Introduction
Worldwide, breast cancer (BRCA) is the second most
common cause of cancer-associated mortality in women, and it has a
high incidence rate in China (1,2).
Triple negative breast cancer (TNBC) refers to a type of BRCA where
patients lack human epidermal growth factor receptor 2 (HER2), ER
estrogen receptors (ER) and progesterone receptors (PR), and this
subtype is characterized by large visceral metastatic spread and
increased rate of nodal invasion (3). Due to the metastasis of BRCA,
particularly in TNBCs, the prognosis remains poor. Therefore, it is
necessary to further identify the molecular mechanism underlying
metastasis in BRCA, particularly in TNBCs.
Checkpoint with FHA and RING finger domains (CHFR)
serves a key role in regulating the cell cycle by regulating the
transition to metaphase in reaction to microtubule stress (4). In a previous study, CHFR was revealed
to be significantly downregulated by promoter methylation or
mutation in gastric cancer (5),
human non-small cell lung carcinoma (NSCLC) (6), and esophageal (7) and colorectal cancer (8). However, aberrant hypermethylation of
the CHFR promoter is uncommon in primary BRCA (9). However, the role of CHFR in metastasis
in BRCA is yet to be characterized.
Cancer cell metastasis is a multistep process
involving proliferation, epithelial-to-mesenchymal transition
(EMT), migration and invasion (10,11).
EMT was originally considered to be a growth-like process, during
which epithelial cells exhibit a migratory and invasive mesenchymal
phenotype (12). A hallmark of EMT
is the functional loss of the epithelial maker E-cadherin and the
upregulation of the mesenchymal markers N-cadherin, vimentin and
fibronectin (13).
In the present study, according to The Cancer Genome
Atlas (TCGA) database, CHFR is upregulated in BRCA tissues compared
with normal tissues. In addition, subclass analysis of BRCA
revealed that CHFR is upregulated in HER2+ and TNBC.
Notably, patients with higher levels of CHFR exhibited poorer
overall survival rates. However, the biological function of CHFR on
the metastasis of BRCA is yet to be elucidated. The current data
revealed that overexpression of CHFR in SKBR3 cells resulted in
enhanced migratory and invasive abilities, and also significant
upregulation of mesenchymal markers, such as N-cadherin, vimentin,
transcription factor Slug and tight junction protein claudin-1.
Furthermore, knockdown of CHFR in MDA-MB-231 cells significantly
inhibited migratory and invasive abilities, and also downregulated
mesenchymal markers, such as N-cadherin, vimentin and tight
junction protein claudin-1. In conclusion, the current results
indicated that CHFR enhanced cell metastasis in BRCA by mediating
EMT. Moreover, the present study indicated that CHFR may provide a
potential therapeutic target for metastatic BRCA treatment.
Materials and methods
Cell culture
All BRCA cell lines cells (SKBR3, MDA-MB-231 and
MCF-7) were purchased from the American Type Culture Collection.
All cells were incubated in Dulbecco's modified Eagle's medium
(DMEM; Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10%
fetal bovine serum (FBS; Hyclone; Cytiva), 2 mM L-glutamine (Gibco;
Thermo Fisher Scientific, Inc.), 1% penicillin (100 U/ml) and
streptomycin (100 µg/ml) (Gibco; Thermo Fisher Scientific, Inc.)
and incubated at 37°C, 5% CO2 in a humidified incubator
and passaged at ≥80% confluence using trypsin (Gibco; Thermo Fisher
Scientific, Inc.).
Western blotting
Cells were lysed in RIPA buffer containing 1%
protease inhibitor cocktail (Sigma-Aldrich; Merck KGaA). The
supernatants of lysates were collected and concentrations of
protein were quantified with the Protein Quantitative Kit (TransGen
Biotech Co., Ltd.) using a microplate reader (Molecular Devices,
LLC). Then, ~50 µg protein was loaded onto a 10% gel, and separated
via SDS-PAGE, then separated proteins were transferred onto
polyvinylidene difluoride membranes. The membranes were blocked
with 5% non-fat milk at room temperature for 2 h, and then
incubated at 4°C overnight with primary antibodies against CHFR
(cat. no. 904S; 1:1,000), N-cadherin (cat. no. 13116; 1:1,000),
β-catenin (cat. no. 8480; 1:1,000), vimentin (cat. no. 5741;
1:1,000), Snail (cat. no. 3879; 1:1,000), Slug (cat. no. 9585;
1:1,000), claudin-1 (cat. no. 4933; 1:1,000) and E-cadherin (cat.
no. 3195; 1:500), all from Cell Signaling Technology, Inc., as well
as β-actin (cat. no. 2228; 1:5,000), which was purchased from
Sigma-Aldrich (Merck KGaA). To determine transfection efficiency
following CHFR knockdown, a different antibody against CHFR was
used (cat. no. 12169-1-AP; 1:500), which was purchased from
ProteinTech Group, Inc. The corresponding anti-rabbit IgG (cat. no.
HS101-01; 1:2,000) and anti-mouse IgG (cat. no. HS201-01; 1:2,000)
horseradish peroxidase (HRP)-conjugated secondary antibodies
(TransGen Biotech Co., Ltd.) was added and incubated at room
temperature for 1 h. Signals were visualized after an
electrochemiluminescence reaction with HRP substrate (cat. no.
P0018S; Beyotime Institute of Biotechnology) and semi-quantified
using ImageJ (version 1.52v; National Institutes of Health).
Transfection and RNA interference of
CHFR
Small interfering (si)RNAs targeting CHFR
(5′-CACCACGCCAUGAAAUUCATT-3′) and non-targeting siRNA negative
controls (5′-UUCUCCGAACGUGUCACGU-3′) were obtained from Santa Cruz
Biotechnology, Inc. MDA-MB-231 cells were seeded into a 6-well
plate at 1×105 and transfected with 4.0 µg siRNA using
Lipofectamine® 2000 reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions.
Prior to any treatment, cells were incubated for 24 h and the
transfection efficiency of the siRNA was determined via western
blotting.
Plasmid construction and
transfection
The coding sequences of human CHFR mRNA were
synthesized and subcloned into the pcDNA3.1 vector (cat. no.
128034; Addgene, Inc.) to construct the CHFR overexpression
plasmid. The integrity of the respective plasmid constructs was
confirmed via DNA sequencing. When SKBR3 cells reached 75%
confluency in the 6-well plate, cells were used to overexpress
CHFR. A complex was formed between the 4.0 µg plasmid and
Lipofectamine for 20 min at room temperature, and transfection was
carried out at 37°C for 24 h. Then, 800 µg/ml G418 was used to
select cells transfected with pcDNA3.1 and CHFR overexpression
plasmid for 48 h. Subsequently, the cells were cultured with 400
µg/ml G418 for maintenance. The cells transfected with pcDNA3.1
vector and CHFR plasmid were defined as the control group and CHFR
group, respectively.
In vitro migration and invasion
assays
For the migration assay, Transwell inserts (24
wells; 8-µm pore size; poly-carbonate membrane; Corning, Inc.) were
used according to the manufacturer's protocol. Cells were
transfected with plasmids (pcDNA3.1 and CHFR plasmids) and siRNA
(siR-control and siR-CHFR), and the cells were seeded into the
upper chambers at 1×105/chamber and cultured in
serum-free DMEM. The lower compartment was filled with DMEM, with
10% FBS used as a chemoattractant. After incubation for 24 h, cells
remaining in the upper chamber were removed, and cells at the
bottom of the insert were fixed with 4% paraformaldehyde for 30 min
at room temperature, stained in 0.5% crystal violet for 20 min at
room temperature and counted under a light microscope
(magnification, ×400; Olympus Corporation). The results were
averaged over three independent experiments. For invasion assays,
the inserts were coated with Matrigel (BD Biosciences) at 37°C for
4 h before the cells were added. The proceeding steps were the same
as migration assay.
Cell proliferation assay
After CHFR overexpression or silencing, cells at a
density of 1,000/well were seeded in a 96-well plate and incubated
for the indicated times (24, 48, 72 and 96 h). The medium was
discarded and cells were incubated with 50 ml of 1 mg/ml MTT
(Sigma-Aldrich; Merck KGaA) in PBS for up to 4 h at 37°C. The
purple formazan was then solubilized by DMSO and absorbance at 570
nm was read by a microplate reader (Molecular Devices, LLC).
Morphological analysis
Cells were transfected with pcDNA3.1 and
pcDNA3.1-CHFR plasmids. Then, 48 h after transfection, the
morphology of the cells was observed with an inverted microscope
(CKX53; Olympus Corporation).
Survival analysis
The samples were divided into two groups based on
the expression of CHFR. The expression of CHFR was listed in
ascending order, the patients in whom expression of CHFR was
<the median were defined as low expression groups; otherwise,
the patients were defined as high expression groups. The clinical
relevance of CHFR in patients with BRCA was analyzed using the
UALCAN database (14) and
Kaplan-Meier plotter (www.KMplot.com). The gene symbol chosen was CHFR
(Affymetrix ID no.223931_s_at). Patients were split by auto select
best cutoff, and to restrict the analysis into subtypes, patients
negative for PR, HER2 and lymph node status were chosen. Then, the
Kaplan-Meier plot was constructed, and the overall survival of
patients with TNBC was obtained using a log-rank test.
Statistical analysis
All data are expressed as the mean ± SD from at
least three independent experiments. All statistical analyses were
performed using GraphPad Prism 5.0 (GraphPad Software, Inc.) and
SPSS 13.0 (SPSS, Inc.) software packages. Statistical significance
between two groups was determined using the two-sided Student's
t-test, and for multiple group comparisons an ANOVA followed by
Bonferroni's post hoc test was performed. P<0.05 was considered
to indicate a statistically significant difference.
Results
CHFR expression analysis in BRCA
dataset
Data from TCGA was used to determine the clinical
relevance of CHFR expression in human BRCA, and the results
revealed that CHFR mRNA was upregulated in BRCA tissues compared
with normal tissues (Fig. 1A).
In addition, subgroup analysis of BRCA revealed that
CHFR expression was upregulated in HER2+ and TNBC types
compared with the normal subclass (Fig.
1B). Notably, patients with higher levels of CHFR exhibited
poorer overall survival rates in patients with TNBC (Fig. 1C). Taken together, these data
indicated that CHFR is significantly upregulated in BRCA, and
exerts a significant pro-tumor effect.
CHFR overexpression enhances migratory
and invasive abilities of BRCA cells, and inhibits cell
proliferation
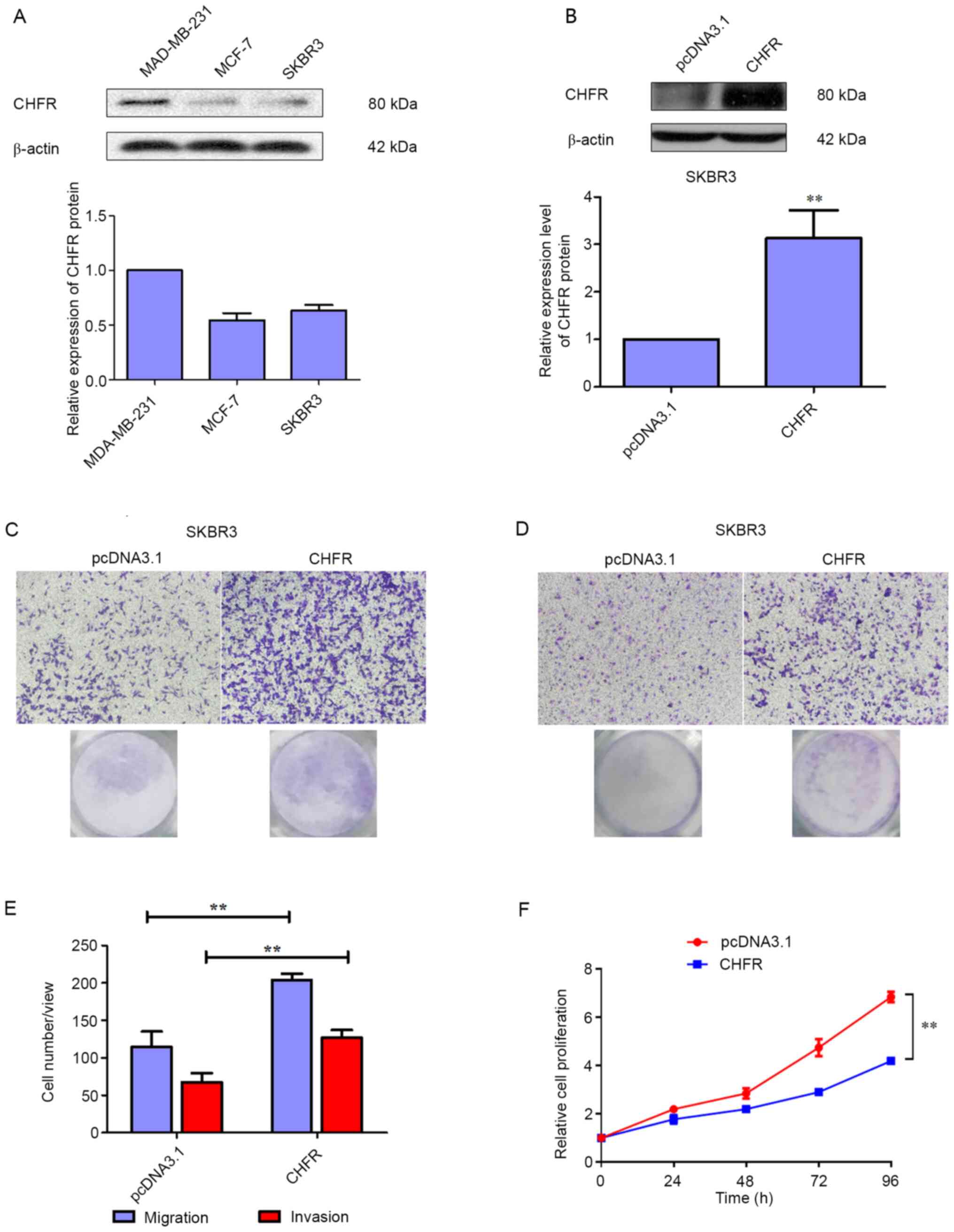
To further investigate the role of CHFR in BRCA,
three human BRCA cell lines were selected, and their basal
expression of CHFR was detected via a western blot assay. As
depicted in Fig. 2A, CHFR
expression was higher in MAD-MB-231 cells, compared with MCF-7 and
SKBR3 cells. Thus, SKBR3 cells were transfected with an expression
plasmid of CHFR to study the biological role of CHFR.
Firstly, the transfection efficiency was
investigated, and the results demonstrated that CHFR levels were
significantly upregulated in SKBR3 cells that were transfected with
a CHFR expression plasmid (Fig.
2B), and overexpression of CHFR significantly increased cell
migration compared with the control group at 24 h (Fig. 2C and E). In addition, as displayed
in Fig. 2D and E, overexpression of
CHFR significantly increased the number of invaded cells compared
with the control group at 24 h. Therefore, the current data
demonstrated that CHFR positively regulates BRCA cell migration and
invasion. However, CHFR overexpression significantly suppressed the
proliferative activity of SKBR3 cells (Fig. 2F).
CHFR knockdown inhibits the migratory
and invasive abilities of BRCA cells, and promotes cell
proliferation
To further verify the effects of CHFR on migration
and invasion in BRCA, MAD-MB-231 cells were transfected with siRNA
to knockdown the expression of CHFR. Firstly, the transfection
efficiency was evaluated and the results revealed that CHFR levels
were significantly decreased in MAD-MB-231 cells that were
transfected with CHFR siRNA (Fig.
3A), and knockdown of CHFR significantly reduced cell migration
compared with the control group at 24 h (Fig. 3B and D). In addition, as indicated
in Fig. 3C and D, knockdown of CHFR
significantly decreased cell invasion compared with the control
group at 24 h. Therefore, the current data also demonstrated that
knockdown of CHFR negatively regulated BRCA cell migration and
invasion. On the other hand, CHFR knockdown promoted the
proliferation of MDA-MB-231 cells (Fig.
3E).
CHFR may promote cell metastasis via
EMT in BRCA cells
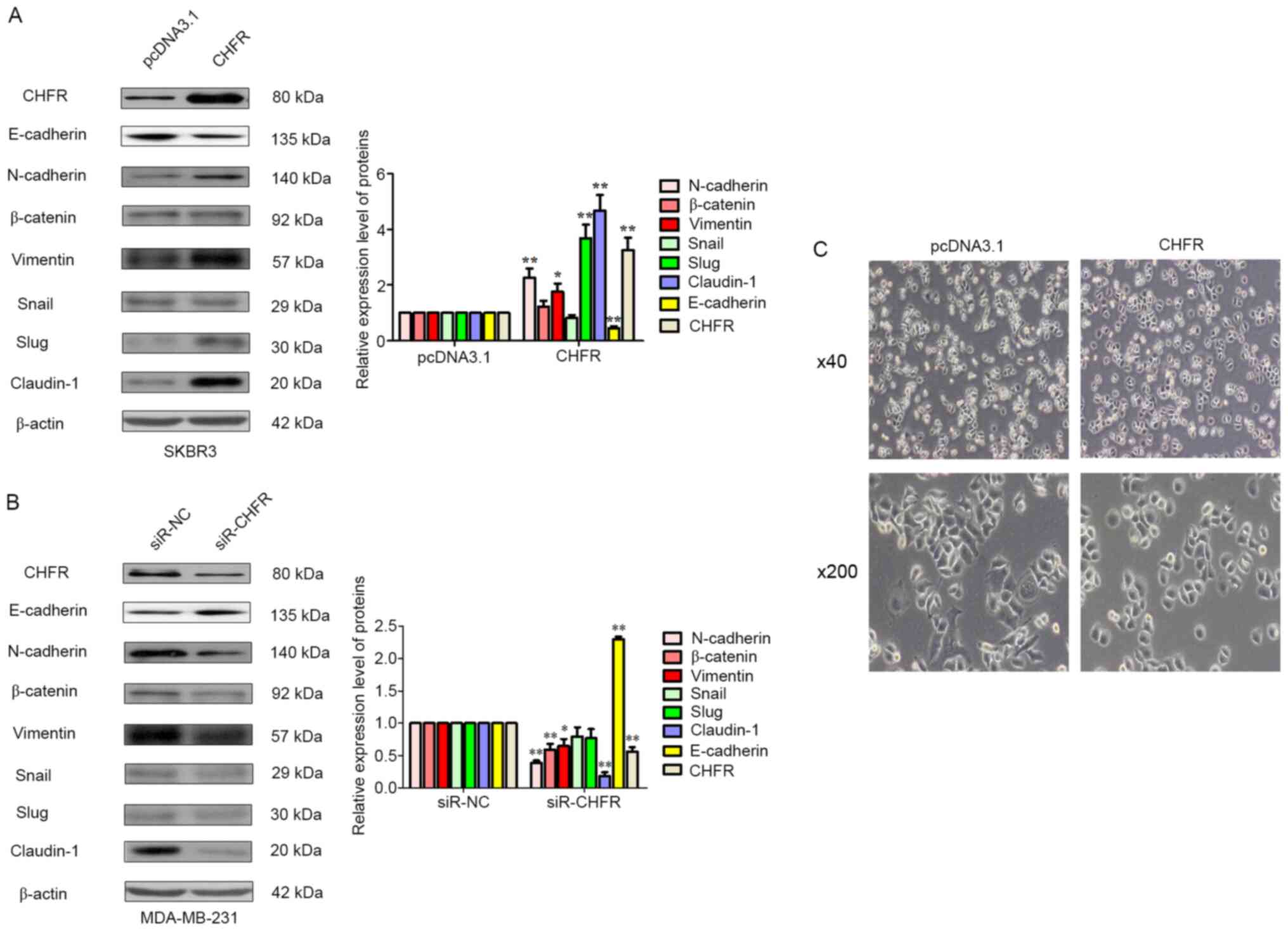
To investigate the underlying mechanisms behind the
role of CHFR in the regulation of cell metastasis in BRCA cells,
mesenchymal markers were examined, such as N-cadherin, vimentin,
transcription factors Slug and Snail, and tight junction proteins
E-cadherin, claudin-1 and β-catenin. Initially, as displayed in
Fig. 4A ectopic expression of CHFR
was evaluated, and the results revealed that overexpression of CHFR
significantly upregulated the mesenchymal markers N-cadherin,
vimentin and its transcription factor Slug, and tight junction
protein claudin-1. But, CHFR overexpression significantly
suppressed the expression of E-cadherin, an epithelial cell marker.
Furthermore, using RNA interference technology, the expression of
CHFR was knocked down, which resulted in the reduction of
N-cadherin, vimentin and claudin-1 expression, and upregulation of
the expression of epithelial marker E-cadherin (Fig. 4B). Finally, the morphological change
of SKBR3 cells following CHFR overexpression was also examined. As
shown in Fig. 4C, after CHFR
overexpression, mesenchymal cells that were rounded became more
polygon, which is more favorable for EMT. In other words, more
mesenchymal characteristics could be observed in SKBR3 cells when
CHFR was overexpressed compared with the control. These results
combined indicated that CHFR-mediated EMT promoted human BRCA cell
metastasis.
Discussion
CHFR is a G2 phase/mitosis checkpoint
protein that works by promoting the degradation of target proteins,
such as PARP-1, to delay entry into metaphase depending on its
E3-ubiquitin ligase activity (4,15).
Inactivation of CHFR in numerous tumors was revealed to result from
methylated CpG islands on its promotor region (16). Although CHFR is a frequent target of
novel promoter hypermethylation in other cancer types, such as
colorectal and esophageal cancer, it is significantly less frequent
in NSCLC, and independently associated with a poor outcome in acute
myeloid leukemia (17–20). However, aberrant hypermethylation of
the CHFR promoter is uncommon in primary BRCA (9).
In the current study, the role of CHFR in the
metastasis of BRCA cells was investigated. According to data
retrieved from TCGA, CHFR was upregulated in BRCA tissues compared
with normal tissues. In addition, CHFR was upregulated in
HER2+ and TNBC subtypes. Notably, patients with TNBC
with higher levels of CHFR exhibited poorer overall survival rates
compared with patients in the low CHFR expression group. Therefore,
the aforementioned summarized data indicated that CHFR expression,
and not its promoter hypermethylation, may represent a biomarker
able to predict a poorer therapeutic response in patients with the
HER2+ or TNBC subtypes of BRCA. However, the effect and
mechanism underlying the role of CHFR expression in the regulation
of TNBC metastasis is yet to be elucidated.
TNBC is a highly aggressive subclass, accounting for
~10–20% of all BRCA diagnoses (21). Due to poor overall survival, early
relapse and distant metastasis, TNBC clinical treatment of BRCA
represents a notable challenge (21,22). A
hallmark of cancer is abnormal activation of EMT, and this is
associated with the metastasis of TNBC (23). EMT was originally speculated to be a
growth process, during which epithelial cells display a migratory
and invasive mesenchymal phenotype (12). From a molecular perspective, EMT is
characterized by downregulation of the epithelial cell marker
E-cadherin, and the upregulation of mesenchymal cell markers
vimentin and N-cadherin (24). The
majority of these regulate various transcription factors implicated
in EMT, such as Snail, Slug and zinc finger E-box-binding homeobox
1 (25). Previous studies have
reported that there are four major epigenetic factors that regulate
EMT in TNBC and are responsible for distant metastases, comprising
long non-coding and microRNAs, and acetylation or methylation of
histones or DNA (22). In the
current study, there were two bands of CHFR in the MDA-MB-231 cells
with siR-CHFR. The CHFR antibody used in the siR-CHFR transfection
was different from the other CHFR antibody batches. It is possible
that the specificity of the antibody was inferior for the siR-CHFR
experiment, which could explain the presence of the two CHFR bands
on the western blots in the siR-CHFR MDA-MB-231 cells.
Overexpression of CHFR in SKBR3 cells significantly upregulated the
expression of mesenchymal markers N-cadherin, vimentin and its
transcription factor Slug, and tight junction protein claudin-1,
while downregulated the expression of epithelial cell marker
E-cadherin. As expected, silencing of CHFR in MDA-MB-231 decreased
the expression of mesenchymal markers N-cadherin, vimentin and
transcription factor Slug, while upregulated the expression of
epithelial cell marker E-cadherin. Although there is little
publication concerning how CHFR influences the EMT of cancer cells,
especially in human BRCA, we speculate that the E3-ubiquitin ligase
activity might contribute this function. Therefore, affinity
purification of CHFR combined with mass spectrometry will be
perform in the future to determine the underlying mechanism for its
regulation in EMT of BRCA cells. Overall, the current data
demonstrated that CHFR modulated the metastasis of BRCA cells via
mediating EMT.
As cell migration and invasion are important
components of cell metastasis, the observed effects of CHFR on BRCA
cell migration and invasion revealed that it may also affect cell
metastasis. One characteristic of malignancy is increased cell
motility. Using a Transwell assay, with or without Matrigel, it was
revealed that overexpression of CHFR significantly promoted BRCA
cell SKBR3 migration and invasion, while knockdown of CHFR notably
inhibited the rate of BRCA cell MDA-MB-231 motility. More
importantly, ectopic expression of CHFR effectively impaired the
cell proliferation of SKBR3 cells, while silencing of CHFR
significantly enhanced the proliferation of MDA-MB-231 cells. These
findings indicated that exogenous CHFR successfully acted as a cell
cycle checkpoint. Taken together, the present data is consistent
with a previous study, which focused on the role of CHFR in human
gastric cancer cells (26). In a
previous study, reduced CHFR expression was found to lead to a
notable increase in population growth and a higher percentage of
mitotic cells when observed in vitro. Importantly, reduced
CHFR expression resulted in an increase in the number of mitotic
(metaphase and anaphase) cells in the population. Reduced CHFR
expression resulted in the acquisition of a number of phenotypes
associated with malignant progression, including increased growth
rate, increased mitotic index, increased invasion, increased
motility, increased aneuploidy and increased colony formation in
soft agar, further supporting the role of CHFR in cancer (27).
In conclusion, the current findings indicated that
CHFR was upregulated in HER2+ and TNBC subclasses of
BRCA. In addition, patients with higher levels of CHFR exhibited
poorer overall survival rates. Notably, CHFR was found to function
as a novel oncogene to regulate the metastasis of BRCA cells via
mediating EMT. Therefore, CHFR may represent a novel molecular
therapeutic target for the treatment of BRCA, via regulation of
metastatic mechanisms.
Acknowledgements
Not applicable.
Funding
The present study was supported by Zhejiang medical
association (grant no. 2018ZYC-A14) and Technology Division of
Taizhou (grant no. 14SF07).
Availability of data and materials
The datasets used and/or analysed during the current
study are available from the corresponding author on reasonable
request. The results published here are in part based upon data
generated by the TCGA Research Network: https://www.cancer.gov/tcga.
Authors' contributions
GJ and FC conceived and designed the experiments.
GJ, XC and FC confirmed the authenticity of all the raw data. GJ,
XS and HF performed the experiments. GJ and XC analyzed the data.
GJ and FC wrote the manuscript. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Al-Bahlani S, Al-Lawati H, Al-Adawi M,
Al-Abri N, Al-Dhahli B and Al-Adawi K: Fatty acid synthase
regulates the chemosensitivity of breast cancer cells to
cisplatin-induced apoptosis. Apoptosis. 22:865–876. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Scolnick DM and Halazonetis TD: Chfr
defines a mitotic stress checkpoint that delays entry into
metaphase. Nature. 406:430–435. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ding Y, Lian HF and Du Y:
Clinicopathological significance of CHFR promoter
methylation in gastric cancer: A meta-analysis. Oncotarget.
9:10083–10090. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mizuno K, Osada H, Konishi H, Tatematsu Y,
Yatabe Y, Mitsudomi T, Fujii Y and Takahashi T: Aberrant
hypermethylation of the CHFR prophase checkpoint gene in human lung
cancers. Oncogene. 21:2328–2333. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shibata Y, Haruki N, Kuwabara Y, Ishiguro
H, Shinoda N, Sato A, Kimura M, Koyama H, Toyama T, Nishiwaki T, et
al: Chfr expression is downregulated by CpG island hypermethylation
in esophageal cancer. Carcinogenesis. 23:1695–1699. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Glinsky GV, Berezovska O and Glinskii AB:
Microarray analysis identifies a death-from-cancer signature
predicting therapy failure in patients with multiple types of
cancer. J Clin Invest. 115:1503–1521. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tokunaga E, Oki E, Nishida K, Koga T,
Yoshida R, Ikeda K, Kojima A, Egashira A, Morita M, Kakeji Y and
Maehara Y: Aberrant hypermethylation of the promoter region of the
CHFR gene is rare in primary breast cancer. Breast Cancer Res
Treat. 97:199–203. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Vanharanta S and Massague J: Origins of
metastatic traits. Cancer Cell. 24:410–421. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Acloque H, Adams MS, Fishwick K,
Bronner-Fraser M and Nieto MA: Epithelial-mesenchymal transitions:
The importance of changing cell state in development and disease. J
Clin Invest. 119:1438–1449. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zheng H, Shen M, Zha YL, Li W, Wei Y,
Blanco MA, Ren G, Zhou T, Storz P, Wang HY and Kang Y: PKD1
phosphorylation-dependent degradation of SNAIL by SCF-FBXO11
regulates epithelial-mesenchymal transition and metastasis. Cancer
Cell. 26:358–373. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chandrashekar DS, Bashel B, Balasubramanya
SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi BVSK and
Varambally S: UALCAN: A portal for facilitating tumor subgroup gene
expression and survival analyses. Neoplasia. 19:649–658. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kashima L, Idogawa M, Mita H, Shitashige
M, Yamada T, Ogi K, Suzuki H, Toyota M, Ariga H, Sasaki Y and
Tokino T: CHFR protein regulates mitotic checkpoint by targeting
PARP-1 protein for ubiquitination and degradation. J Biol Chem.
287:12975–12984. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Derks S, Cleven AH, Melotte V, Smits KM,
Brandes JC, Azad N, van Criekinge W, de Bruïne AP, Herman JG and
van Engeland M: Emerging evidence for CHFR as a cancer biomarker:
from tumor biology to precision medicine. Cancer Metastasis Rev.
33:161–171. 2014.PubMed/NCBI
|
|
17
|
Kawasaki T, Ohnishi M, Nosho K, Suemoto Y,
Kirkner GJ, Meyerhardt JA, Fuchs CS and Ogino S: CpG island
methylator phenotype-low (CIMP-low) colorectal cancer shows not
only few methylated CIMP-high-specific CpG islands, but also
low-level methylation at individual loci. Mod Pathol. 21:245–255.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Soutto M, Peng D, Razvi M, Ruemmele P,
Hartmann A, Roessner A, Schneider-Stock R and El-Rifai W:
Epigenetic and genetic silencing of CHFR in esophageal
adenocarcinomas. Cancer. 116:4033–4042. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pillai RN, Brodie SA, Sica GL, Shaojin Y,
Li G, Nickleach DC, Yuan L, Varma VA, Bonta D, Herman JG, et al:
CHFR protein expression predicts outcomes to taxane-based first
line therapy in metastatic NSCLC. Clin Cancer Res. 19:1603–1611.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gao L, Liu F, Zhang H, Sun J and Ma Y:
CHFR hypermethylation, a frequent event in acute myeloid leukemia,
is independently associated with an adverse outcome. Genes
Chromosomes Cancer. 55:158–168. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
He MY, Rancoule C, Rehailia-Blanchard A,
Espenel S, Trone JC, Bernichon E, Guillaume E, Vallard A and Magné
N: Radiotherapy in triple-negative breast cancer: Current situation
and upcoming strategies. Crit Rev Oncol Hematol. 131:96–101. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Khaled N and Bidet Y: New insights into
the implication of epigenetic alterations in the EMT of triple
negative breast cancer. Cancers (Basel). 11:5592019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Neelakantan D, Zhou H, Oliphant MUJ, Zhang
X, Simon LM, Henke DM, Shaw CA, Wu MF, Hilsenbeck SG, White LD, et
al: EMT cells increase breast cancer metastasis via paracrine GLI
activation in neighbouring tumour cells. Nat Commun. 8:157732017.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nieto MA, Huang RY, Jackson RA and Thiery
JP: Emt: 2016. Cell. 166:21–45. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hinz S and LaBarge MA: Hijacking EMT:
Better fat than dead. Cancer Cell. 35:1–2. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yang S, He F, Dai M, Pan J, Wang J and Ye
B: CHFR promotes the migration of human gastric cancer cells by
inducing epithelial-to-mesenchymal transition in a HDAC1-dependent
manner. OncoTargets Ther. 12:1075–1084. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Privette LM, González ME, Ding L, Kleer CG
and Petty EM: Altered expression of the early mitotic checkpoint
protein, CHFR, in breast cancers: Implications for tumor
suppression. Cancer Res. 13:6064–6074. 2007. View Article : Google Scholar : PubMed/NCBI
|