Introduction
Acute lung injury (ALI) causes high morbidity and
when aggravated further, develops into acute respiratory distress
syndrome (ARDS) with a fatality rate ≤40% worldwide (1). Pulmonary and extrapulmonary factors
are responsible for causing ARDS (2). The common pathophysiologic features of
ARDS are injury to the pulmonary alveoli epithelial cells and
pulmonary capillaries, with refractory hypoxemia being a
conspicuous feature (3). Bluish
skin coloration, shortness of breath and rapid breathing are also
prominent symptoms of ARDS (4). The
high mortality of ARDS has received increasing attention, with
several studies indicating that ARDS may be closely associated with
conditions such as pancreatitis (5,6),
sepsis (7), aspiration (8), pneumonia (9) and trauma (10). ARDS pathogenesis comprises diffuse
cell damage, including diffuse cell injury, pulmonary microballoon
barrier formation, surfactant dysfunction, immune system activation
and coagulation regulation dysfunction (11,12).
Lipopolysaccharide (LPS) is found on the cytoderm of
Gram-negative bacteria and significantly contributes to pulmonary
infection and sepsis by protecting the bacterial cell wall
(13,14). As indicated by several studies, LPS
is a common pathogenic factor of ALI/ARDS (15–17);
therefore, it can be used to establish ARDS cell models.
Penehyclidine hydrochloride (PHC) is an
anticholinergic drug that is widely used in the clinic for
organophosphorus poisoning, visceral smooth muscle spasms and as a
reversal agent for anesthesia (18). PHC may participate in overcoming
microcirculation disorders and aid in organ protection (19). PHC is beneficial in pharmacology
compared with other anticholinergic agents, including atropine and
scopolamine (20). In addition, PHC
possesses anti-inflammatory, antioxidative and antiapoptotic
features, which serve protective effects in various organs such as
the lungs (21), brain (22) and heart (23). In ARDS, excessive release of
acetylcholine in the body results in acute pulmonary
microcirculation disorder, leading to pulmonary microcirculation
spasm (24). Thus, by preventing
pulmonary permeability via decreasing the damage of ARDS to the
microvascular barrier, PHC can improve the pulmonary inflammatory
response and ventilation function in ARDS (25). The protective role of PHC in ARDS
has been reported in several studies (26,27);
however, the mechanisms underlying the effects of PHC on autophagy
have not been previously reported.
The present study aimed to investigate the effects
of PHC on cell activities, such as proliferation, inflammation,
apoptosis and autophagy, in LPS-induced BEAS-2B cells. The effect
of PHC on the expression levels of apoptosis- and autophagy-related
proteins was assessed, and the reversal effect of autophagy
inhibitor (3-methyladenine (3-MA)) on PHC in ARDS was also
investigated.
Materials and methods
Cell culture
Lung alveolar epithelial cells (BEAS-2B; American
Type Culture Collection) were cultured in DMEM medium (cat. no.
31600-026; Gibco; Thermo Fisher Scientific, Inc.) with 10% fetal
bovine serum (FBS; cat. no. SH30071.03; HyClone; GE Healthcare Life
Sciences) and penicillin/streptomycin (cat. no. 10378-016;
Invitrogen; Thermo Fisher Scientific, Inc.). Cells were cultured in
an incubator with 5% CO2 at 37°C. Cells in the
logarithmic phase were used for subsequent experiments.
Grouping and modeling
To establish a cell model of ARDS, BEAS-2B cells in
the logarithmic stage of growth were treated with 10 ng/ml LPS
(Sigma-Aldrich; Merck KGaA). Following incubation for 12 h at 37°C,
the ARDS cell model was treated with 0.01, 0.05 and 1.0 µM PHC
(cat. no. BP0704-YTX; Beijing Baiaolaibo Technology Co., Ltd.) for
1 h at 37°C. And the ARDS cells in the control group were also
given an equal amount of double distilled water. ARDS model cells
were divided into the following groups: i) Control (normal saline);
ii) LPS; iii) LPS-PHC-low concentration (LPS and 0.01 µM PHC); iv)
LPS-PHC-medium concentration (LPS and 0.05 µM PHC); and v)
LPS-PHC-high concentration (1.0 µM). Alternatively, following
treatment with 0.05 µM PHC, LPS-induced BEAS-2B cells were treated
with 100 nM autophagy agonist rapamycin (Rapa; cat. no.
IMT1001-100MG; Gene Operation) or 10 mM autophagy inhibitor (3-MA;
cat. no. S2767-1; Selleck Chemicals).
Western blot analysis
BEAS-2B cells from each group were lysed using RIPA
Lysis buffer (Beyotime Institute of Biotechnology) on ice. The BCA
protein assay kit (Pierce; Thermo Fisher Scientific, Inc.) was used
to quantify the total protein concentration. Protein samples (30 µg
per lane) were separated via 10% SDS-PAGE (Beijing Solarbio Science
& Technology Co., Ltd.) and transferred to PVDF membranes
(Sigma-Aldrich; Merck KGaA). The membranes were blocked with 5%
non-fat dry milk (cat. no. 198-10605; Wako Chemicals GmbH) at room
temperature for 1.5 h to block nonspecific protein binding sites.
Subsequently, the membranes were incubated at 4°C overnight with
primary antibodies targeted against: Bax (cat. no. SAB2108447;
1:1,000; Sigma-Aldrich; Merck KGaA), Bcl-2 (cat. no. SAB5500014;
1:1,000; Sigma-Aldrich; Merck KGaA), caspase-3 (cat. no. ZRB1221;
1:1,000; Sigma-Aldrich; Merck KGaA), Beclin-1 (cat. no. ab62557;
1:1,000; Abcam), light chain 3 (LC3B; cat. no. ab168831; 1:1,000,
Abcam), p62 (cat. no. ab91526; 1:1,000; Abcam) and GAPDH (cat. no.
G5262; 1:100; Sigma-Aldrich; Merck KGaA). Following primary
incubation, the membranes were incubated with goat anti-rabbit IgG
H&L (cat. no. ab6721; 1:5,000; Abcam) and goat anti-mouse IgG
H&L (cat. no. ab205719; 1:5,000; Abcam) secondary antibodies at
room temperature for 1 h. Protein bands were visualized using an
enhanced chemiluminescence kit (cat. no. 32106; Invitrogen; Thermo
Fisher Scientific, Inc.). Protein expression was semi-quantified
using Image-pro Plus analysis software (version 6.0; Media
Cybernetics, Inc.) with GAPDH as the loading control.
Immunofluorescence (IF) assay
BEAS-2B cells (5×104 cells/ml) from each
group were harvested and mounted on coverslips. Cells were fixed
using 4% paraformaldehyde (cat. no. P6148l; Sigma Aldrich; Merck
KGaA) at room temperature for 10 min. After washing with PBS,
permeabilization was conducted with 0.3% Triton X-100
(Sigma-Aldrich; Merck KGaA) at room temperature for 10 min.
Following rinsing with PBS, cells were blocked using 5% bovine
serum albumin (cat. no. A7906; Sigma-Aldrich; Merck KGaA) at room
temperature for 2 h and incubated with an anti-LC3B primary
antibody (cat. no. ab51520; 1:500; Abcam) at 4°C overnight. Cells
were subsequently washed and incubated with a secondary
fluorescein-conjugated antibody (cat. no. 4414; 1:200; Cell
Signaling Technology, Inc.) at room temperature for 1 h. Cells were
washed with PBS and stained with DAPI (cat. no. D9542;
Sigma-Aldrich; Merck KGaA) at room temperature for 5 min. Stained
cells were visualized using an SZX12 fluorescent microscope
(Olympus Corporation) at ×200 magnification.
Cell counting Kit-8 (CKK-8) assay
BEAS-2B cells were seeded (3.0×103
cells/well) into 96-well plates. Cells were treated with varying
concentrations of PHC, 100 nM Rapa or 10 mM 3-MA in an incubator at
37°C with 5% CO2 for 24, 48 or 72 h. Subsequently, 10 µl
CCK-8 solution (5 mg/ml; Dojindo Molecular Technologies, Inc.) was
added to each well. After incubation at 37°C for 2 h, the
absorbance of each well was measured at a wavelength of 450 nm
using a microplate reader.
ELISA
As previously described (28), The concentrations of factors in the
culture medium of each group, such as interferon (IFN)-γ, tumor
necrosis factor (TNF)-α, interleukin (IL)-1β and IL-6, were
measured via ELISA. The following ELISA kits were used: IFN-γ (cat.
no. ab46048; Abcam), TNF-α (cat. no. E-EL-H0109c; Elabscience),
IL-1β (cat. no. E-EL-H0149c; Elabscience) and IL-6 (cat. no.
E-EL-H0102c; Elabscience). Nitric oxide (NO) levels were assessed
using a Nitrite Colorimetric kit (cat. no. E-BC-K070-S;
Elabscience).
Flow cytometry analysis
To measure the early + late apoptosis, the Annexin
V/FITC kit (cat. no. PHN1008; Invitrogen; Thermo Fisher Scientific,
Inc.) was used. Briefly, cells were collected at a density of
5.0×105 cells/ml and resuspended in PBS. Subsequently,
cells were transferred to flow tubes and incubated with 5 µl
Annexin V/FITC at room temperature for 5 min and then rinsed with
ice-cold PBS. Fluorescence intensity was determined using a FACScan
flow cytometer (BD Biosciences), and the data was analyzed using
Cell Quest 3.1 software (BD Biosciences).
Hoechst staining
BEAS-2B cells were harvested and the cell density
was calibrated to 6×104 cells/ml and cultured in 6-well
plates at 37°C with 5% CO2 for 24 h. Subsequently, cells
were washed with PBS and fixed with 4% paraformaldehyde (cat. no.
abx082483; Abbexa Ltd.) at room temperature for 20 min. Cells were
rinsed with PBS and stained using 1 ml Hoechst 33342 (20 µg/ml;
Invitrogen; Thermo Fisher Scientific, Inc.) at room temperature for
10 min. Stained cells were observed using an A1R/A1 confocal laser
scanning microscope (Nikon Corporation) at ×200 magnification.
Transmission electron microscopy
(TEM)
Cell autophagy was assessed by TEM. BEAS-2B cells
(5×105 cells) were resuspended in PBS and fixed with
2.5% glutaraldehyde (cat. no. G7776; Sigma-Aldrich; Merck KGaA) for
2 h at 4°C. Subsequently, cells were rinsed with PBS three times,
and treated with 1% uranyl acetate (cat. no. 0509-25G; Amresco,
LLC) at room temperature for 20 mins. Cells were dehydrated using
50, 70, 80 and 90% acetone (cat. no. 650501; Sigma-Aldrich; Merck
KGaA). The cells were embedded with the epoxy resin mixture at room
temperature for 20 mins. The prepared cells were observed under a
H-7650 transmission electron microscope (Hitachi High-Technologies
Corporation) at ×40,000 magnification to study the morphology of
BEAS-2B cells.
Animal experiments
Male SD rats (n=40; age, 8 weeks; weight, 200–250 g;
SPF grade) purchased from the Experimental Animal Center of
Shanghai Jiaotong University, China were randomly divided into five
groups (n=8 per group). Rats were intraperitoneally injected with 5
mg/kg LPS (cat. no. L8880; Beijing Solarbio Science &
Technology Co., Ltd.) or an equal volume of normal saline
(control). Prior to the experiment, all rats were kept adaptively
for one week. Rats were housed at 18–22°C, 50–60% humidity, with a
12 h light/dark cycle and had free access to food and water. The
present study was approved by the Ethics Committee of the Shanghai
Jiaotong University Affiliated Sixth People's Hospital. Rats were
intraperitoneally injected with 0.3, 1.0 or 3.0 mg/kg PHC 30 min
before LPS injection. After treatment with LPS for 24 h, the rats
were sacrificed using a Quietek CO2 Delivery Systems
(HEAD Beijing Biotechnology Co., Ltd.). Briefly, 100%
CO2 was delivered from a purified compressed air
cylinder with a special valve (Guangzhou Yigas Gases, Co., Ltd.) to
the cage at a volume displacement rate of 30% per min (~5 l/min)
using a flowmeter (Hua Yun Meter Co., Ltd.). The lung tissues of
each group were fixed with 4% paraformaldehyde at room temperature
for 20 min.
Hematoxylin and eosin (H&E)
staining
The lung tissues were dehydrated using a graded
series (100, 95, 80 and 75%) of ethanol and cleared using
dimethylbenzene. Following paraffin embedding, the tissues were cut
into 4-µm sections. The sections were heated, dewaxed, hydrated and
stained using the hematoxylin and eosin staining kit (cat. no.
C0105; Beyotime Institute of Biotechnology) according to the
manufacturer's instructions. Subsequently, the sections were placed
in 1% hydrochloric acid alcohol for 30 sec. Following dehydration,
clearing and mounting, five random fields of the stained sections
were observed under a CKX41 light microscope (Olympus Corporation)
at ×200 magnification.
TUNEL staining
After the tissue sections had been dewaxed with
dimethylbenzene, hydrated with 100, 90 and 70% ethanol and washed
with PBS, the tissue sections were incubated with 20 µg/ml protease
K at 37°C for 15 mins to remove the tissue proteins. Subsequently,
the sections were treated with 100 µl TUNEL reaction mixture (cat.
no. C1086; Beyotime Institute of Biotechnology) at 37°C for 1 h,
followed by incubation with 100 µl DNase at room temperature for 5
min. Following washing with PBS, the sections were treated with 3
ml/l H2O2 for 5 min at room temperature to
block endogenous peroxidase. Subsequently, 100 µl
streptavidin-labeled HRP (1:50) was added to the sections at room
temperature for 30 min. Following washing with PBS, the sections
were treated with 100 µl DAB for at room temperature 10 min in the
dark. The sections were re-stained with hematoxylin at room
temperature for 3 min, dehydrated with gradient alcohol (80, 90 and
100%), cleared with dimethylbenzene and sealed with neutral balsam.
Apoptotic cells were observed and the images from five random
fields were captured using an IX73 fluorescence microscope (Olympus
Corporation) at ×200 magnification.
Statistical analysis
Statistical analyses were performed using SPSS
software (version 19.0; IBM Corp.). All experiments were performed
at least three times. Data are presented as the mean ± SD. The data
between two groups were analyzed using the unpaired Student's
t-test, and the data between the multiple groups were analyzed by a
one-way ANOVA followed by Tukey's post hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Results
PHC accelerates proliferation and
inhibits apoptosis in LPS-induced BEAS-2B cells
First, the effect of PHC on ARDS proliferation and
apoptosis was investigated. As indicated in Fig. 1A, LPS significantly reduced BEAS-2B
cell proliferation compared with the control group, which was
rescued by PHC in a dose-dependent manner. Hoechst staining and
flow cytometry demonstrated that the rate of cell apoptosis was
increased in the LPS group compared with the control group; whereas
PHC treatment inhibited LPS-induced BEAS-2B cell apoptosis, with
increasing PHC concentrations significantly decreasing the rate of
LPS-induced apoptosis (Fig. 1B and
C). Based on the aforementioned findings, it was hypothesized
that PHC could inhibit LPS-induced apoptosis in a dose-dependent
manner. Western blotting was performed to verify the expression
levels of apoptosis-related proteins (Bax, caspase-3 and Bcl-2). As
demonstrated in Fig. 1D, the
expression levels of proapoptosis genes (Bax and caspase-3) were
significantly increased in the LPS group compared with the control
group. LPS-induced Bax and caspase-3 expression levels were
inhibited by PHC treatment in a dose-dependent manner. By contrast,
antiapoptotic factor Bcl-2 expression was significantly decreased
in the LPS group compared with the control group, and PHC treatment
significantly increased Bcl-2 expression in a dose-dependent manner
compared with the LPS group. Therefore, the results indicated that
PHC inhibited LPS-induced apoptosis.
PHC inhibits inflammatory mediators in
LPS-induced BEAS-2B cells
The effect of PHC-induced ARDS injury was further
investigated. As shown in Fig. 2,
the levels of IFN-γ, TNF-α, IL-1β, IL-6 and NO were significantly
elevated in the LPS group compared with the control group, whereas
PHC treatment significantly reduced LPS-mediated effects in BEAS-2B
cells. Thus, PHC attenuated the LPS-induced inflammatory response
in BEAS-2B cells.
 | Figure 2.PHC downregulates ARDS injury-related
markers in LPS-induced BEAS-2B cells. Following PHC treatment, the
levels of (A) IFN-γ, (B) TNF-α, (C) IL-1β, (D) IL-6 and (E) NO were
analyzed by ELISA in the medium of the LPS-induced BEAS-2B cells.
**P<0.01 and ***P<0.001 vs. control group;
#P<0.05 and ##P<0.01 vs. LPS group.
PHC, penehyclidine hydrochloride; ARDS, acute respiratory distress
syndrome; LPS, lipopolysaccharide; IFN, interferon; TNF, tumor
necrosis factor; IL, interleukin; NO, nitric oxide. |
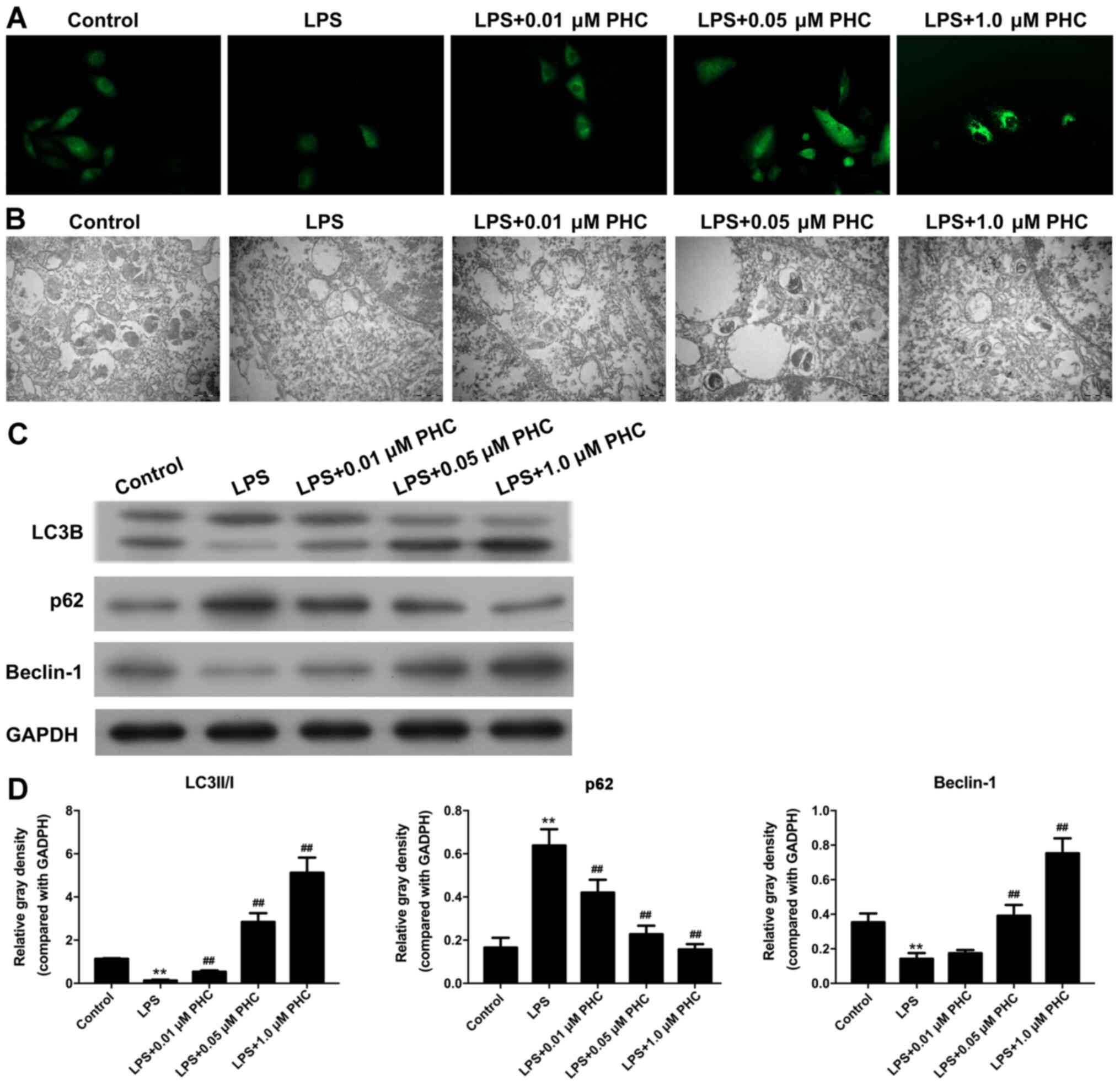
PHC facilitates autophagy in
LPS-induced BEAS-2B cells
IF assays and TEM observations were conducted to
study the effect of PHC on LPS autophagy. The IF assay revealed
weaker fluorescence intensity in the LPS group compared with the
control group, indicating lower levels of autophagy in the model
group. The fluorescence intensity was enhanced by PHC treatment in
a concentration-dependent (Fig.
3A). The number of autophagosomes was decreased in the LPS
group compared with the control group, and PHC treatment
ameliorated the LPS-induced reduction in autophagosomes, with the
number of autophagosomes increasing gradually with increasing PHC
concentrations (Fig. 3B). Beclin-1
and LC3B expression levels were significantly decreased, and p62
expression levels were significantly increased in the LPS group
compared with the control group. LPS-induced alterations to the
expression levels of LC3B, p62 and Beclin-1 were reversed by PHC in
a dose-dependent manner (Fig. 3C and
D).
3-MA reverses the effects of PHC on
LPS-induced BEAS-2B cell proliferation and apoptosis
Subsequently, the influence of PHC on LPS-induced
BEAS-2B cell proliferation and apoptosis was further investigated.
The CCK-8 assay results indicated that PHC reversed the inhibitory
effect of LPS treatment on BEAS-2B cell proliferation, whereas 3-MA
attenuated the effects of PHC on LPS-treated BEAS-2B cell
proliferation (Fig. 4A). In
addition, RAPA also enhanced the proliferation of LPS-induced
BEAS-2B cells (Fig. 4A). PHC
significantly suppressed LPS-induced BEAS-2B cell apoptosis,
whereas the 3-MA reversed PHC-mediated apoptosis suppression. The
Rapa also significantly inhibited LPS-induced BEAS-2B cell
apoptosis (Fig. 4B and C). PHC in
combination with 3-MA significantly upregulated the expression
levels of proapoptosis genes (Bax and caspase-3) and downregulated
antiapoptotic factor Bcl-2 expression compared with PHC alone.
Additionally, Rapa treatment also significantly reduced Bax and
caspase-3 expression levels, and significantly increased Bcl-2
expression levels in LPS-treated BEAS-2B cells compared with the
LPS group (Fig. 4D).
PHC weakens the inflammatory state by
regulating the autophagy pathway in BEAS-2B cells
Whether the effect of PHC on ARDS damage was related
to autophagy-related pathways was further investigated. In
accordance with the aforementioned results, 3-MA reversed
PHC-mediated inhibition of IFN-γ, TNF-α, IL-1β, IL-6 and NO levels
in LPS-treated BEAS-2B cells, which suggested that PHC decreased
inflammation by regulating the autophagy pathway (Fig. 5).
 | Figure 5.PHC weakens ARDS damage by regulating
the autophagy pathway. LPS-induced BEAS-2B cells were treated with
PHC, Rapa or 3-MA. ELISAs were performed to analyze alterations to
(A) IFN-γ, (B) TNF-α, (C) IL-1β, (D) IL-6 and (E) NO levels.
*P<0.05 and **P<0.01 vs. control group; #P<0.05
and ##P<0.01 vs. LPS group; &P<0.05
and &&P<0.01 vs. LPS + PHC group. PHC,
penehyclidine hydrochloride; ARDS, acute respiratory distress
syndrome; LPS, lipopolysaccharide; Rapa, rapamycin; IFN,
interferon; TNF, tumor necrosis factor; IL, interleukin; NO, nitric
oxide. |
3-MA reverses the effects of PHC on
LPS-induced autophagy in BEAS-2B cells
IF assays and TEM observations were conducted to
further investigate the effects of PHC on LPS-induced autophagy in
BEAS-2B cells. As demonstrated in Fig.
6A, PHC in combination with 3-MA suppressed the fluorescence
intensity of LC3B in LPS-induced BEAS-2B cells compared with PHC
treatment alone. In addition, Rapa promoted LPS-induced BEAS-2B
cell autophagy. Moreover, the combination of PHC and 3-MA markedly
decreased the number of autophagosomes compared with PHC treatment
alone (Fig. 6B), which indicated
that 3-MA inhibited PHC-mediated autophagy in LPS-induced BEAS-2B
cells. Furthermore, Beclin-1 and LC3B expression levels were
significantly decreased, whereas p62 expression was significantly
increased in the LPS + PHC + 3-MA group compared with the LPS + PHC
group. Similarly, Rapa also significantly increased Beclin-1 and
LC3B expression levels, but decreased p62 expression levels in
LPS-induced BEAS-2B cells compared with the LPS + PHC group,
suggesting a crucial role for autophagy (Fig. 6C and D).
PHC suppresses apoptosis and induces
autophagy in a rat model of ALI
In addition, in vivo experiments were
performed to further investigate the effects of PHC on the
morphology, apoptosis and autophagy of lung tissues in ALI model
rats. The H&E staining results demonstrated that the structure
of the lung tissues presented as an intact alveolar cavity with no
secretions in the alveolar cavity, almost no pulmonary edema and
complete alveolar wall structure in the control group. In the model
group, the lung tissues displayed a disordered arrangement of
alveolar wall cells and a reduced alveolar space accompanied by
diffuse hyperemia and small focal pulmonary hemorrhage. In the PHC
groups, especially the 3 mg/kg PHC treatment group, the lung
tissues displayed a decrease in the blood-red area on the surface
of the lung, and the majority of edema and congestion had
disappeared (Fig. 7A). The TUNEL
staining results demonstrated that cell apoptosis was markedly
enhanced in the LPS group compared with the control group, whereas
PHC evidently weakened LPS-induced effects in rats (Fig. 7B). The results from the IF assay
revealed that LC3B expression was dramatically downregulated in the
LPS group compared with the control group, but PHC reversed
LPS-induced suppression of LC3B expression in rats (Fig. 7C). Overall, the results demonstrated
that PHC inhibited apoptosis and promoted autophagy in ALI model
rats.
 | Figure 7.PHC suppresses apoptosis and induces
autophagy in a rat model of ALI. The ALI rat model was established
by intraperitoneal injection of 5 mg/kg LPS. ALI model rats were
intraperitoneally injected with 0.3, 1.0 or 3.0 mg/kg PHC 30 min
before LPS injection. (A) Pathological alterations to the lung
tissues were assessed by hematoxylin and eosin staining
(magnification, ×200; scale bar, 100 µm). (B) Cell apoptosis was
assessed by conducting TUNEL staining (magnification, ×400; scale
bar, 50 µm). (C) The expression of LC3B was confirmed by performing
immunofluorescence assays (magnification, ×200; scale bar, 100 µm).
PHC, penehyclidine hydrochloride; ALI, acute lung injury; LPS,
lipopolysaccharide; LC3, light chain 3. |
Discussion
ARDS is typically characterized by respiratory
frequency, distress and progressive hypoxemia, and the clinical
mortality rate is 32–55% worldwide (12). Nonetheless, the mechanism underlying
ARDS has yet to be elucidated. ARDS is closely linked to
inflammatory reactions and oxidative/antioxidant disorders
(29). An increasing number of
studies have revealed that apoptosis serves a crucial role in the
occurrence and progression of ARDS (30). Apoptosis is an autonomous programmed
cell death mediated by multiple genes and is an active process that
maintains homeostasis in the body; once the cell apoptosis balance
is altered, the individual may experience the development of
abnormalities, distortion and even mortality (31,32).
Therefore, apoptosis is closely linked to the occurrence of various
diseases, such as cancer (33,34),
diabetes (35), Parkinson's disease
(36) and cardiovascular diseases
(37). At present, Bax, Bcl-2 and
caspase-3 have been established to be associated with ARDS
(38). ARDS is caused by
alveolar-capillary membrane injury triggering pulmonary edema and
atelectasis (39). During this
process, progressive apoptosis occurs in the alveolar epithelium
and pulmonary vascular endothelial cells (40). Bax and Bcl-2 participate in the
regulation of apoptosis (41), and
caspase-3, a key factor, also participates in the occurrence and
development of ARDS (42,43).
PHC is an anticholinergic drug possessing both
antimuscarinic and antinicotinic activities (27). PHC has been ascertained to improve
microcirculation, reduce the permeability of capillaries and exert
a cell protective effect in ARDS (44). In addition, PHC has been reported to
serve an important role as a protective agent for the lungs,
wherein PHC preconditioning imparts pulmonary and systemic
protection in a rat model of lung ischemia reperfusion injury
(18). PHC interaction with the M
receptor diminishes endothelial injury in LPS-stimulated human
pulmonary microvascular endothelial cells (45). The protective effect of PHC in rats
with myocardial ischemia or reperfusion injury has also been
studied (46,47). The present study established an
LPS-induced ARDS model. In ARDS model cells, LPS inhibited cell
proliferation and promoted apoptosis. Bax and caspase-3 are
proapoptotic proteins, and Bcl-2 is an antiapoptotic protein
(48). In the present study, Bax
and caspase-3 were highly expressed in the model group, whereas
Bcl-2 displayed significantly decreased expression in the model
group compared with the control group. The results indicated that
cell apoptosis was induced by LPS. The protective effects of
various doses of PHC on the ARDS model regarding cell proliferation
and apoptosis were also explored. A dose-dependent study was also
conducted, which indicated that cell proliferation was inhibited
and cell apoptosis was increased by PHC in vitro and in
vivo in a dose-dependent manner. In addition, higher doses of
PHC resulted in more significant reductions in the expression
levels of proapoptotic proteins compared with the LPS group.
Inflammatory response to stimulation is a normal
response of the body (49).
Nevertheless, it can induce the uncontrolled release of
inflammatory mediators, such as IFN-γ, TNF-α, IL-1β, IL-6 and NO,
in severe pathological conditions due to excessive systemic
inflammatory response, which can even lead to multiple organ
dysfunction syndrome (50). Thus,
preventing the overexpression of inflammatory mediators is an
effective strategy to prevent ARDS injury. In the present study,
PHC significantly reduced the levels of IFN-γ, TNF-α, IL-1β, IL-6
and NO in LPS-induced BEAS-2B cells, which indicated that PHC could
suppress the inflammatory response of LPS-induced BEAS-2B cells,
suggesting that PHC could relieve ARDS injury.
Autophagy is an intracellular chronic mild stress
response that involves transportation of damaged proteins to
lysosomes for degradation and circulation for maintaining the
stability of cell structure, function and metabolism (51). Rapa acts as an autophagy promoter,
with the ability to accelerate the formation and maturation of
autophagosomes (52); however, 3-MA
behaves as an autophagy inhibitor, essentially inhibiting the
formation of autophagy bodies (53). In lung diseases, lung tissues
frequently display varying degrees of autophagy abnormalities
(54) and a moderate degree of
autophagy is conducive to the improvement in the disease condition
(55,56). Autophagy is closely linked to
LPS-induced ALI (57), which can be
reversed by Rapa via autophagy promotion (58). A previous study has testified that
saturated hydrogen saline improves LPS-induced ALI by regulating
autophagy (59). Besides, a
previous study has also demonstrated that ethyl pyruvate protects
against LPS-induced ALI via regulating autophagy (60). In the present study, the effect of
LPS was observed to be associated with cell autophagy. The
protective effects of various doses of PHC against cell autophagy
in the ARDS model were also explored. p62 expression in the LPS +
3-MA group was the highest among the different groups, but LC3B and
Beclin-1 were expressed at the lowest levels in the LPS + 3-MA
group among the different groups. By contrast, p62 expression in
the LPS + Rapa group was lowest among the different groups, whereas
LC3B and Beclin-1 expression levels highest in the LPS + Rapa group
compared with the other groups, which indicated that Rapa reversed
LPS-induced autophagy. In addition, 3-MA reversed the effects of
PHC on LPS-induced proliferation, apoptosis and the inflammatory
response in BEAS-2B cells.
PHC evidently facilitated proliferation and
autophagy, and suppressed apoptosis and the inflammatory response
in LPS-induced BEAS-2B cells and ALI model rats in a dose-dependent
manner. In addition, PHC also protected against LPS-induced ARDS
injury by influencing the autophagy pathway.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XW participated in designing the study and
performing the experiments. FL and MX helped perform the
experiments and analyze the data. XW and LW wrote the manuscript
and analyzed data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the Shanghai Jiaotong University Affiliated Sixth
People's Hospital.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ARDS
|
Acute respiratory distress
syndrome
|
|
PHC
|
penehyclidine hydrochloride
|
|
LPS
|
lipopolysaccharide
|
|
3-MA
|
3-methyladenine
|
|
TEM
|
transmission electron microscopy
|
|
Rapa
|
rapamycin
|
|
IF
|
immunofluorescence
|
References
|
1
|
Matthay MA and Zemans RL: The acute
respiratory distress syndrome: Pathogenesis and treatment. Annu Rev
Pathol. 6:147–163. 2011. View Article : Google Scholar
|
|
2
|
Li BQ, Sun HC, Nie SN, Shao DB, Liu HM and
Qian XM: Effect of penehyclidine hydrochloride on patients with
acute lung injury and its mechanisms. Chin J Traumatol. 13:329–335.
2010.
|
|
3
|
Saguil A and Fargo M: Acute respiratory
distress syndrome: Diagnosis and management. Am Fam Physician.
85:352–358. 2012.
|
|
4
|
Lee-Chiong T Jr and Matthay RA:
Drug-induced pulmonary edema and acute respiratory distress
syndrome. Clin Chest Med. 25:95–104. 2004. View Article : Google Scholar
|
|
5
|
Shah J and Rana SS: Acute respiratory
distress syndrome in acute pancreatitis. Indian J Gastroenterol.
39:123–132. 2020. View Article : Google Scholar
|
|
6
|
Ibadov RA, Arifjanov AS, Ibragimov SK and
Abdullajanov BR: Acute respiratory distress-syndrome in the general
complications of severe acute pancreatitis. Ann Hepatobiliary
Pancreat Surg. 23:359–364. 2019. View Article : Google Scholar
|
|
7
|
Iriyama H, Abe T, Kushimoto S, Fujishima
S, Ogura H, Shiraishi A, Saitoh D, Mayumi T, Naito T, Komori A, et
al: Risk modifiers of acute respiratory distress syndrome in
patients with non-pulmonary sepsis: A retrospective analysis of the
FORECAST study. J Intensive Care. 8:72020. View Article : Google Scholar
|
|
8
|
Rambaud J, Lidouren F, Sage M, Kohlhauer
M, Nadeau M, Fortin-Pellerin É, Micheau P, Zilberstein L, Mongardon
N, Ricard JD, et al: Hypothermic total liquid ventilation after
experimental aspiration-associated acute respiratory distress
syndrome. Ann Intensive Care. 8:572018. View Article : Google Scholar
|
|
9
|
Tambe A, Gentile T, Ramadas P, Tambe V and
Badrinath M: Cytomegalovirus pneumonia causing acute respiratory
distress syndrome after brentuximab vedotin therapy. Am J Ther.
26:e794–e795. 2019. View Article : Google Scholar
|
|
10
|
Killien EY, Huijsmans RLN, Ticknor IL,
Smith LS, Vavilala MS, Rivara FP and Watson RS: Acute respiratory
distress syndrome following pediatric trauma: Application of
pediatric acute lung injury consensus conference criteria. Crit
Care Med. 48:e26–e33. 2020. View Article : Google Scholar
|
|
11
|
Fan E, Brodie D and Slutsky AS: Acute
respiratory distress syndrome: Advances in diagnosis and treatment.
JAMA. 319:698–710. 2018. View Article : Google Scholar
|
|
12
|
Fanelli V and Ranieri VM: Mechanisms and
clinical consequences of acute lung injury. Ann Am Thorac Soc. 12
(Suppl 1):S3–S8. 2015. View Article : Google Scholar
|
|
13
|
Su BC, Huang HN, Lin TW, Hsiao CD and Chen
JY: Epinecidin-1 protects mice from LPS-induced endotoxemia and
cecal ligation and puncture-induced polymicrobial sepsis. Biochim
Biophys Acta Mol Basis Dis. 1863:3028–3037. 2017. View Article : Google Scholar
|
|
14
|
Chen M, Chen C, Yuan X, Chen X, Zheng F,
Shao L and Zhang Z: Angiotensin II aggravates lipopolysaccharide
induced human pulmonary microvascular endothelial cells
permeability in high glucose status. Endocr J. 65:717–725. 2018.
View Article : Google Scholar
|
|
15
|
Wang H, Wang T, Yuan Z, Cao Y, Zhou Y, He
J, Shen Y, Zeng N, Dai L, Wen F and Chen L: Role of receptor for
advanced glycation end products in regulating lung fluid balance in
lipopolysaccharide-induced acute lung injury and infection-related
acute respiratory distress syndrome. Shock. 50:472–482. 2018.
View Article : Google Scholar
|
|
16
|
Wu DQ, Wu HB, Zhang M and Wang JA: Effects
of zinc finger protein A20 on lipopolysaccharide (LPS)-induced
pulmonary inflammation/anti-inflammatory mediators in an acute lung
injury/acute respiratory distress syndrome rat model. Med Sci
Monit. 23:3536–3545. 2017. View Article : Google Scholar
|
|
17
|
Shi L, Dong N, Ji D, Huang X, Ying Z, Wang
X and Chen C: Lipopolysaccharide-induced CCN1 production enhances
interleukin-6 secretion in bronchial epithelial cells. Cell Biol
Toxicol. 34:39–49. 2018. View Article : Google Scholar
|
|
18
|
Wang Y, Lin D, Tan H, Gao Y and Ma J:
Penehyclidine hydrochloride preconditioning provides pulmonary and
systemic protection in a rat model of lung ischaemia reperfusion
injury. Eur J Pharmacol. 839:1–11. 2018. View Article : Google Scholar
|
|
19
|
Xiao HT, Liao Z and Tong RS: Penehyclidine
hydrochloride: A potential drug for treating COPD by attenuating
Toll-like receptors. Drug Des Devel Ther. 6:317–22. 2012.
View Article : Google Scholar
|
|
20
|
Wang Y, Gao Y and Ma J: Pleiotropic
effects and pharmacological properties of penehyclidine
hydrochloride. Drug Des Devel Ther. 12:3289–3299. 2018. View Article : Google Scholar
|
|
21
|
Lin D, Ma J, Xue Y and Wang Z:
Penehyclidine hydrochloride preconditioning provides
cardioprotection in a rat model of myocardial ischemia/reperfusion
injury. PLoS One. 10:e01380512015. View Article : Google Scholar
|
|
22
|
Shen W, Gan J, Xu S, Jiang G and Wu H:
Penehyclidine hydrochloride attenuates LPS-induced acute lung
injury involvement of NF-kappaB pathway. Pharmacol Res. 60:296–302.
2009. View Article : Google Scholar
|
|
23
|
Wang YA, Zhou WX, Li JX, Liu YQ, Yue YJ,
Zheng JQ, Liu KL and Ruan JX: Anticonvulsant effects of
phencynonate hydrochloride and other anticholinergic drugs in soman
poisoning: neurochemical mechanisms. Life Sci. 78:210–223. 2005.
View Article : Google Scholar
|
|
24
|
Guoshou Z, Chengye Z, Zhihui L and Jinlong
L: Effects of high dose of anisodamine on the respiratory function
of patients with traumatic acute lung injury. Cell Biochem Biophys.
66:365–369. 2013. View Article : Google Scholar
|
|
25
|
Zhan J, Xiao F, Li JJ, Zhang ZZ, Chen K,
Wang YP and Wang YL: Penehyclidine hydrochloride decreases
pulmonary microvascular permeability by upregulating beta arrestins
in a murine cecal ligation and puncture model. J Surg Res.
193:391–398. 2015. View Article : Google Scholar
|
|
26
|
Krenn K, Lucas R, Croizé A, Boehme S,
Klein KU, Hermann R, Markstaller K and Ullrich R: Inhaled AP301 for
treatment of pulmonary edema in mechanically ventilated patients
with acute respiratory distress syndrome. A phase IIa randomized
placebo-controlled trial. 21:1942017.
|
|
27
|
Zheng F, Xiao F, Yuan QH, Liu QS, Zhang
ZZ, Wang YL and Zhan J: Penehyclidine hydrochloride decreases
pulmonary microvascular endothelial inflammatory injury through a
beta-arrestin-1-dependent mechanism. Inflammation. 41:1610–1620.
2018. View Article : Google Scholar
|
|
28
|
Dong L, Yu WM, Zheng H, Loh ML, Bunting
ST, Pauly M, Huang G, Zhou M, Broxmeyer HE, Scadden DT and Qu CK:
Leukaemogenic effects of Ptpn11 activating mutations in the stem
cell microenvironment. Nature. 539:304–308. 2016. View Article : Google Scholar
|
|
29
|
Huang X, Kong G, Li Y, Zhu W, Xu H, Zhang
X, Li J, Wang L, Zhang Z, et al: Decitabine and 5-azacitidine both
alleviate LPS induced ARDS through anti-inflammatory/antioxidant
activity and protection of glycocalyx and inhibition of MAPK
pathways in mice. Biomedicine & Pharmacotherapy. 84:447–453.
2016. View Article : Google Scholar
|
|
30
|
Kellner M..Noonepalle S..Lu Q..Srivastava
A..Zemskov E..Black S. M: ROS Signaling in the Pathogenesis of
Acute Lung Injury (ALI) and Acute Respiratory Distress Syndrome
(ARDS). Wang YX: Pulmonary Vasculature Redox Signaling in Health
and Disease. Advances in Experimental Medicine and Biology.
Springer; Cham: 967. pp. 105–137. 2017, View Article : Google Scholar
|
|
31
|
Kartlaşmış K, Kökbaş U and Kayrin L:
Biochemistry of apoptosis. Arch Med Rev J. 25:52–69. 2016.
|
|
32
|
Redza-Dutordoir M and Averill-Bates DA:
Activation of apoptosis signalling pathways by reactive oxygen
species. Biochim Biophys Acta. 1863:2977–2992. 2016. View Article : Google Scholar
|
|
33
|
Ekizceli G, Uluer ET and Inan S:
Investigation of the effects of rapamycin on the mTOR pathway and
apoptosis in metastatic and non-metastatic human breast cancer cell
lines. Bratisl Lek Listy. 121:308–315. 2020.
|
|
34
|
Diwanji N and Bergmann A: An unexpected
friend-ROS in apoptosis-induced compensatory proliferation:
Implications for regeneration and cancer. Semin Cell Dev Biol.
80:74–82. 2018. View Article : Google Scholar
|
|
35
|
El-Shafey ES and Elsherbiny ES: The role
of apoptosis and autophagy in the insulin-enhancing activity of
oxovanadium(IV) bipyridine complex in streptozotocin-induced
diabetic mice. Biometals. 33:123–135. 2020. View Article : Google Scholar
|
|
36
|
Liu J, Liu W and Yang H: Balancing
apoptosis and autophagy for parkinson's disease therapy: Targeting
BCL-2. ACS Chem Neurosci. 10:792–802. 2019. View Article : Google Scholar
|
|
37
|
Yang Y, Gao H, Zhou H, Liu Q, Qi Z, Zhang
Y and Zhang J: The role of mitochondria-derived peptides in
cardiovascular disease: Recent updates. Biomed Pharmacother.
117:1090752019. View Article : Google Scholar
|
|
38
|
Kiraz Y, Adan A, Kartal Yandim M and Baran
Y: Major apoptotic mechanisms and genes involved in apoptosis.
Tumour Biol. 37:8471–8486. 2016. View Article : Google Scholar
|
|
39
|
Wang J, Oppenheimer L, Fata P, Pintin J,
Stimpson R and Mantsch HH: Spectroscopic approach to
capillary-alveolar membrane damage induced acute lung injury. Can
Respir J. 6:499–506. 1999. View Article : Google Scholar
|
|
40
|
Kiser TH, Burnham EL, Clark B, Ho PM,
Allen RR, Moss M and Vandivier RW: Half-dose versus full-dose
alteplase for treatment of pulmonary embolism. Crit Care Med.
46:1617–1625. 2018. View Article : Google Scholar
|
|
41
|
Li W, Qiu X, Jiang H, Han Y, Wei D and Liu
J: Downregulation of miR-181a protects mice from LPS-induced acute
lung injury by targeting Bcl-2. Biomed Pharmacother. 84:1375–1382.
2016. View Article : Google Scholar
|
|
42
|
Li B, Zeng M, Zheng H, Huang C, He W, Lu
G, Li X, Chen Y and Xie R: Effects of ghrelin on the apoptosis of
human neutrophils in vitro. Int J Mol Med. 38:794–802. 2016.
View Article : Google Scholar
|
|
43
|
Wang L, Wang T, Li H, Liu Q, Zhang Z, Xie
W, Feng Y, Socorburam T, Wu G, Xia Z and Wu Q: Receptor interacting
protein 3-mediated necroptosis promotes lipopolysaccharide-induced
inflammation and acute respiratory distress syndrome in mice. PLoS
One. 11:e01557232016. View Article : Google Scholar
|
|
44
|
Zhan J, Wang Y, Wang C, Li J, Zhang Z and
Jia B: Protective effects of penehyclidine hydrochloride on septic
mice and its mechanism. Shock. 28:727–732. 2007.
|
|
45
|
Yuan Q, Xiao F, Liu Q, Zheng F, Shen S, He
Q, Chen K, Wang Y, Zhang Z and Zhan J: M3 receptor is
involved in the effect of penehyclidine hydrochloride reduced
endothelial injury in LPS-stimulated human pulmonary microvascular
endothelial cell. Pulm Pharmacol Ther. 48:144–150. 2018. View Article : Google Scholar
|
|
46
|
Feng M, Wang L, Chang S and Yuan P:
Penehyclidine hydrochloride regulates mitochondrial dynamics and
apoptosis through p38MAPK and JNK signal pathways and provides
cardioprotection in rats with myocardial ischemia-reperfusion
injury. Eur J Pharm Sci. 121:243–250. 2018. View Article : Google Scholar
|
|
47
|
Lin D, Cui B, Ren J and Ma J: Regulation
of VDAC1 contributes to the cardioprotective effects of
penehyclidine hydrochloride during myocardial ischemia/reperfusion.
Exp Cell Res. 367:257–263. 2018. View Article : Google Scholar
|
|
48
|
Liu ZY, Guo L, Xiao G, Dong GY, Zhang YX,
Cheng H, Wang XY and Yang C: Significance and expression of
c-erBb-2, p53, and caspase-3 in breast cancer tissue in different
age groups. Eur J Gynaecol Oncol. 39:430–432. 2018.
|
|
49
|
Bosmann M and Ward PA: The inflammatory
response in sepsis. Trends Immunol. 34:129–236. 2013. View Article : Google Scholar
|
|
50
|
Johnson CL, Soeder Y and Dahlke MH:
Concise review: Mesenchymal stromal cell-based approaches for the
treatment of acute respiratory distress and sepssis syndromes. Stem
Cells Transl Med. 6:1141–1151. 2017. View Article : Google Scholar
|
|
51
|
Monteleon CL, Agnihotri T, Dahal A, Liu M,
Rebecca VW, Beatty GL, Amaravadi RK and Ridky TW: Lysosomes support
the degradation, signaling, and mitochondrial metabolism necessary
for human epidermal differentiation. J Invest Dermatol.
138:1945–1954. 2018. View Article : Google Scholar
|
|
52
|
Ko S, Gu MJ, Kim CG, Kye YC, Lim Y, Lee
JE, Park BC, Chu H, Han SH and Yun CH: Rapamycin-induced autophagy
restricts porcine epidemic diarrhea virus infectivity in porcine
intestinal epithelial cells. Antiviral Res. 146:86–95. 2017.
View Article : Google Scholar
|
|
53
|
Lv C, Wang L, Zhu X, Lin W, Chen X, Huang
Z, Huang L and Yang S: Glucosamine promotes osteoblast
proliferation by modulating autophagy via the mammalian target of
rapamycin pathway. 99:271–277. 2018.
|
|
54
|
Zeng M, Sang W, Chen S, Chen R, Zhang H,
Xue F, Li Z, Liu Y and Gong Y: 4-PBA inhibits LPS-induced
inflammation through regulating ER stress and autophagy in acute
lung injury models. Toxicol. 271((5)): 26–37. 2017.
|
|
55
|
Ren J and Zhang Y: Targeting autophagy in
aging and aging-related cardiovascular diseases. Trends Pharmacol
Sci. 39:1064–1076. 2018. View Article : Google Scholar
|
|
56
|
Decuypere JP, Ceulemans LJ, Agostinis P,
Monbaliu D, Naesens M, Pirenne J and Jochmans I: Autophagy and the
kidney: Implications for ischemia-reperfusion injury and therapy.
Am J Kidney Dis. 66:699–709. 2015. View Article : Google Scholar
|
|
57
|
Gao Y, Wang N, Liu L, Liu Y and Zhang J:
Relationship between mammalian target of rapamycin and autophagy in
lipopolysaccharide-induced lung injury. J Surg Res. 201:356–363.
2016. View Article : Google Scholar
|
|
58
|
Zhao H and Luo F: Rapamycin reverse
lipopolysaccharide-induced acute lung injury through activating
autophagy flux. J Pharm Univ. 2015.
|
|
59
|
Liu Y and Zhang J: Saturated hydrogen
saline ameliorates lipopolysaccharide-induced acute lung injury by
reducing excessive autophagy. Exp Ther Med. 13:2609–2615. 2017.
|
|
60
|
Zhu Q, Wang H, Wang H, Luo Y, Yu Y, Du Q,
Fei A and Pan S: Protective effects of ethyl pyruvate on
lipopolysaccharide-induced acute lung injury through inhibition of
autophagy in neutrophils. Mol Med Rep. 15:1272–1278. 2017.
View Article : Google Scholar
|