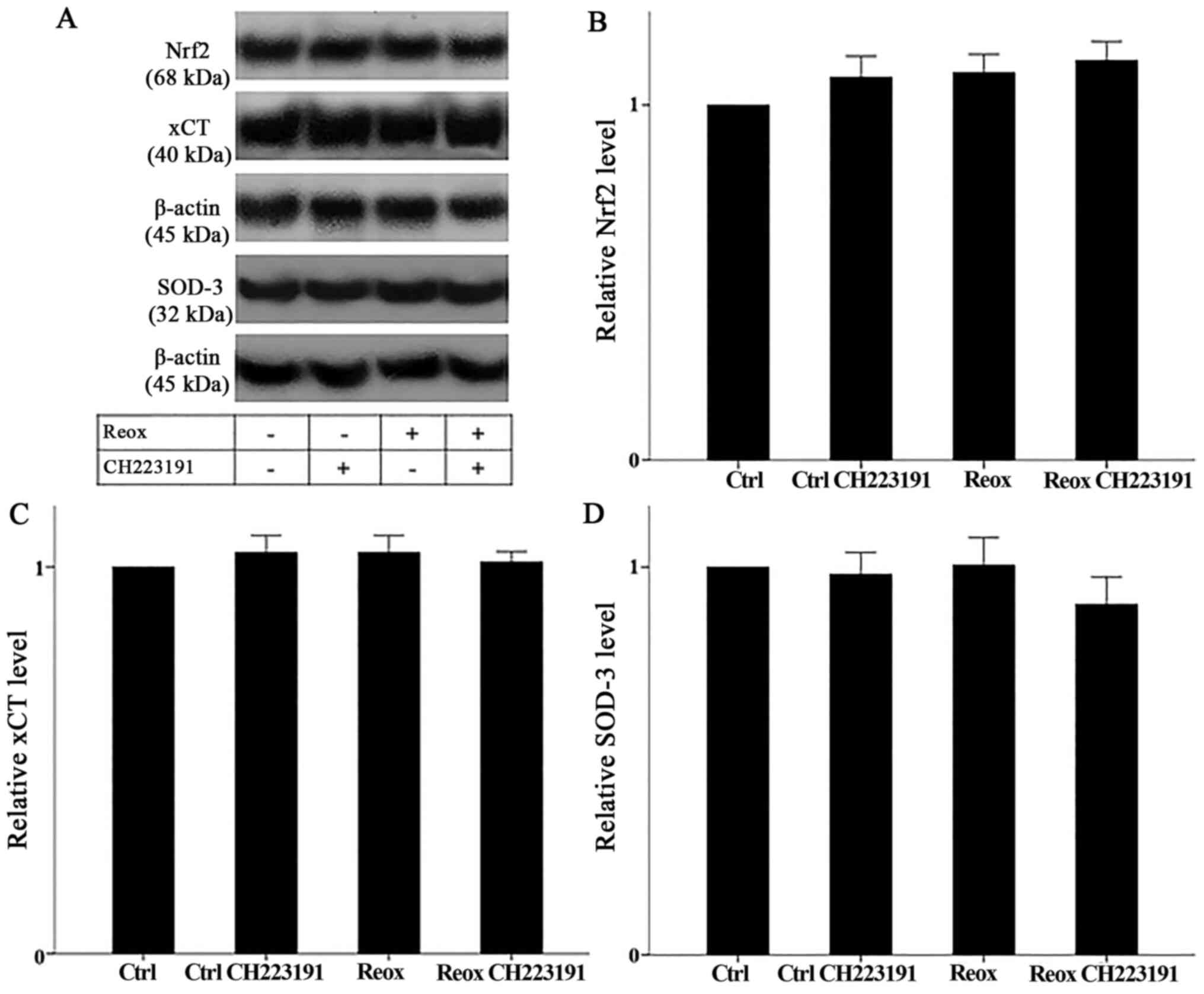
AhR activation status does not affect
Nrf2 activation or transcriptional activity
Neither reoxygenation nor the AhR inhibitor CH223191
affected Nrf2 levels (which correspond to its activation). In
RPTECs subjected to reoxygenation, Nrf2 levels equaled 1.01±0.07 of
the control (P not significant). In RPTECs subjected to
reoxygenation and treated with CH223191, Nrf2 levels equaled
0.97±0.03 of the control (P not significant, compared with control
cells). Under normoxia, RPTECs treated with CH223191, Nrf2 levels
equaled 0.98±0.06 of the control (P not significant; Fig. 5A and B). Thus, AhR does not affect
Nrf2 activation status.
The stable expression of the Nrf2 transcriptional
targets xCT and SOD-3 confirmed the aforementioned conclusion.
Under reoxygenation, xCT and SOD-3 expression remained stable and
equaled 1.04±0.04 and 1.01±0.07 of the control, respectively (P not
significant in both cases). In RPTECs subjected to reoxygenation
and treated with CH223191, xCT and SOD-3 expression also remained
stable. In this case, xCT and SOD-3 levels equaled 1.01±0.03 and
0.91±0.07 of the control, respectively (P not significant in both
cases). Moreover, under normoxic conditions, CH223191 did not alter
the expression of the above proteins. xCT and SOD-3 expression
levels equaled 1.04±0.04 and 0.98±0.06 of the control, respectively
(P not significant in both cases; Fig.
5A, C and D).
AhR activation status does not affect
HIF-1α levels or its transcriptional targets
Neither reoxygenation nor CH223191 affected HIF-1α
levels (which correspond to its activation). In cells treated with
the AhR inhibitor CH223191, cells subjected to reoxygenation, and
cells subjected to reoxygenation and treated with CH223191, HIF-1α
levels were 1.10±0.07, 0.92±0.11, and 1.12±0.12 of the control,
respectively (P not significant in any case; Fig. 6A and B). Thus, AhR does not alter
HIF-1α activity.
The AhR inhibitor CH223191 did not affect the
expression of the HIF-1α transcriptional target LDH-A, confirming
the aforementioned conclusion. Compared to the control cells, in
cells treated with CH223191, LDH-A expression remained stable
(1.12±0.02 of the control; P not significant). Reoxygenation
increased LDH-A expression to 1.56±0.16 of the control (P=0.008).
However, CH223191 did not result in any further change to LDH-A
expression, (1.62±0.15 of the control; P not significant compared
with reoxygenation only; Fig. 6A and
C).
Roxadustat increases HIF-1α level and
transcriptional activity but does not affect ROS production, lipid
peroxidation, or cell survival
Under normoxia, roxadustat increased HIF-1α to
1.68±0.09 of the control (P<0.001). Reoxygenation did not alter
HIF-1α, compared with the control cells (0.95±0.07 of the control;
P not significant). In RPTECs subjected to reoxygenation and
treated with roxadustat, HIF-1α increased to 1.77±0.03 of the
control (P<0.001, compared with the control and cells subjected
to reoxygenation alone; Fig. 7A and
B). Thus, the HIF-1α activator roxadustat increased HIF-1α
level both in control cells and in cells subjected to
reoxygenation.
 | Figure 7.Roxadustat increases HIF-1α level and
transcriptional activity but does not affect ROS production, lipid
peroxidation, or cell necrosis. RPTECs were cultured under ctrl
conditions or subjected to Reox with or without the HIF-1α
activator roxadustat. (A) Representative western blot images of
HIF-1α levels (corresponding to its activation status) and its
transcriptional target LDH-A. (B and C) Statistical analysis of the
western blots. Roxadustat enhanced HIF-1α levels in both ctrl cells
and cells subjected to Reox. Roxadustat also upregulated LDH-A
expression in both ctrl cells and cells subjected to Reox. Compared
to ctrl cells, LDH-A levels increased in cells subjected to Reox.
(D) ROS production, (E) lipid peroxidation and (F) cell necrosis
were also assessed. Roxadustat did not affect Reox-induced ROS
production, lipid peroxidation, and cell necrosis. Data are
presented as the mean ± SEM. *P<0.05 vs. ctrl;
#P<0.05 vs. ctrl roxadustat; ^P<0.05
vs. Reox; +P<0.05 vs. Reox roxadustat. RPTEC, renal
proximal tubular epithelial cell; HIF-1α, hypoxia-inducible factor
1α; LDH-A, lactate dehydrogenase-A; ROS, reactive oxygen species;
MDA, malondialdehyde; ctrl, control; Reox, reoxygenation. |
Similarly, roxadustat increased the expression of
the HIF-1α transcriptional target LDH-A, confirming that this
compound activates HIF-1α at the used concentration of 10 µg/ml.
Under normoxic conditions, roxadustat enhanced LDH-A expression to
1.88±0.04 of the control (P<0.001, compared with control cells).
Reoxygenation increased LDH-A to 1.75±0.10 of the control
(P<0.001, compared with control cells), although roxadustat
treatment of RPTECs subjected to reoxygenation further increased
LDH-A expression to 2.51±0.18 of the control (P<0.001, compared
to control cells and cells subjected to reoxygenation alone;
Fig. 7A and C).
Roxadustat did not affect ROS production or lipid
peroxidation. Treatment of control cells with roxadustat did not
alter relative ROS signal intensity. Considering the ROS production
in the control cells as 100%, ROS levels reached 95.6±5.9% of the
control in cells under normoxia and treated with roxadustat (P not
significant). Reoxygenation increased ROS signal intensity to
199.9±9.6% (P<0.001, compared with control cells), a value that
remained stable when roxadustat was administered (195.7±12.3%, P
not significant, compared with cells subjected to reoxygenation
alone; Fig. 7D). In RPTECs treated
with roxadustat, cellular MDA did not change significantly,
compared with the control group (3.0±0.2 µM and 3.6±0.6,
respectively; P not significant). Reoxygenation increased MDA
concentration to 42.6±0.5 µM (P<0.001, compared to the control
cells). When roxadustat was administered at the onset of
reoxygenation, MDA did not alter considerably (45.8±1.8 µM, P not
significant., compared to cells subjected to reoxygenation only;
Fig. 7E).
As regards the hard end-point of cell survival, LDH
release assay revealed that in control cells, roxadustat did not
alter the percentage of cell necrosis compared with the control
(10.7±0.6 vs. 11.7±0.4%, respectively; P not significant).
Reoxygenation increased cell necrosis to 23.3±0.6% (P<0.001,
compared to the control cells), while roxadustat did not affect
necrosis (23.7±0.2%; P not significant compared with cells
subjected to reoxygenation only; Fig.
7F). Cell imaging confirmed the above LDH release assay results
since it showed that roxadustat treatment did not offer any
survival benefit in RPTECs subjected to reoxygenation. In all
performed experiments, both treated and untreated cells died within
4 h of reoxygenation (P not significant; Fig. 8).
Discussion
I-R injury contributes to the pathogenesis of many
diseases, including the acute kidney injury (1–4). As a
consequence, clarifying the pathogenesis of I-R injury is decisive
in developing novel therapeutic strategies. This study aimed to
evaluate the role of AhR during the reoxygenation phase of I-R
injury.
Some data support a detrimental role of AhR during
the anoxic phase of I-R injury. Inhibition of AhR/CYP1A1 protects
murine hippocampal cells from anoxia-induced apoptosis (44). However, no data are available
concerning the role of AhR during the subsequent reoxygenation. To
evaluate the role of AhR in RPTECs subjected to reoxygenation, a
cell culture system designed to study anoxia and reoxygenation
separately was established.
In the present study, reoxygenation decreased AhR
levels. The decrease in AhR level indicates its activation since
activated AhR becomes vulnerable to proteasomal degradation,
leading to decreased AhR cellular levels (20–24).
Accordingly, reoxygenation increased the expression of CYP1A1, an
AhR transcriptional target (18,19).
The specific AhR inhibitor CH223191 increased AhR level and
decreased CYP1A1 expression. Thus, AhR was activated during
reoxygenation, and CH223191 successfully inhibited AhR activation.
The exact endogenous AhR ligands, which results in its activation
during reoxygenation, remain to be defined. Initially, AhR was
considered a critical factor for the CYP-mediated metabolism of
xenobiotics, such as dioxins, which are also AhR activators.
However, endogenous AhR activators were identified subsequently
(45,46). Examples of exogenous AhR activators
include natural plant flavonoids, polyphenolics, and indoles, as
well as dioxins-like and other synthetic compounds, collectively
characterized as xenobiotics. Examples of endogenous AhR ligands
include tryptophan derivatives, steroids, eicosanoids, and heme
metabolites (45).
Observation of cell cultures under reoxygenation
suggested that mouse RPTECs were particularly vulnerable to
reoxygenation injury since they died within 4 hours. However, when
the AhR inhibitor CH223191 was added at the onset of reoxygenation,
it prevented cell death, with the cells remaining alive after 24 h
of culture.
In accordance with previous experiments with the
same cell type and similar experimental conditions (5), reoxygenation-induced cell death was
not apoptotic since the levels of the activated CC3, in which all
the apoptotic pathways converge (47), was not altered. Of note, CH223191
did not enhance CC3, indicating that at the used concentration,
this inhibitor does not induce apoptosis in RPTECs.
The primary factor that induces cell injury during
the reperfusion phase of I-R is the burst of ROS production, which
follows the restoration of oxygen supply to the ischemic tissues
(1–3). CYPs play a significant role in ROS
production during I-R injury. In experimental models of heart and
liver I-R injury, inhibition of CYPs ameliorates ROS production and
organ dysfunction (13–17). AhR transcribes certain CYP genes
(18,19), and consequently, AhR activation
during reoxygenation may contribute to cell injury. Indeed, in
experimental models of heart and lung I-R injury, inhibition of AhR
was beneficial (25,35). However, in the aforementioned
studies, AhR and CYPs were investigated separately, and not as a
part of a pathway. Importantly, the aforementioned studies did not
discriminate between events taking place under the anoxic phase and
from those occurring during the reoxygenation phase of I-R injury.
The effect of AhR on ROS production was evaluated in RPTECs
subjected to reoxygenation. Reoxygenation induced ROS production,
which was prevented by the AhR inhibitor CH223191. Thus, AhR may
play a pivotal role in the burst of ROS production that follows
reoxygenation.
With regards reoxygenation-induced cell death,
previous studies using primary RPTECs and similar experimental
conditions have demonstrated that reoxygenation leads to lipid
peroxidation and ferroptotic cell death (5,6).
Elegant research using isolated murine renal tubules detected the
same (7). Interestingly, besides
ROS production, CYPs can directly induce lipid peroxidation
(48), which causes ferroptosis
(43). In our model, reoxygenation
increased lipid peroxidation considerably, as identified by
cellular MDA. Nevertheless, when AhR was inhibited,
reoxygenation-induced lipid peroxidation decreased significantly.
In accordance with microscopic observation of cell cultures, the
LDH release assay confirmed the reoxygenation-induced cell necrosis
biochemically. Administration of the ferroptosis inhibitor
α-tocopherol prevented reoxygenation-induced cell necrosis
completely, indicating the ferroptotic nature of cell death. The
inhibition of AhR attenuated reoxygenation-induced cell death
considerably, suggesting great therapeutic potential. Of note, in
control RPTECs, CH223191 did not induce cell necrosis, proving
non-toxic for RPTECs at the used concentration. Thus, during
reoxygenation, AhR is activated and increases certain CYPs
expression, which in turn, by increasing ROS, induces ferroptotic
cell death. The endogenous AhR activators that participate in
reoxygenation-induced cell injury remain to be identified.
Interestingly, indoleamine 2,3-dioxygenase metabolizes tryptophan
to kynurenine, an endogenous AhR activator. Inhibition of this
enzyme was protective for murine kidneys subjected to I-R injury.
However, whether the beneficial effect of indoleamine
2,3-dioxygenase inhibition was mediated by the suppression of
AhR-CYPs pathway was not evaluated (49). The exact impact of indoleamine
2,3-dioxygenase on this pathway deserves further investigation.
Several studies have suggested that AhR induces the
expression of Nrf2 (30–32), a transcription factor implicated in
the expression of many antioxidant and anti-ferroptotic proteins
(33,34). From a teleological point of view,
this achieves a balance between AhR-induced CYP-mediated oxidative
stress and AhR-induced Nrf2-mediated antioxidant defense (31,32).
However, in our model, and despite the reoxygenation-induced AhR
activation, the cellular Nrf2 level remained stable. Since
activated Nrf2 is released from the ubiquitin ligase cullin-3 and
avoids proteasomal degradation, its cellular level corresponds to
its activity (33,34). The nuclear Nrf2 levels were not
assessed in order to better determine its activation status;
instead, the expression of the Nrf2 transcriptional targets SOD-3
and xCT were measured (33). SOD-3
catalyzes the dismutation of superoxide radical into either
molecular oxygen or hydrogen peroxide (50), while xCT enters the necessary for
glutathione synthesis cystine into the cell and prevents
ferroptosis (43). In the present
study, the expression of both Nrf2 transcriptional targets SOD-3
and xCT remained stable, also indicating that reoxygenation left
Nrf2 activation status unaffected. This was in agreement with a
previous study showing that reoxygenation failed to enhance Nrf2
activity in mouse RPTECs (51).
Interestingly, in RPTECs from the hibernator Syrian hamster,
reoxygenation activates Nrf2 contributing to the resistance of
these cells to reoxygenation-induced ferroptosis (51). Moreover, in the present study, the
specific AhR inhibitor CH223191 was used. Cell treatment with the
AhR inhibitor did not alter Nrf2, SOD-3, and xCT level, offering
additional support to the idea that in the context of
reoxygenation, AhR does not affect the Nrf2 pathway.
Another transcription factor implicated in I-R is
HIF-1α. HIF-1α is upregulated during the ischemic phase, forms a
complex with HIF-1β, and transcribes many genes required for
adaptation to anoxic conditions. For instance, LDH upregulation
promotes the anaerobic catabolism of glucose (36,37).
However, activated AhR also associates with HIF-1β (33,34).
In certain experimental models, this competition between AhR and
HIF-α for HIF-1β results in reduced HIF-1α transcriptional activity
in the case of AhR activation, or to decreased AhR transcriptional
activity in the case of HIF-1α activation (38–40).
However, the interaction between AhR and HIF-1 has not been
evaluated during the reperfusion phase of I-R injury. In the
present experimental model, inhibition of AhR during reoxygenation
did not affect either HIF-1α levels (36,37),
or the expression of its transcriptional target LDH-A.
Interestingly, in RPTECs under reoxygenation, HIF-1α level was
similar to the control cells, whereas the expression of LDH-A was
increased. The latter may result from rapid HIF-1α degradation when
oxygen is available. By contrast, the increased LDH-A levels may be
the result of a slower turnover of this enzyme, which is
upregulated during the anoxic phase.
To delineate the role of HIF-1α further, RPTECs were
subjected to reoxygenation with the HIF-1α activator roxadustat. As
expected, roxadustat increased HIF-1α levels and LDH-A expression
in both control cells and cells subjected to reoxygenation.
However, roxadustat did not affect ROS production, lipid
peroxidation or cell death. Thus, during reoxygenation, HIF-1α does
not play a pivotal role in cell survival.
Certainly, the in vitro nature of our study
is a limitation. However, this approach allowed us to apply strict
experimental conditions and to evaluate the effect of AhR on cell
survival solely during the reoxygenation phase. As already noted,
I-R injury consists of two consecutive but pathophysiologically
distinct phases, anoxia and reoxygenation (1–3). A
better understanding of the molecular mechanisms that govern the
two stages of I-R injury may result in an efficient combination of
therapeutic approaches targeting both stages.
In conclusion, in RPTECs, AhR is activated during
reoxygenation and induces ROS production, lipid peroxidation and
ferroptotic cell death. These detrimental effects may be mediated
by AhR-induced CYP overexpression, while AhR does not affect the
Nrf2 or the HIF-1α pathways. Thus, the AhR pathway may represent a
promising therapeutic target for the prevention of reperfusion
injury.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
TE designed the study. GP and TE performed the
experiments. TE, GP, GF, VL and IS analyzed the results. TE wrote
the manuscript with help from GP. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Neri M, Riezzo I, Pascale N, Pomara C and
Turillazzi E: Ischemia/reperfusion injury following acute
myocardial infarction: a critical issue for clinicians and forensic
pathologists. Mediators Inflamm. 2017:70183932017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bakthavachalam P and Shanmugam PST:
Mitochondrial dysfunction - silent killer in cerebral ischemia. J
Neurol Sci. 375:417–423. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tsukamoto T, Chanthaphavong RS and Pape
HC: Current theories on the pathophysiology of multiple organ
failure after trauma. Injury. 41:21–26. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bonventre JV and Yang L: Cellular
pathophysiology of ischemic acute kidney injury. J Clin Invest.
121:4210–4221. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Eleftheriadis T, Pissas G, Antoniadi G,
Liakopoulos V and Stefanidis I: Cell death patterns due to warm
ischemia or reperfusion in renal tubular epithelial cells
originating from human, mouse, or the native hibernator hamster.
Biology (Basel). 7:482018.
|
|
6
|
Eleftheriadis T, Pissas G, Liakopoulos V
and Stefanidis I: Factors that may protect the native hibernator
syrian hamster renal tubular epithelial cells from ferroptosis due
to warm anoxia-reoxygenation. Biology (Basel). 8:222019.
|
|
7
|
Linkermann A, Skouta R, Himmerkus N, Mulay
SR, Dewitz C, De Zen F, Prokai A, Zuchtriegel G, Krombach F, Welz
PS, et al: Synchronized renal tubular cell death involves
ferroptosis. Proc Natl Acad Sci USA. 111:16836–16841. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Galluzzi L, Bravo-San Pedro JM, Vitale I,
Aaronson SA, Abrams JM, Adam D, Alnemri ES, Altucci L, Andrews D,
Annicchiarico-Petruzzelli M, et al: Essential versus accessory
aspects of cell death: Recommendations of the NCCD 2015. Cell Death
Differ. 22:58–73. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hrycay EG and Bandiera SM: Monooxygenase,
peroxidase and peroxygenase properties and reaction mechanisms of
cytochrome P450 enzymes. 851:1–61. 2015.PubMed/NCBI
|
|
10
|
Lewis DFV: Oxidative stress: The role of
cytochromes P450 in oxygen activation. J Chem Technol Biotechnol.
77:1095–1100. 2002. View
Article : Google Scholar
|
|
11
|
Zeng Q, Han Y, Bao Y, Li W, Li X, Shen X,
Wang X, Yao F, O'Rourke ST and Sun C: 20-HETE increases NADPH
oxidase-derived ROS production and stimulates the L-type Ca2+
channel via a PKC-dependent mechanism in cardiomyocytes. Am J
Physiol Heart Circ Physiol. 299:H1109–H1117. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Han Y, Zhao H, Tang H, Li X, Tan J, Zeng Q
and Sun C: 20-Hydroxyeicosatetraenoic acid mediates isolated heart
ischemia/reperfusion injury by increasing NADPH oxidase-derived
reactive oxygen species production. Circ J. 77:1807–1816. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Granville DJ, Tashakkor B, Takeuchi C,
Gustafsson AB, Huang C, Sayen MR, Wentworth P Jr, Yeager M and
Gottlieb RA: Reduction of ischemia and reperfusion-induced
myocardial damage by cytochrome P450 inhibitors. Proc Natl Acad Sci
USA. 101:1321–1326. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ishihara Y, Sekine M, Nakazawa M and
Shimamoto N: Suppression of myocardial ischemia-reperfusion injury
by inhibitors of cytochrome P450 in rats. Eur J Pharmacol.
611:64–71. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ishihara Y, Sekine M, Hamaguchi A,
Kobayashi Y, Harayama T, Nakazawa M and Shimamoto N: Effects of
sulfaphenazole derivatives on cardiac ischemia-reperfusion injury:
Association of cytochrome P450 activity and infarct size. J
Pharmacol Sci. 113:335–342. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shaik IH and Mehvar R: Cytochrome P450
induction by phenobarbital exacerbates warm hepatic
ischemia-reperfusion injury in rat livers. Free Radic Res.
44:441–453. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shaik IH and Mehvar R: Effects of
cytochrome p450 inhibition by cimetidine on the warm hepatic
ischemia-reperfusion injury in rats. J Surg Res. 159:680–688. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nebert DW, Dalton TP, Okey AB and Gonzalez
FJ: Role of aryl hydrocarbon receptor-mediated induction of the
CYP1 enzymes in environmental toxicity and cancer. J Biol Chem.
279:23847–23850. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lindsey S and Papoutsakis ET: The evolving
role of the aryl hydrocarbon receptor (AHR) in the normophysiology
of hematopoiesis. Stem Cell Rev Rep. 8:1223–1235. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pollenz RS: The mechanism of AH receptor
protein down-regulation (degradation) and its impact on AH
receptor-mediated gene regulation. Chem Biol Interact. 141:41–61.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ma Q and Baldwin KT:
2,3,7,8-tetrachlorodibenzo-p-dioxin-induced degradation of aryl
hydrocarbon receptor (AhR) by the ubiquitin-proteasome pathway.
Role of the transcription activaton and DNA binding of AhR. J Biol
Chem. 275:8432–8438. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Forrester AR, Elias MS, Woodward EL,
Graham M, Williams FM and Reynolds NJ: Induction of a chloracne
phenotype in an epidermal equivalent model by
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) is dependent on aryl
hydrocarbon receptor activation and is not reproduced by aryl
hydrocarbon receptor knock down. J Dermatol Sci. 73:10–22. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang SC, Wu CH, Tu YK, Huang SY and Chou
PC: Exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin increases the
activation of aryl hydrocarbon receptor and is associated with the
aggressiveness of osteosarcoma MG-63 osteoblast-like cells. Oncol
Lett. 16:3849–3857. 2018.PubMed/NCBI
|
|
24
|
Moretti S, Nucci N, Menicali E, Morelli S,
Bini V, Colella R, Mandarano M, Sidoni A and Puxeddu E: The Aryl
Hydrocarbon Receptor Is Expressed in Thyroid Carcinoma and Appears
to Mediate Epithelial-Mesenchymal-Transition. Cancers (Basel).
12:1452020. View Article : Google Scholar
|
|
25
|
Couroucli XI, Liang YW, Jiang W, Barrios R
and Moorthy B: Attenuation of oxygen-induced abnormal lung
maturation in rats by retinoic acid: Possible role of cytochrome
P4501A enzymes. J Pharmacol Exp Ther. 317:946–954. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kopf PG, Scott JA, Agbor LN, Boberg JR,
Elased KM, Huwe JK and Walker MK: Cytochrome P4501A1 is required
for vascular dysfunction and hypertension induced by
2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol Sci. 117:537–546.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kopf PG and Walker MK:
2,3,7,8-tetrachlorodibenzo-p-dioxin increases reactive oxygen
species production in human endothelial cells via induction of
cytochrome P4501A1. Toxicol Appl Pharmacol. 245:91–99. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lee HJ, Pyo MC, Shin HS, Ryu D and Lee
K-W: Renal toxicity through AhR, PXR, and Nrf2 signaling pathway
activation of ochratoxin A-induced oxidative stress in kidney
cells. Food Chem Toxicol. 122:59–68. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pei J, Juni R, Harakalova M, Duncker DJ,
Asselbergs FW, Koolwijk P, Hinsbergh VV, Verhaar MC, Mokry M and
Cheng C: Indoxyl sulfate stimulates angiogenesis by regulating
reactive oxygen species production via CYP1B1. Toxins (Basel).
11:4542019. View Article : Google Scholar
|
|
30
|
Furue M, Takahara M, Nakahara T and Uchi
H: Role of AhR/ARNT system in skin homeostasis. Arch Dermatol Res.
306:769–779. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yagishita Y, Uruno A and Yamamoto M:
NRF2-mediated gene regulation and glucose homeostasis. Molecular
nutrition and diabetes. Elsevier Inc.; pp. 331–348. 2016,
View Article : Google Scholar
|
|
32
|
Dietrich C: Antioxidant functions of the
aryl hydrocarbon receptor. Stem Cells Int. 2016:79434952016.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ma Q: Role of nrf2 in oxidative stress and
toxicity. Annu Rev Pharmacol Toxicol. 53:401–426. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Corsello T, Komaravelli N and Casola A:
Role of hydrogen sulfide in NRF2- and sirtuin-dependent maintenance
of cellular redox balance. Antioxidants. 7:1292018. View Article : Google Scholar
|
|
35
|
Cuartero MI, Ballesteros I, de la Parra J,
Harkin AL, Abautret-Daly A, Sherwin E, Fernández-Salguero P, Corbí
AL, Lizasoain I and Moro MA: L-kynurenine/aryl hydrocarbon receptor
pathway mediates brain damage after experimental stroke.
Circulation. 130:2040–2051. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kaluz S, Kaluzová M and Stanbridge EJ:
Regulation of gene expression by hypoxia: Integration of the
HIF-transduced hypoxic signal at the hypoxia-responsive element.
Clin Chim Acta. 395:6–13. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Myllyharju J: Prolyl 4-hydroxylases,
master regulators of the hypoxia response. Acta Physiol (Oxf).
208:148–165. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Vorrink SU, Sarsour EH, Olivier AK,
Robertson LW, Goswami PC and Domann FE: PCB 126 perturbs
hypoxia-induced HIF-1α activity and glucose consumption in human
HepG2 cells. Exp Toxicol Pathol. 66:377–382. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Vorrink SU, Severson PL, Kulak MV,
Futscher BW and Domann FE: Hypoxia perturbs aryl hydrocarbon
receptor signaling and CYP1A1 expression induced by PCB 126 in
human skin and liver-derived cell lines. Toxicol Appl Pharmacol.
274:408–416. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Eleftheriadis T, Pissas G, Antoniadi G,
Liakopoulos V and Stefanidis I: Kynurenine, by activating aryl
hydrocarbon receptor, decreases erythropoietin and increases
hepcidin production in HepG2 cells: A new mechanism for anemia of
inflammation. Exp Hematol. 44:60–7.e1. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kim SH, Henry EC, Kim DK, Kim YH, Shin KJ,
Han MS, Lee TG, Kang JK, Gasiewicz TA, Ryu SH, et al: Novel
compound 2-methyl-2H-pyrazole-3-carboxylic acid
(2-methyl-4-o-tolylazo-phenyl)-amide (CH-223191) prevents
2,3,7,8-TCDD-induced toxicity by antagonizing the aryl hydrocarbon
receptor. Mol Pharmacol. 69:1871–1878. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dhillon S: Roxadustat: First Global
Approval. Drugs. 79:563–572. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Stockwell BR, Friedmann Angeli JP, Bayir
H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascón S, Hatzios SK,
Kagan VE, et al: Ferroptosis: A regulated cell death nexus linking
metabolism, redox biology, and disease. Cell. 171:273–285. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Rzemieniec J, Wnuk A, Lasoń W, Bilecki W
and Kajta M: The neuroprotective action of 3,3′-diindolylmethane
against ischemia involves an inhibition of apoptosis and autophagy
that depends on HDAC and AhR/CYP1A1 but not ERα/CYP19A1 signaling.
Apoptosis. 24:435–452. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Stejskalova L, Dvorak Z and Pavek P:
Endogenous and exogenous ligands of aryl hydrocarbon receptor:
Current state of art. Curr Drug Metab. 12:198–212. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wu PY, Chuang PY, Chang GD, Chan YY, Tsai
TC, Wang BJ, Lin KH, Hsu WM, Liao YF and Lee H: Novel endogenous
ligands of aryl hydrocarbon receptor mediate neural development and
differentiation of neuroblastoma. ACS Chem Neurosci. 10:4031–4042.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Fadeel B and Orrenius S: Apoptosis: A
basic biological phenomenon with wide-ranging implications in human
disease. J Intern Med. 258:479–517. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Huang B, Bao J, Cao YR, Gao HF and Jin Y:
Cytochrome P450 1A1 (CYP1A1) catalyzes lipid peroxidation of oleic
acid-induced HepG2 cells. Biochemistry (Mosc). 83:595–602. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Mohib K, Wang S, Guan Q, Mellor AL, Sun H,
Du C and Jevnikar AM: Indoleamine 2,3-dioxygenase expression
promotes renal ischemia-reperfusion injury. Am J Physiol Renal
Physiol. 295:F226–F234. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Fang YZ, Yang S and Wu G: Free radicals,
antioxidants, and nutrition. Nutrition. 18:872–879. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Eleftheriadis T, Pissas G, Nikolaou E,
Liakopoulos V and Stefanidis I: The H2S-Nrf2-antioxidant proteins
axis protects renal tubular epithelial cells of the native
hibernator syrian hamster from reoxygenation-induced cell death.
Biology (Basel). 8:742019.
|