Introduction
Heart failure (HF) has become a major public health
problem that affects 23 million people worldwide (1), and due to increases in life expectancy
over time, the prevalence of HF is continually increasing (2). Myocardial fibrosis (MF), a common
pathological manifestation of various cardiovascular diseases at a
certain stage, can disrupt the myocardial structure, leading to
myocardial disarray and vasomotor dysfunction, thereby promoting HF
progression (3). MF involves
cardiac interstitial remodeling characterized by excessive cardiac
interstitial fibroblast proliferation, as well as excessive
deposition and abnormal distribution of collagen (4). It has been reported that the severity
of MF is associated with higher long-term mortality in patients
with heart disease, especially in patients with HF (5,6).
Therefore, identifying therapeutic strategies to prevent and
eliminate MF is important to improve the outcomes of HF.
Nowadays, MF detection primarily occurs via imaging
or measuring the expression of related biomarkers; however,
biomarkers that are directly associated with MF have not been
identified (7,8). Diuretics, Digitalis and
angiotensin-converting enzyme inhibitors are used to delay chronic
HF in clinical settings, but display side effects and are not
suitable for long-term treatment (9,10).
Therefore, the identification of novel drugs and therapeutic
targets to delay MF and prevent HF is important. Previous research
has demonstrated that myocardial cell death triggers an
inflammatory response, leading to fibroblast activation and
replacement of dead myocardial cells with fibrous tissue (4). Multiple studies have demonstrated that
TGF-β, IL-11, α-smooth muscle actin (α-SMA), Collagen I and
Collagen III were all upregulated in MF and served as important
determinants of the condition (11–13).
It has been reported that inhibition of myocardial apoptosis,
fibrosis and inflammation could protect against myocardial injury
in diabetic cardiomyopathy (14),
and oxidative stress has also been reported to be closely related
to the progression of MF (15). AXL
receptor tyrosine kinase (Axl) has been utilized to predict the
adverse pathology of MF (16).
Moreover, angiotensin II receptor type 1 (AT1R) has been reported
to interact with angiotensin II (AngII) and promote the formation
of tissue fibrosis (17). In
addition, Zachariah et al (18) reported that circulating matrix
metalloproteinase (MMP)3 may serve as a marker of MF and
ventricular arrhythmia in adolescents with hypertrophic
cardiomyopathy. It was also demonstrated that MMP3 was upregulated
during AngII-induced cardiac remodeling and was associated with the
regulation of angiogenesis and immune responses via RNA sequencing
analysis (19). MMP3 is a type of
stromelysin that serves an essential role in tissue remodeling by
degrading extracellular matrix (ECM) (20,21).
Tuncer et al (22) reported
that MMP3 expression was significantly higher in patients with
rheumatoid arthritis compared with control patients, and high MMP3
expression levels were related to high disease activity, as
demonstrated by C-reactive protein levels, Health Assessment
Questionnaire scores and erythrocyte sedimentation rates. However,
the effects of MMP3 on MF are not completely understood.
A previous study demonstrated that activation of the
renin-angiotensin system (RAS) is a key mediator in HF progression
(23). AngII is a central signaling
molecule of RAS that serves a vital role in MF (24). Therefore, in the present study,
AngII was used to induce fibrosis in rat myocardial cells, and MMP3
was knocked down to investigate its effects on MF progression. The
results of the present study may improve the current understanding
of HF, and provide novel therapeutic targets for the prevention and
reversal of MF.
Materials and methods
Cell culture
H9C2 rat myocardial cells (The Cell Bank of Type
Culture Collection of Chinese Academy of Sciences) were cultured in
DMEM (Wuhan Boster Biological Technology, Ltd.) supplemented with
10% FBS (Thermo Fisher Scientific, Inc.), 100 kU/l penicillin
(Thermo Fisher Scientific, Inc.) and 100 mg/l streptomycin (Thermo
Fisher Scientific, Inc.) at 37°C with 5% CO2.
AngII concentration screening
H9C2 cells were seeded (5×105 cells/well)
into 12-well plates and cultured overnight. Cells were treated with
different concentrations (0, 10−8, 10−7 or
10−6 M) of AngII (Shanghai YuanYe Biotechnology Co.,
Ltd.) for 6, 12 and 24 h at 37°C. Subsequently, reverse
transcription-quantitative PCR (RT-qPCR) was performed to measure
Axl expression levels.
Cell transfection
H9C2 cells were resuspended in complete culture
medium and seeded (5×105 cells/well) into 6-well plates.
Following culture overnight, the culture medium was replaced with
serum-free medium. Cells were divided into the following five
groups: i) blank control; ii) small interfering (si)RNA-negative
control (NC); iii) siRNA-MMP3 1; iv) siRNA-MMP3 2; and v)
siRNA-MMP3 3. siRNA-NC (non-targeting; forward,
5′-UUCUCCGAACGUGUCACGUTT-3′ and reverse,
5′-ACGUGACACGUUCGGAGAATT-3′) and siRNA-MMP3 (1/2/3) were prepared
and obtained from Yanzai Biotechnology (Shanghai) Co., Ltd. The
sequences of siRNA-MMP3 were as follows: siRNA-MMP3-1,
5′-GAAGCAGTTTACTAAGAAA-3′; siRNA-MMP3-2, 5′-GAGAAGTCTTGTTCTTTAA-3′;
and siRNA-MMP3-3, 5′-GATGCAGCCATTTCTTTAA-3′. Cell transfection was
performed as previously described (25). Briefly, H9C2 cells were seeded
(5×105 cells/well) into 6-well plates and cultured
overnight. Subsequently, cell medium was changed to serum-free
medium, and cells were transfected with 100 nM siRNA-NC or 100 nM
siRNA-MMP3 (1/2/3) at 24±2°C for 20 min using
Lipofectamine® 3000 (Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. Cells in the blank
control group were cultured in medium. Following transfection for 6
h, the medium was replaced with complete culture medium. After
culture for another 24 h, transfection efficiency was assessed via
RT-qPCR.
Cell viability assay
The effects of MMP3 on fibrotic H9C2 cell viability
were determined by performing the Cell Counting Kit-8 (CCK-8) assay
(Beyotime Institute of Biotechnology). H9C2 cells were seeded
(5×105 cells/well) into 6-well plates. Following culture
overnight, cells were divided into the following five groups: i)
Control; ii) MF; iii) MF + levocarnitine; iv) MF + siRNA-NC; and v)
MF + siRNA-MMP3. To induce cellular fibrosis, cells in the MF, MF +
levocarnitine, MF + siRNA-NC and MF + siRNA-MMP3 groups were
treated with 10−8 M AngII at 25°C for 24 h.
Subsequently, cells in the MF + levocarnitine group were treated
with 100 µM levocarnitine (Shanghai YuanYe Biotechnology Co., Ltd.)
at room temperature for 24 h, and cells in the MF + siRNA-NC and MF
+ siRNA-MMP3 groups were transfected with siRNA-NC or siRNA-MMP3,
respectively, according to the aforementioned protocol. Cells in
the control group were untreated. At 24 h post-transfection, cells
were collected and seeded (1×104 cells/well) into a
96-well plate. Following culture for 24, 48, 72 or 96 h, 10 µl
CCK-8 reagent was added to each well and incubated for 2.5 h at
37°C. The absorbance was measured at a wavelength of 450 nm using a
microplate reader.
Cell apoptosis assay
The Annexin V-FITC Apoptosis Detection kit (Beyotime
Institute of Biotechnology) was used to measure H9C2 cell apoptosis
according to the manufacturer's protocol. H9C2 cells were seeded
(1×104 cells/well) into a 24-well plate and cultured for
24 h at 37°C. H9C2 cells were harvested and centrifuged at 1000 × g
for 5 min at room temperature. Subsequently, cells were resuspended
with 195 µl Annexin V-FITC. Subsequently, cells were double-stained
with 5 µl Annexin V-FITC and 10 µl PI in the dark at 20°C for 20
min. Cell apoptosis was assessed using a FACSCalibur flow cytometer
(Becton-Dickinson and Company) and the rate of apoptosis (early and
late apoptosis) was calculated using FCS Express software (version
4; De Novo Software).
Cell migration assay
H9C2 cell migration was evaluated using Transwell
chambers (pore size, 8 µm; Guangzhou Jet Bio-Filtration Co., Ltd.).
Cells were seeded (6×104 cells/well) into the upper
chamber with complete medium. Medium supplemented with 10% FBS was
loaded into the lower chamber. Following incubation at 37°C for 24
h, cells were fixed with 4% paraformaldehyde (Sinopharm Chemical
Reagent Co., Ltd.) at room temperature for 10 min, and then stained
with 0.5% crystal violet (Beyotime Institute of Biotechnology) at
room temperature for 10 min. Stained cells were visualized using a
light microscope (Olympus Corporation; magnification, ×200).
RT-qPCR
Total RNA was extracted from cells using
TRIzol® reagent (Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. Total RNA was reverse
transcribed into cDNA using the PrimeScript™ II 1st Strand cDNA
Synthesis kit (Takara Bio, Inc.). at 37°C for 60 min and 85°C for 5
sec. Subsequently, qPCR was performed using SYBR Premix EX Taq
(Thermo Fisher Scientific, Inc.). The primers used for qPCR are
listed in Table I. The following
thermocycling conditions were used for qPCR: 95°C for 3 min; 95°C
for 10 sec; followed by 40 cycles at 60°C for 30 sec and 60°C for
30 sec. mRNA expression levels were quantified using the
2−ΔΔCq method (26) and
normalized to the internal reference gene GAPDH.
 | Table I.Sequences of primers used in the
present study. |
Table I.
Sequences of primers used in the
present study.
| Gene | Sequence (5→3) |
|---|
| GAPDH | F:
AGACAGCCGCATCTTCTTGT |
|
| R:
CTTGCCGTGGGTAGAGTCAT |
| MMP3 | F:
CAGGCATTGGCACAAAGGTG |
|
| R:
GTGGGTCACTTTCCCTGCAT |
| Axl | F:
GCCCAGTGAGTGAACCCC |
|
| R:
TCTCCTTCAGCTCTTCGCTG |
| AT1R | F:
GGAAACAGCTTGGTGGTGAT |
|
| R:
CACACTGGCGTAGAGGTTGA |
| MMP9 | F:
GCATCTGTATGGTCGTGGCT |
|
| R:
CTGTAGGGGCCTCAGAAGGA |
| Bcl-2 | F:
GACTGAGTACCTGAACCGGCATC |
|
| R:
CTGAGCAGCGTCTTCAGAGACA |
| Caspase-3 | F:
GAATCCACGAGCAGAGTC |
|
| R:
TCAACAAGCCAACCAAGT |
| p53 | F:
ACAGTTAGGGGGTACCTGGC |
|
| R:
GCTGTGGTGGGCAGAATATCAT |
| α-SMA | F:
GGAGATGGCGTGACTCACAA |
|
| R:
CGCTCAGCAGTAGTCACGAA |
| Collagen I | F:
ACTTAACATCCAAGGCCGCT |
|
| R:
ACAATATTTGCCTCAGTTTGTGC |
| STAT3 | F:
CCTTGGATTGAGAGCCAAGAT |
|
| R:
ACCAGAGTGGCGTGTGACT |
| p22Phox | F:
TTGTTGCAGGAGTGCTCATC |
|
| R:
CACGGACAGCAGTAAGTGGA |
| p47Phox | F:
TCCCTGCATCCTATTTGGAG |
|
| R:
GGGACACCTCATCCTCTTCA |
Western blotting
Total protein was isolated from cells using
radioimmunoprecipitation assay protein lysis buffer (Beyotime
Institute of Biotechnology). Protein concentrations were measured
using a Bicinchoninic Acid Protein Assay kit (Wuhan Boster
Biological Technology, Ltd.) according to the manufacturer's
protocol. Proteins (20 µg) were separated via 10% SDS-PAGE and
transferred to PVDF membranes, which were blocked with 5% skimmed
milk for 2 h at 37°C. Subsequently, the membranes were incubated
overnight at 4°C with the following primary antibodies: Anti-MMP3
(1:4,000; cat. no. 66338-1-Ig; ProteinTech Group, Inc.),
anti-caspase-3 (1:1,000; cat. no. 66470-2-Ig; ProteinTech Group,
Inc.), anti-p53 (1:2,000; cat. no. 60283-2-Ig; ProteinTech Group,
Inc.) and anti-GAPDH (1:10,000; cat. no. 60004-1-Ig; ProteinTech
Group, Inc.). After washing three times with PBST (0.05% Tween-20
in PBS) the membranes were incubated with a goat anti-mouse IgG
(1:10,000; cat. no. 115-035-003; Jackson ImmunoResearch
Laboratories, Inc.) secondary antibody at 37°C for 2 h. Following
washing three times with PBST, protein bands were visualized using
the ECL assay kit (Beyotime Institute of Biotechnology) and
chemiluminescence apparatus (Shanghai Tanon Science &
Technology Co., Ltd.). Protein expression was semi-quantified using
Image-Pro Plus software (version 6.0; Media Cybernetics, Inc.)
Statistical analysis
Each experiment was performed in triplicate. Data
are presented as the mean ± standard deviation. Statistical
analyses were performed using GraphPad Prism software (version 7.0;
GraphPad Software, Inc.). Prior to statistical analysis, the
homogeneity of variance test for all data was performed to obtain
the F-value and P-value (Table
SI). If P>0.50, one-way ANOVA followed by Tukey's post hoc
test was performed. If P≤0.05, Brown-Forsythe and Welch ANOVA tests
followed by Dunnett's T3 post hoc test were performed. P<0.05
was considered to indicate a statistically significant
difference.
Results
Selection of optimal conditions for
AngII
To construct a myocardial fibroblast cell model,
H9C2 cells were treated with different concentrations of AngII.
Following treatment with different concentrations of AngII for 6 h,
there was no significant difference in the relative expression
levels of Axl compared with the control group (P>0.05; Fig. 1A). Following treatment for 12 h, the
relative expression levels of Axl were significantly increased in
cells treated with 10−8 M AngII compared with the
control group (P<0.05; Fig. 1B).
However, following treatment for 24 h, the relative expression
levels of Axl in cells treated with 10−8 M,
10−7 M or 10−6 M AngII were significantly
higher compared with the control group (P<0.05), with
10−8 M AngII resulting in the most significant increase
in Axl expression levels (P<0.01; Fig. 1C). Therefore, in subsequent
experiments, H9C2 cells were treated with 10−8 M AngII
for 24 h to induce fibrosis in myocardial cells.
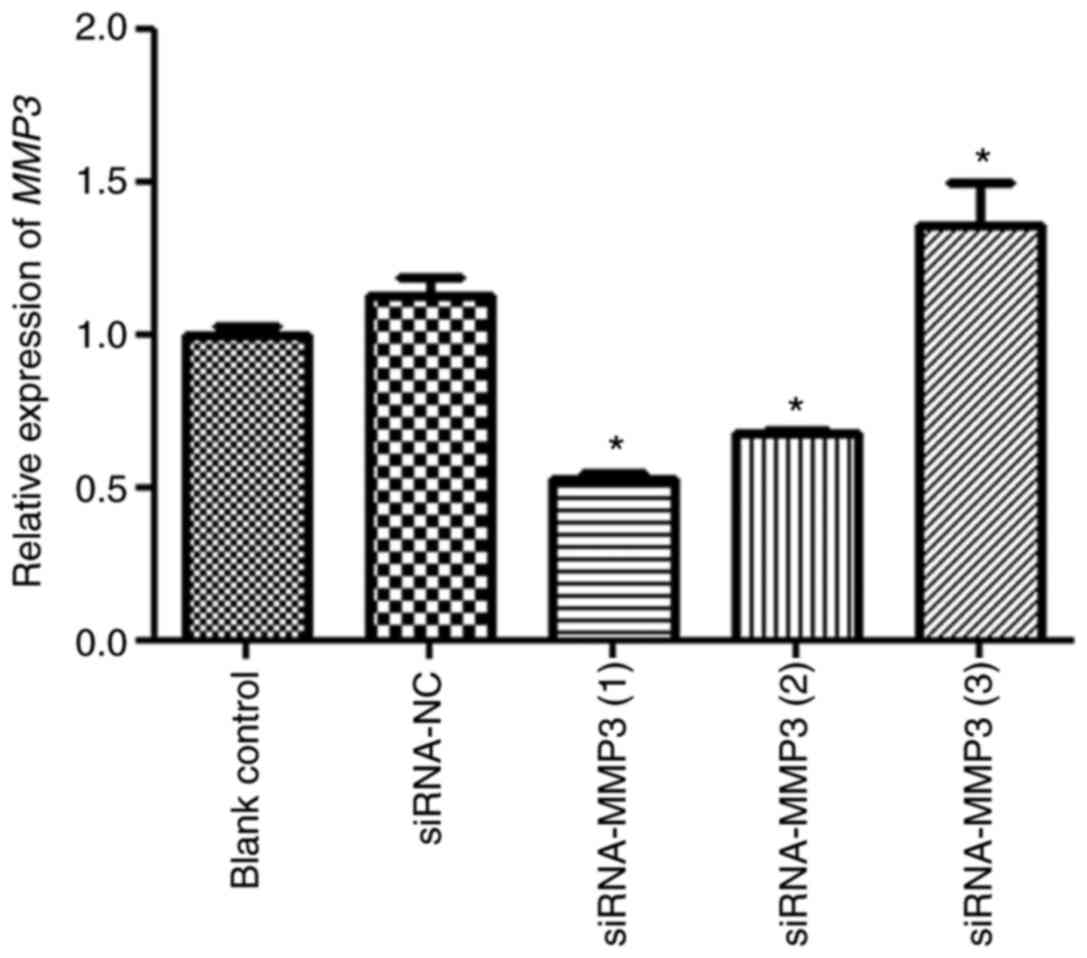
Transfection efficiency
Cell transfection efficiency was evaluated by
measuring MMP3 expression levels (Fig.
2). There was no significant difference in MMP3 expression
levels between the blank control and siRNA-NC groups (P>0.05).
MMP3 expression levels in the siRNA-MMP (3) group were significantly higher compared
with the siRNA-NC group (P<0.05). Additionally, MMP3 expression
levels in the siRNA-MMP3 (1) and
siRNA-MMP3 (2) groups were
0.52±0.04 and 0.68±0.02, respectively, which were significantly
decreased compared with the siRNA-NC group (P<0.05). Therefore,
siRNA-MMP3 (1) was selected to
establish MMP3 knockdown in H9C2 cells in subsequent
experiments.
Effects of MMP3 on cell viability
To investigate the effects of MMP3 on H9C2
fibroblast cells, cellular fibrosis was induced using
10−8 M AngII and the CCK-8 assay was performed to assess
cell viability. Following culture for 24 h, cell viability in the
MF group was significantly decreased compared with the control
group (P<0.05; Fig. 3A).
Following culture for 24 h, there was no significant difference in
cell viability between the MF and MF + siRNA-NC groups (P>0.05;
Fig. 3A). Compared with the MF
group, cell viability in the MF + levocarnitine and MF + siRNA-MMP3
groups was significantly inhibited (P<0.05; Fig. 3A). In addition, cell viability after
culture for 48, 72 and 96 h was similar compared with cell
viability after culture for 24 h (Fig.
3B-D). The results suggested that MMP3 knockdown and
levocarnitine treatment inhibited myocardial fibroblast cell
viability.
Effects of MMP3 on cell apoptosis
The effects of AngII on H9C2 cell apoptosis were
detected via flow cytometry (Fig.
4A). Cell apoptosis in the control, MF and MF + siRNA-NC groups
were 3.49±0.27, 7.80±0.05 and 7.58±0.33%, respectively. The results
demonstrated that the rate of cell apoptosis in the MF group was
significantly increased compared with the control group (P<0.05;
Fig. 4B), whereas there was no
significant difference in the rate of cell apoptosis between the MF
and MF + siRNA-NC groups (P>0.05; Fig. 4B). Compared with the MF group, the
rate of cell apoptosis was significantly increased in the MF +
levocarnitine and MF + siRNA-MMP3 groups (P<0.05; Fig. 4B). Therefore, the results indicated
that compared with control cells, AngII increased H9C2 cell
apoptosis and MMP3 knockdown further enhanced AngII-induced H9C2
cell apoptosis.
Effects of MMP3 on cell migration
The Transwell assay was performed to evaluate the
effects of MMP3 on cell migration (Fig.
5A). Compared with the control group, the number of migratory
cells was significantly decreased in the MF group (P<0.05;
Fig. 5B). There was no significant
difference in the number of migratory cells between the MF and MF +
siRNA-NC groups (P>0.05). However, the number of migratory cells
in the MF + levocarnitine and MF + siRNA-MMP3 groups was
significantly lower compared with the MF group (P<0.05). The
results suggested that MMP3 knockdown further inhibited cell
migration in AngII-treated H9C2 cells.
Effects of MMP3 on mRNA expression
levels
RT-qPCR was performed to investigate the molecular
mechanisms underlying the effects of MMP3 knockdown on myocardial
fibroblast cell viability, apoptosis and migration. Compared with
the control group, the expression levels of Axl and AT1R were
significantly increased in the MF group (P<0.05), but
significantly decreased in the MF + levocarnitine and MF +
siRNA-MMP3 groups compared with the MF group (P<0.05; Fig. 6A and B). Bcl-2 expression levels
were significantly lower in the MF group compared with the control
group (P<0.05), which were further decreased in the MF +
levocarnitine and MF + siRNA-MMP3 groups compared with the MF group
(P<0.05; Fig. 6C). However, the
trends in caspase-3 and p53 expression levels in the different
groups were opposite compared with Bcl-2 expression levels
(Fig. 6D and E). Moreover, the
trends in α-SMA and Collagen I expression levels in the different
groups were similar to Axl expression levels (Fig. 6F and G). Additionally, compared with
the control group, the expression levels of MMP3, MMP9, STAT3,
p22Phox and p47Phox were significantly upregulated in the MF group,
but significantly downregulated in the MF + levocarnitine and MF +
siRNA-MMP3 groups compared with the MF group (P<0.05; Fig. 6H-L). The expression level of MMP3 in
the MF + siRNA-MMP3 group was significantly lower compared with the
control and MF + levocarnitine groups (P<0.05; Fig. 6H).
 | Figure 6.Effects of MMP3 on mRNA expression
levels in H9C2 cells. (A) Axl, (B) AT1R, (C) Bcl-2, (D) Caspase-3,
(E) p53, (F) α-SMA, (G) Collagen I, (H) MMP3, (I) MMP9, (J) STAT3,
(K) p22Phox and (L) p47Phox mRNA expression levels in H9C2 cells
were measured via reverse transcription-quantitative PCR.
*P<0.05 vs. control; #P<0.05 vs. MF;
$P<0.05 vs. MF+ levocarnitine. MMP, matrix
metalloproteinase; Axl, AXL receptor tyrosine kinase; AT1R,
angiotensin II receptor type 1; α-SMA, α-smooth muscle actin; MF,
myocardial fibrosis; siRNA, small interfering RNA; NC, negative
control. |
Effects of MMP3 on protein expression
levels
Furthermore, the protein expression levels of MMP3,
caspase-3 and p53 were determined via western blotting (Fig. 7A). There was no significant
difference in the protein expression levels of MMP3, caspase-3 and
p53 between the MF and MF + siRNA-NC groups (P>0.05; Fig. 7B-D). MMP3 protein expression levels
in the MF group were significantly higher compared with the control
group (P<0.05); however, in the MF + levocarnitine and MF +
siRNA-MMP3 groups, MMP3 protein expression levels were
significantly downregulated compared with the MF group (P<0.05;
Fig. 7B). Caspase-3 and p53
expression levels were significantly upregulated in the MF group
compared with the control group (P<0.05), and further increased
in the MF + levocarnitine and MF + siRNA-MMP3 groups compared with
the MF group (P<0.05; Fig. 7C and
D). The trends in MMP3, caspase-3 and p53 protein expression
levels were consistent with the trends in MMP3, caspase-3 and p53
mRNA expression levels.
Discussion
Fibrosis is a terminal process that may also target
the liver and kidneys, and several pathogenetic aspects that may
translate into novel therapeutic approaches have been previously
reported (27–29). Fagone et al (30) performed a meta-analysis of datasets
including whole-genome transcriptional data on hepatic stellate
cells (HSCs), which identified that several genes associated with
HSC activation could be used to assess liver disease progression.
MF serves an important role in the pathogenesis of heart and
vascular remodeling, and is closely associated with pathological
processes of numerous clinical diseases, including hypertension,
myocardial infarction, atherosclerosis and diabetes (1,31,32).
MF can contribute to HF progression, seriously affecting the life
and health of elderly patients (33). MMP3 is related to certain diseases
characterized by extensive tissue destruction and collagen
degradation (34), but few studies
have investigated the effects of MMP3 on MF. In the present study,
H9C2 myocardial cells were treated with AngII to induce MF,
followed by transfection with siRNA-MMP3. The results demonstrated
that MMP3 knockdown and levocarnitine treatment inhibited
AngII-induced myocardial fibroblast cell viability and migration,
and promoted AngII-induced cell apoptosis. The RT-qPCR results
indicated that compared with the control group, AngII treatment
significantly upregulated Axl and AT1R expression levels, which
were significantly downregulated by MMP3 knockdown. MMP3 knockdown
also significantly decreased α-SMA, Collagen I and Bcl-2 expression
levels, and significantly increased caspase-3 and p53 expression
levels in AngII-treated cells. Additionally, MMP3, MMP9, STAT3,
p22Phox and p47Phox expression levels were significantly decreased
by MMP3 knockdown in AngII-treated cells. Collectively, the results
indicated that MMP3 altered myocardial fibroblast cell viability,
migration and apoptosis by regulating the expression levels of
apoptosis- and oxidative stress-related genes.
Previous studies have reported that AngII is
associated with MF formation and development (35,36).
Related to its ability to degrade ECM components, MMP3 has been
reported to serve key roles in ECM remodeling and promote tumor
progression (37,38). In the present study, MMP3 knockdown
inhibited myocardial fibroblast cell viability and migration, and
promoted myocardial fibroblast cell apoptosis. Compared with the
control group, AngII treatment significantly increased Axl, AT1R,
α-SMA and Collagen I expression levels, which were significantly
reversed by MMP3 knockdown. Bufu et al (39) reported that celastrol suppressed
colorectal cancer cell proliferation and migration by
downregulating MMP3 and MMP7 expression levels. Furthermore, Axl,
which is activated by growth arrest-specific 6, was upregulated in
patients with HF and MF, and was used to predict the adverse
pathology of cardiovascular disease (16,40).
AT1R can interact with AngII and promote tissue fibrotic formation
(17). Previous studies have also
demonstrated that pharmacological inhibition of AT1R may
significantly reduce cardiac fibrosis and improve cardiac function
(41,42). MMP3 knockdown significantly
downregulated Axl and AT1R expression levels in AngII-treated
cells, which suggested that MF progression was inhibited.
Additionally, α-SMA and Collagen I, which are biomarkers of
fibrosis, were upregulated during MF progression (43). Li et al (44) reported that astragaloside IV
effectively decreased α-SMA and Collagen I expression levels in
silica-induced rats, thus inhibiting the transformation to MF.
Based on the aforementioned studies and the results of the present
study, it was hypothesized that MMP3 knockdown may downregulate
biomarkers of MF (α-SMA and Collagen I) and relieve MF progression
by mediating cell viability, migration and apoptosis.
To further understand the molecular mechanisms
underlying the effects of MMP3 on MF, RT-qPCR was performed to
determine the expression levels of apoptosis- (Bcl-2, caspase-3 and
p53) and oxidative stress- (MMP3, MMP9, STAT3, p22Phox and p47Phox)
related genes. In the present study, MMP3 knockdown significantly
downregulated Bcl-2, MMP3, MMP9, STAT3, p22Phox and p47Phox
expression levels, but upregulated caspase-3 and p53 expression
levels in AngII-treated cells. Bcl-2, an antiapoptotic factor, has
been reported to be associated with the occurrence of fibrosis
(45). Caspase-3, which belongs to
the Caspase family, links the internal and external apoptosis
signaling pathways, and is the primary executor of cell apoptosis
(46). p53, another
apoptosis-related gene, is an important essential regulator of the
cell cycle and DNA repair that induces cell apoptosis (47). A study conducted by Wang et
al (48) demonstrated that IL-6
downregulated Bcl-2 expression levels and upregulated caspase-3
expression levels, thereby promoting myocardial cell apoptosis
under hypoxic incubation and delaying MF progression. Another study
reported that Boschniakia rossica polysaccharide induced
laryngeal cancer cell apoptosis by regulating Bcl-2, caspase-3 and
p53 expression levels, and displayed potential anticancer activity
(49). Based on the aforementioned
studies and the results of the present study, it was hypothesized
that MMP3 knockdown may facilitate fibrotic myocardial cell
apoptosis by downregulating Bcl-2 expression levels, and
upregulating caspase-3 and p53 expression levels.
In addition, oxidative stress has been reported to
participate in MF remodeling and serves an important function in
the occurrence and development of HF (15). Oxidative stress is defined as
excessive reactive oxygen species (ROS) relative to antioxidant
defenses, and is advanced in HF (50). ROS can mediate cell apoptosis and
MMPs, leading to ECM remodeling (51). MMP3 and MMP9 are extracellular zinc
proteases that serve vital roles in the development and progression
of atherosclerosis (52). Zhang
et al (53) reported that
resveratrol downregulated MMP3 and MMP9 expression levels in
lipopolysaccharide (LPS)-treated human umbilical vein endothelial
cells, and reduced LPS-induced MMP3 and MMP9 secretion. The present
study indicated that MMP3 and MMP9 expression levels were
downregulated by MMP3 knockdown in AngII-treated cells, which
suggested that MMP3 knockdown influenced ECM remodeling by
regulating MMP3 and MMP9 expression. STAT3, a potential nuclear
transcription factor, is involved in oxidative stress, and
endogenous STAT3 serves an essential roles in preventing heart
diseases, such as age-associated HF and ischemia/reperfusion injury
(54). Han et al (55) reported that AngII induced STAT3
activation, and STAT3 inhibition in mice protected against
AngII-induced fibrosis and cardiac dysfunction. In addition,
p22Phox and p47Phox, oxidative stress-related genes, are associated
with HF (56). In the present
study, MMP3 knockdown significantly decreased STAT3, p22Phox and
p47Phox expression levels in AngII-treated cells. Collectively, the
results indicated that MMP3 knockdown may relieve oxidative stress
by downregulating MMP3, MMP9, STAT3, p22Phox and p47Phox expression
levels, thus suppressing MF progression and preventing HF.
However, the present study had a number of
limitations. For example, the relationship between MMP3 and
oxidative stress requires further investigation, and the roles of
MMP3 in MF should be further verified in vivo.
In conclusion, compared with the control group,
AngII successfully induced fibrosis in rat myocardial cells,
inhibited H9C2 cell viability and migration, and promoted H9C2 cell
apoptosis. Therefore, MMP3 knockdown may downregulate MF biomarkers
and delay MF progression by regulating fibrotic cell viability,
migration and apoptosis. Additionally, the results indicated that
MMP3 knockdown may alter myocardial fibroblast cell proliferation
by mediating the expression levels of apoptosis- and oxidative
stress-related genes. The present study provided novel insights for
the development of therapeutic strategies for HF and other
cardiovascular diseases based on MF, indicating that MMP3 may serve
as a potential therapeutic target to promote cardiovascular health
and prolong the lives of elderly patients.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the Medical
Research Project of the Jing'an District Science and Technology
Commission of Shanghai (grant no. 2020MS12).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HW and LJ conceived the study, edited and reviewed
the manuscript, and supervised the project. JS and YG performed the
experiments, analyzed and interpreted the data, and wrote the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
MMP3
|
matrix metalloproteinase 3
|
|
AngII
|
angiotensin II
|
|
MF
|
myocardial fibrosis
|
|
HF
|
heart failure
|
|
ECM
|
extracellular matrix
|
References
|
1
|
Talman V and Ruskoaho H: Cardiac fibrosis
in myocardial infarction-from repair and remodeling to
regeneration. Cell Tissue Res. 365:563–581. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hao G, Wang X, Chen Z, Zhang L, Zhang Y,
Wei B, Zheng C, Kang Y, Jiang L, Zhu Z, et al China Hypertension
Survey Investigators, : Prevalence of heart failure and left
ventricular dysfunction in China: The China Hypertension Survey,
2012–2015. Eur J Heart Fail. 21:1329–1337. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
González A, Schelbert EB, Díez J and
Butler J: Myocardial interstitial fibrosis in heart failure:
Biological and translational perspectives. J Am Coll Cardiol.
71:1696–1706. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kong P, Christia P and Frangogiannis NG:
The pathogenesis of cardiac fibrosis. Cell Mol Life Sci.
71:549–574. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Aoki T, Fukumoto Y, Sugimura K, Oikawa M,
Satoh K, Nakano M, Nakayama M and Shimokawa H: Prognostic impact of
myocardial interstitial fibrosis in non-ischemic heart failure.
-Comparison between preserved and reduced ejection fraction heart
failure. Circ J. 75:2605–2613. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gyöngyösi M, Winkler J, Ramos I, Do QT,
Firat H, McDonald K, González A, Thum T, Díez J, Jaisser F, et al:
Myocardial fibrosis: Biomedical research from bench to bedside. Eur
J Heart Fail. 19:177–191. 2017. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Helm PA, Caravan P, French BA, Jacques V,
Shen L, Xu Y, Beyers RJ, Roy RJ, Kramer CM and Epstein FH:
Postinfarction myocardial scarring in mice: Molecular MR imaging
with use of a collagen-targeting contrast agent. Radiology.
247:788–796. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
López B, González A, Ravassa S, Beaumont
J, Moreno MU, San José G, Querejeta R and Díez J: Circulating
biomarkers of myocardial fibrosis: The need for a reappraisal. J Am
Coll Cardiol. 65:2449–2456. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ellison DH and Felker GM: Diuretic
treatment in heart failure. N Engl J Med. 377:1964–1975. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Schupp T, Behnes M, Weiss C, Nienaber C,
Reiser L, Bollow A, Taton G, Reichelt T, Ellguth D, Engelke N, et
al: Digitalis therapy and risk of recurrent ventricular
tachyarrhythmias and ICD therapies in atrial fibrillation and heart
failure. Cardiology. 142:129–140. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schafer S, Viswanathan S, Widjaja AA, Lim
WW, Moreno-Moral A, DeLaughter DM, Ng B, Patone G, Chow K, Khin E,
et al: IL-11 is a crucial determinant of cardiovascular fibrosis.
Nature. 552:110–115. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shinde AV, Humeres C and Frangogiannis NG:
The role of α-smooth muscle actin in fibroblast-mediated matrix
contraction and remodeling. Biochim Biophys Acta Mol Basis Dis.
1863:298–309. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang Y, Luo G, Zhang Y, Zhang M, Zhou J,
Gao W, Xuan X, Yang X, Yang D, Tian Z, et al: Critical effects of
long non-coding RNA on fibrosis diseases. Exp Mol Med. 50:e4282018.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang X, Pan L, Yang K, Fu Y, Liu Y, Chi
J, Zhang X, Hong S, Ma X and Yin X: H3 relaxin protects against
myocardial injury in experimental diabetic cardiomyopathy by
inhibiting myocardial apoptosis, fibrosis and inflammation. Cell
Physiol Biochem. 43:1311–1324. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kazakov A, Hall RA, Werner C, Meier T,
Trouvain A, Rodionycheva S, Nickel A, Lammert F, Maack C, Böhm M,
et al: Raf kinase inhibitor protein mediates myocardial fibrosis
under conditions of enhanced myocardial oxidative stress. Basic Res
Cardiol. 113:422018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Batlle M, Recarte-Pelz P, Roig E, Castel
MA, Cardona M, Farrero M, Ortiz JT, Campos B, Pulgarín MJ, Ramírez
J, et al: AXL receptor tyrosine kinase is increased in patients
with heart failure. Int J Cardiol. 173:402–409. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zheng RH, Bai XJ, Zhang WW, Wang J, Bai F,
Yan CP, James EA, Bose HS, Wang NP and Zhao ZQ: Liraglutide
attenuates cardiac remodeling and improves heart function after
abdominal aortic constriction through blocking angiotensin II type
1 receptor in rats. Drug Des Devel Ther. 13:2745–2757. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zachariah JP, Colan SD, Lang P, Triedman
JK, Alexander ME, Walsh EP, Berul CI and Cecchin F: Circulating
matrix metalloproteinases in adolescents with hypertrophic
cardiomyopathy and ventricular arrhythmia. Circ Heart Fail.
5:462–466. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shu J, Liu Z, Jin L and Wang H: An RNA
sequencing study identifies candidate genes for angiotensin II
induced cardiac remodeling. Mol Med Rep. 17:1954–1962.
2018.PubMed/NCBI
|
|
20
|
Craig VJ, Zhang L, Hagood JS and Owen CA:
Matrix metalloproteinases as therapeutic targets for idiopathic
pulmonary fibrosis. Am J Respir Cell Mol Biol. 53:585–600. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kitanaka N, Nakano R, Sakai M, Kitanaka T,
Namba S, Konno T, Nakayama T and Sugiya H: ERK1/ATF-2 signaling
axis contributes to interleukin-1β-induced MMP-3 expression in
dermal fibroblasts. PLoS One. 14:e02228692019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tuncer T, Kaya A, Gulkesen A, Kal GA,
Kaman D and Akgol G: Matrix metalloproteinase-3 levels in relation
to disease activity and radiological progression in rheumatoid
arthritis. Adv Clin Exp Med. 28:665–670. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Patel VB, Zhong JC, Grant MB and Oudit GY:
Role of the ACE2/Angiotensin 1–7 axis of the renin-angiotensin
system in heart failure. Circ Res. 118:1313–1326. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jin HY, Song B, Oudit GY, Davidge ST, Yu
HM, Jiang YY, Gao PJ, Zhu DL, Ning G, Kassiri Z, et al: ACE2
deficiency enhances angiotensin II-mediated aortic profilin-1
expression, inflammation and peroxynitrite production. PLoS One.
7:e385022012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sahoo S, Meijles DN, Al Ghouleh I, Tandon
M, Cifuentes-Pagano E, Sembrat J, Rojas M, Goncharova E and Pagano
PJ: MEF2C-MYOCD and leiomodin1 suppression by miRNA-214 promotes
smooth muscle cell phenotype switching in pulmonary arterial
hypertension. PLoS One. 11:e01537802016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xu G, Ao R, Zhi Z, Jia J and Yu B: miR-21
and miR-19b delivered by hMSC-derived EVs regulate the apoptosis
and differentiation of neurons in patients with spinal cord injury.
J Cell Physiol. 234:10205–10217. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fagone P, Mangano K, Pesce A, Portale TR,
Puleo S and Nicoletti F: Emerging therapeutic targets for the
treatment of hepatic fibrosis. Drug Discov Today. 21:369–375. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gungor O, Unal HU, Guclu A, Gezer M,
Eyileten T, Guzel FB, Altunoren O, Erken E, Oguz Y, Kocyigit I, et
al: IL-33 and ST2 levels in chronic kidney disease: Associations
with inflammation, vascular abnormalities, cardiovascular events,
and survival. PLoS One. 12:e01789392017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ke B, Zhu N, Luo F, Xu Y and Fang X:
Targeted inhibition of endoplasmic reticulum stress: New hope for
renal fibrosis (Review). Mol Med Rep. 16:1014–1020. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fagone P, Mangano K, Mammana S, Pesce A,
Pesce A, Caltabiano R, Giorlandino A, Portale TR, Cavalli E,
Lombardo GA, et al: Identification of novel targets for the
diagnosis and treatment of liver fibrosis. Int J Mol Med.
36:747–752. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ambale-Venkatesh B, Liu CY, Liu YC,
Donekal S, Ohyama Y, Sharma RK, Wu CO, Post WS, Hundley GW, Bluemke
DA, et al: Association of myocardial fibrosis and cardiovascular
events: The multi-ethnic study of atherosclerosis. Eur Heart J
Cardiovasc Imaging. 20:168–176. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chin CWL, Everett RJ, Kwiecinski J, Vesey
AT, Yeung E, Esson G, Jenkins W, Koo M, Mirsadraee S, White AC, et
al: Myocardial fibrosis and cardiac decompensation in aortic
stenosis. JACC Cardiovasc Imaging. 10:1320–1333. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gulati A, Japp AG, Raza S, Halliday BP,
Jones DA, Newsome S, Ismail NA, Morarji K, Khwaja J, Spath N, et
al: Absence of myocardial fibrosis predicts favorable long-term
survival in new-onset heart failure. Circ Cardiovasc Imaging.
11:e0077222018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mirastschijski U, Lupše B, Maedler K,
Sarma B, Radtke A, Belge G, Dorsch M, Wedekind D, McCawley LJ,
Boehm G, et al: Matrix metalloproteinase-3 is key effector of
TNF-α-induced collagen degradation in skin. Int J Mol Sci.
20:202019. View Article : Google Scholar
|
|
35
|
Wang Q, Sui X, Chen R, Ma PY, Teng YL,
Ding T, Sui DJ and Yang P: Ghrelin ameliorates angiotensin
ii-induced myocardial fibrosis by upregulating peroxisome
proliferator-activated receptor gamma in young male rats. BioMed
Res Int. 2018:98975812018.PubMed/NCBI
|
|
36
|
Wu P, Liu Z, Zhao T, Xia F, Gong L, Zheng
Z, Chen Z, Yang T and Duan Q: Lovastatin attenuates angiotensin II
induced cardiovascular fibrosis through the suppression of YAP/TAZ
signaling. Biochem Biophys Res Commun. 512:736–741. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Berg G, Barchuk M and Miksztowicz V:
Behavior of metalloproteinases in adipose tissue, liver and
arterial wall: An update of extracellular matrix remodeling. Cells.
8:82019. View Article : Google Scholar
|
|
38
|
Taha EA, Sogawa C, Okusha Y, Kawai H, Oo
MW, Elseoudi A, Lu Y, Nagatsuka H, Kubota S, Satoh A, et al:
Knockout of MMP3 weakens solid tumor organoids and cancer
extracellular vesicles. Cancers (Basel). 12:122020. View Article : Google Scholar
|
|
39
|
Bufu T, Di X, Yilin Z, Gege L, Xi C and
Ling W: Celastrol inhibits colorectal cancer cell proliferation and
migration through suppression of MMP3 and MMP7 by the PI3K/AKT
signaling pathway. Anticancer Drugs. 29:530–538. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
McShane L, Tabas I, Lemke G,
Kurowska-Stolarska M and Maffia P: TAM receptors in cardiovascular
disease. Cardiovasc Res. 115:1286–1295. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhang LH, Pang XF, Bai F, Wang NP, Shah
AI, McKallip RJ, Li XW, Wang X and Zhao ZQ: Preservation of
glucagon-like peptide-1 level attenuates angiotensin ii-induced
tissue fibrosis by altering AT1/AT2 receptor expression and
angiotensin-converting enzyme 2 activity in rat heart. Cardiovasc
Drugs Ther. 29:243–255. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhang WW, Bai F, Wang J, Zheng RH, Yang
LW, James EA and Zhao ZQ: Edaravone inhibits pressure
overload-induced cardiac fibrosis and dysfunction by reducing
expression of angiotensin II AT1 receptor. Drug Des Devel Ther.
11:3019–3033. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Harikrishnan V, Titus AS, Cowling RT and
Kailasam S: Collagen receptor cross-talk determines α-smooth muscle
actin-dependent collagen gene expression in angiotensin
II-stimulated cardiac fibroblasts. J Biol Chem. 294:19723–19739.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Li N, Feng F, Wu K, Zhang H, Zhang W and
Wang W: Inhibitory effects of astragaloside IV on silica-induced
pulmonary fibrosis via inactivating TGF-beta1/Smad3 signaling.
Biomed Pharmacother. 119:1093872019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Weder B, Mamie C, Rogler G, Clarke S,
McRae B, Ruiz PA and Hausmann M: BCL2 regulates differentiation of
intestinal fibroblasts. Inflamm Bowel Dis. 24:1953–1966. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lossi L, Castagna C and Merighi A:
Caspase-3 mediated cell death in the normal development of the
mammalian cerebellum. Int J Mol Sci. 19:192018. View Article : Google Scholar
|
|
47
|
Wang X, Simpson ER and Brown KA: p53:
Protection against tumor growth beyond effects on cell cycle and
apoptosis. Cancer Res. 75:5001–5007. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wang JH, Zhao L, Pan X, Chen NN, Chen J,
Gong QL, Su F, Yan J, Zhang Y and Zhang SH: Hypoxia-stimulated
cardiac fibroblast production of IL-6 promotes myocardial fibrosis
via the TGF-β1 signaling pathway. Lab Invest. 96:839–852. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Yao C, Cao X, Fu Z, Tian J, Dong W, Xu J,
An K, Zhai L and Yu J: Boschniakia rossica polysaccharide
triggers laryngeal carcinoma cell apoptosis by regulating
expression of Bcl-2, Caspase-3, and P53. Med Sci Monit.
23:2059–2064. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Tsutsui H, Kinugawa S and Matsushima S:
Oxidative stress and heart failure. Am J Physiol Heart Circ
Physiol. 301:H2181–H2190. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Singh S and Torzewski M: Fibroblasts and
their pathological functions in the fibrosis of aortic valve
sclerosis and atherosclerosis. Biomolecules. 9:92019. View Article : Google Scholar
|
|
52
|
Wang M, Kim SH, Monticone RE and Lakatta
EG: Matrix metalloproteinases promote arterial remodeling in aging,
hypertension, and atherosclerosis. Hypertension. 65:698–703. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Zhang M, Xue Y, Chen H, Meng L, Chen B,
Gong H, Zhao Y and Qi R: Resveratrol inhibits MMP3 and MMP9
expression and secretion by suppressing TLR4/NF-κB/STAT3 activation
in Ox-LDL-treated HUVECs. Oxid Med Cell Longev.
2019:90131692019.PubMed/NCBI
|
|
54
|
Szczepanek K, Chen Q, Larner AC and
Lesnefsky EJ: Cytoprotection by the modulation of mitochondrial
electron transport chain: The emerging role of mitochondrial STAT3.
Mitochondrion. 12:180–189. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Han J, Ye S, Zou C, Chen T, Wang J, Li J,
Jiang L, Xu J, Huang W, Wang Y, et al: Angiotensin II causes
biphasic STAT3 activation through TLR4 to initiate cardiac
remodeling. Hypertension. 72:1301–1311. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Yim J, Cho H and Rabkin SW: Gene
expression and gene associations during the development of heart
failure with preserved ejection fraction in the Dahl salt sensitive
model of hypertension. Clin Exp Hypertens. 40:155–166. 2018.
View Article : Google Scholar : PubMed/NCBI
|